医疗器械质量手册

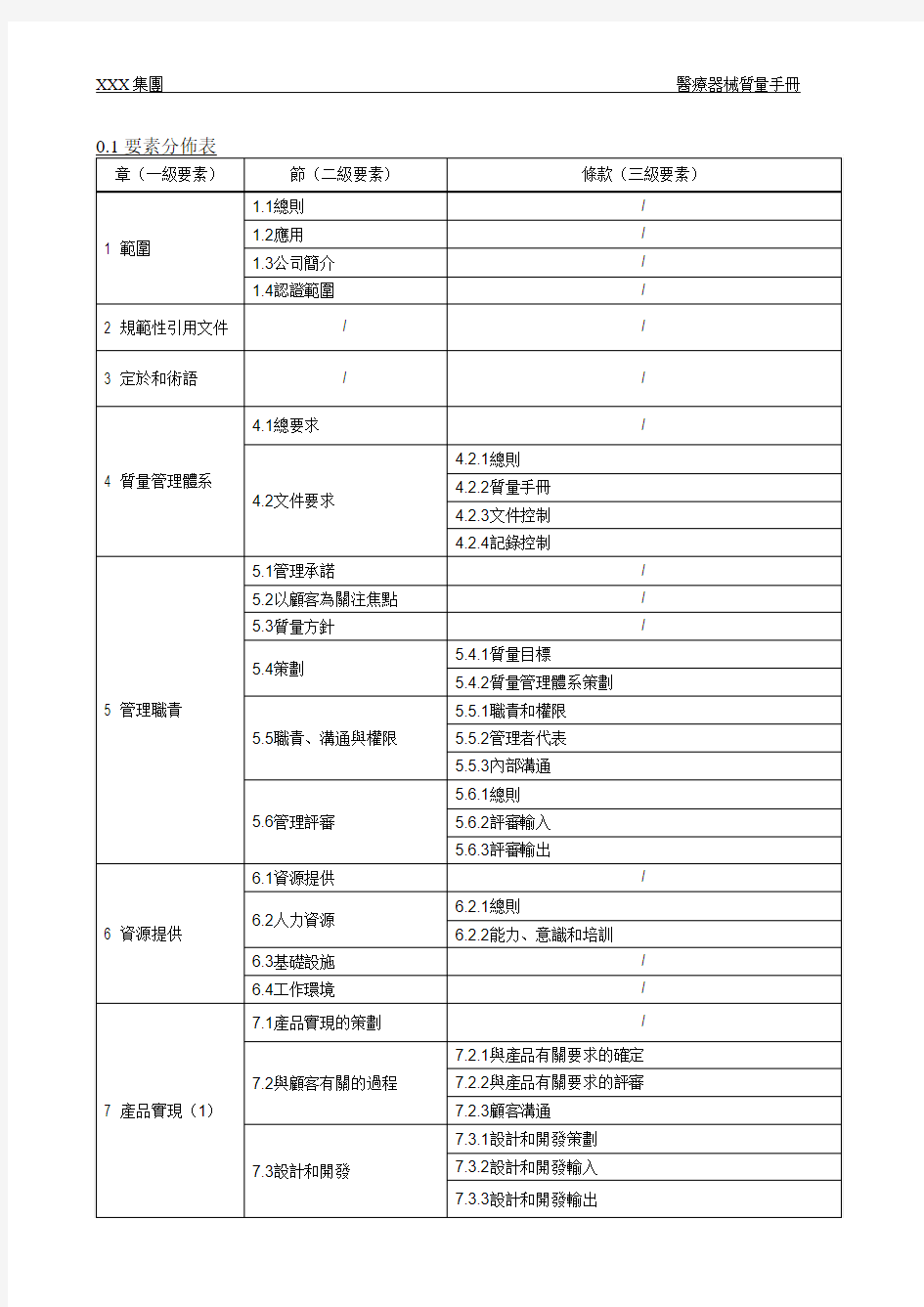
1 範圍(ISO13485第1章)
1.1總則(ISO13485第1.1節)
本質量手冊作為除無菌醫療器械、有源植入性醫療器械和植入性醫療器械以外的其他一般醫療器械的設計和製造的總要求,本手冊旨在向本公司各級人員和相關方明確傳達和明確要求關於醫療器械的設計和製造有關的質量管理體系要求。
1.2 應用(ISO13485第1.2節)
本公司設計和製造的產品屬於一、二類醫療器械,為了滿足法律法規和客戶的要求,本公司根據【ISO13485醫療器械質量管理體系用於法規的要求】建立醫療器械質量管理體系,並在公司內貫徹實施、保持醫療器械質量管理體系。
本公司設計和製造的產品不屬於無菌醫療器械、有源植入性醫療器械和植入性醫療器械,故本手冊刪減標準中的無菌醫療器械專用要求(7.5.1.3、7.5.2.2)、有源植入性醫療器械和植入性醫療器械專用要求(7.5.3.2.2、8.2.4.2)。
1.3公司簡介
XXX有限公司成立於1977年,擁有大約240名員工。成立初期是一間以玩具製造為主的生產型企業,現在已發展成一間以提供多元化的玩具、嬰兒用品及家用電器的設計開發、工程控制、採購、提供產品的質量管理及生產計畫控制的管理型企業。今日的XXX集團擁有包括:搪膠、注塑、車發、植毛、電鍍、噴油、移印、絲印、電子、SMT、邦定、裝配、嬰兒用品生產、制衣、啤塑、雕刻、精密加工等各類型生產車間,可為客人提供多元化的玩具、家庭用品、醫療器械、模具的製造及服務。本集團最高管理者鄭重承諾:嚴格遵守並執行國家及產品銷售地的法律法規和相關的行業標準及規範。公司通過對質量目標、設計和開發、加工商評估、採購產品驗證、生產和服務等過程進行監視和測量(詳見本質量手冊及工作指引等相關文件),以確保向顧客提供高質量的產品達至顧客的滿意並實現產品質量的不斷持續改進。
2 規範性引用文件(ISO13485第2章)
本公司的醫療器械質量管理體系(MDQMS),及【醫療器械質量手冊】(MDQM)主要引用的文件包括以下:
a. ISO13485:2003 Medical devices-Quality management systems-Requirements for
regulatory purposes
b. BS EN ISO13485:2012 Medical devices-Quality management systems-Requirements for
regulatory purposes
c. YY/T 0287-2003/ISO 13485:2003醫療器械質量管理體系用於法規的要求
d. YY/T 0316-2008/ISO 14791:2007醫療器械風險管理對醫療器械的應用
e. ISO9001:2008 質量管理體系要求
f. ISO9000:2008 質量管理體系基礎和術語
g.XXX集團質量手冊
MDQM、MDOP所引標準號ISO13485,其等同於ISO13485:2003、BS EN ISO13485:2012和YY/T 0287-2003。
MDQM、MDOP所引標準號ISO9001、ISO9000,其等同於ISO9001:2008、ISO9001:2008。
3.術語和定義(ISO13485第3章)
本手冊的術語和定義包括下列各款以及本公司的質量手冊和ISO9000標準含有的其他術語和定義
3.1.有源植入性醫療器械
任何通過外科或內科手段,擬部份或全部插入人體,或通過醫療手段介入自然腔口且擬留在體內的有源醫療器械。
3.2.有源醫療器械
任何依靠電能或其他能源而不是直接由人體或重力產生的能源來發揮其功能的醫療器械。
3.3.植入性醫療器械
任何通過外科手段來達到下列目的的醫療器械:
--- 全部或部份插入人體或自然腔口中;或
--- 為替代上表皮或眼表皮用的;
並且使其在體內至少存留30天,且只能通過內科或外科的手段取出。
注:該定義適用於植入性醫療器械,而不適用於有源植入性醫療器械。
3.4.醫療器械
產品的預期用途是為下列一個或多個特定目的的用於人類的,不論單獨使用或組合使用的儀器、設備、器具、機器、用具、植入物、體外試劑或校準物、軟體、材料或者其他相似或相關物品。
這些目的是:
--- 疾病的診斷、預防、監護、治療或者緩解;
--- 損傷的診斷、監護、治療、緩解或者補償;
--- 解剖或生理過程的研究、替代、調節或者支持;
--- 支援或維持生命;
--- 妊娠控制;
--- 醫療器械的消毒;
--- 通過對取自人體的樣本進行體外檢查的方式來提供醫療信息。
其作用於人體體表或體內的主要設計作用不是用藥理學。免疫學或代謝的手段獲得,但可能有這些手段參與並起一定輔助作用
3.5.無菌醫療器械
旨在滿足無菌要求的醫療器械類別。
注:對醫療器械無菌的要求,可按國家或地區的法規或標準執行。
3.6. MDQMS
醫療器械質量管理體系medical devices-quality management system的簡稱。
3.7. MDQM
醫療器械質量手冊medical devices-quality manual的簡稱。
3.8. MDOP
醫療器械操作程序medical devices-operation procedure的簡稱。
3.9. MDWI
醫療器械工作指引medical devices-work instruction的簡稱。
3.10.S&C部
集團系統管理部system and compliance department的簡稱
4.質量管理體系(ISO13485第4章)
4.1.總要求(ISO13485第4.1節)
本公司按ISO13485標準建立公司的MDQMS(醫療器械質量管理體系)。銷售目的地為中國大陸地區的3C產品,除了應滿足ISO13485的要求外,還應滿足我國有關3C產品的法律法規、部門規章、國家標準和行業標準等要求,如中國強制性產品認證工廠保證能力要求。
本公司及各職能部門應按照本手冊的要求實施、保持MDQMS的有效性,並確保滿足以下要求:
a. MDQMS所需的過程包括管理職責、資源管理、產品實現、測量分析改進等過程,這些過
程的適用範圍和應用本手冊第1.2條。
b. 在本MDQMS所需的管理職責、資源管理、產品實現、測量分析改進等四大過程中:管理
職責和資源管理為產品實現提供軟體和硬體上的支援,是產品實現的基本保障;產品實現過
程基於管理職責和資源管理的支援,是質量管理體系運行的核心過程,其也需要從測量、分
析和改進過程提供可靠性的資訊進行改進。
與產品有關的過程由工程部根據產品設計開發階段根據本手冊第7.1~7.2節的要求以及產
品的實際情況作出確定,這些過程的順序和相互作用根據不同的產品而定。產品實現過程的
一般流程:設計開發→原材料和零配件採購→來料驗證→注塑或/和搪膠或/和車縫或/和部份
工序外包→制程巡檢或/和半製品檢驗或/和外包件驗證→噴油或/和移印或/和電鍍→制程巡
檢或/和半製品檢驗→SMT或/和邦定或/和電子裝配→制程巡檢或/和半製品檢驗→裝配→制
程巡檢或/和半製品檢驗→成品包裝→成品驗證→顧客QC驗證→產品付運。這些流程的順
序可能會根據不同產品以及實際生產情況作出調整,這些過程構成鏈條的生產模式。前工序
直接形成下一道工序的輸入,其產品質量和產量直接影響後工序的產品滿足要求以及影響過
程的流暢程度。
c. 為支持質量管理體系各過程的運行和控制的有效性,本手冊各章節、MDOP(醫療器械操作
程序)和MDWI(醫療器械工作指引)對所需的準則和方法作出規定。與產品有關的準則
和方法,由工程部或/和品管部根據顧客要求、本手冊第7.1~7.2節的要求確定產品工藝要求以及質量控制要求。 d.
總經理、管理者代表、系統管理部等按照本手冊第4?6章的要求採取適宜的措施確保獲得必要的資源和資訊。這些資源和資訊是為了支援質量管理體系過程的運行和監視。 e.
管理者代表、品管部和其他職能部門應根據本手冊第7?8章的要求對質量管理體系以及產品實現所需的過程在適宜的時間、區域、地點、流程作出監視、測量(適用時)和分析這些過程。 f.
總經理、管理者代表和系統管理部按本手冊第5章的要求採取適宜的措施實現質量管理體系的策劃結果和持續改進質量管理體系各過程的有效性。
各職能部門因產品特性和生產計畫的需要,選擇把影響產品符合要求的任何過程外包,品管部、PPC 部按照本手冊第7.4節、第7.6節、MDOP7.4.1a 及相關文件對這些外包過程進行控制。外包過程包括產品外發加工、監視和測量裝置送外校準和檢定、計算機硬件和軟件的維護保養、生産設備的維護保養等過程。
4.2文件要求(ISO13485第4.2節)
4.2.1總則(ISO13485 第4.2.1條)
本集團的MDQMS 文件包括:
a. 質量方針和質量目標;質量方針的具體要求見本手冊第5.3節,質量目標的具體要求見本
手冊第5.4.1條。
b. 質量手冊,全稱為醫療器械質量手冊,簡稱MDQM 。具體要求見本手冊第4.2.2條。
c. 操作程序,全稱為醫療器械操作程序,簡稱MDOP ,在ISO13485標準中要求形成文件的
程序,以及根據公司實際情況編寫。MDOP 由S&C 部相關人員根據ISO13485標準、法
律法規要求、顧客要求、公司要求進行編寫和更新,S&C 部董事批准生效,由管理者代表
安排人員發放、回收和儲存管理。具體要求見【MDOP4.2.1a MD 文件程序管理規定】。
d. 文件架構,本公司的MDQMS 的文件層次可透過以下圖形進行表述:
一級文件: 質量方針、質量手冊 二級文件: 操作程序、質量目標 質量方針 質量手冊 質量目標 工作指引 操作程序 四級文件: 1、一般文件,為確保過程的有效策劃、運行和控制所需的文件,包括產品品質要求、產品生產要求、與產品實現有關的顧客要求和內部要求。 2、記錄,管理體系有效運行的證據 3. 適用的法規及相關文件,國家及地區的法規及相關文件 三級文件: 工作指引,簡述跨部門、部門內部的操作要求。 一般文件 記錄 適用的法規
e. 公司確定的為確保MDQMS過程有效策劃、運行和控制所需的文件。
這類文件除質量方針、質量目標、質量手冊、標準要求的程序文件和記錄以外,應按本手冊第4.2.3條進行控制。一般包括但不限於:
a) 工作指引:包括系統管理部發佈的集團工作指引,以及品管部、工程部、生產部、
PPC部、貨倉部、廠部、行政部以及其他部門編寫的工作指引,這些工作指引直接
與產品實現過程有關。各部門可根據實際情況編寫適合於本部門的工作指引,如果
編寫的工作指引影響其他部門,則需得到受影響部門的同意才能生效。
b) 產品質量要求:包括玩具安全標準,顧客提供的通用質量文件、產品規格、一般測
試規格、樣板,品管部發出的QC檢驗規格/產品質量規格、樣板。
c) 產品生產要求:包括工程部發出的產品生產流程圖、表面加工工藝流程、特殊工位
指導書、工夾模具表、EDM、Tool Plan、New Toy Schedule、樣板,PPC部發出
的生產/設備負荷表、兩/三周排期表、MRP物料計算等。
d) 一般文件:這類文件是各部門在實施質量管理體系要求以及產品要求時,需獲取的
外來文件或編制的專項文件以支持過程的有效運行。
這類記錄除標準要求的記錄外,為證明質量管理體系運行有效和產品滿足要求的證據所需的記錄,這些記錄應按照本手冊4.2.4條進行控制。
f. ISO13485標準要求形成記錄。標準要求形成的記錄應有書面記錄,記錄可採用紙類文件
和電子文件,無論採用哪種形式的記錄都應符合本手冊第4.2.4條。
g. 國家和地區法規規定的其他文件,主要指產品銷售地國家及其地方政府發出的法規和政府
文件,包括中國內地、香港地區、澳門地區、臺灣地區以及其他付運目的的國家和地區。
按照ISO13485標準規定將要求、程序、活動或特殊安排形成的程序文件、工作指引,各部門應按照文件要求進行體系的實施和保持。
工程部、品管部、生產部、PPC及貨倉等部門應按本手冊及【MDOP4.2.1b MD產品技術文件建檔規定】要求分類建立和保持醫療器械產品技術文文件,技術文文件應包括產品質量規格、生產工藝要求、產品防護要求、檢驗記錄、產品質量改進記錄等。這些文件應規定完整的生產過程,適用時,還包括安裝和服務過程。文件類型包括電子文件、紙質文件、光碟和樣品等。
4.2.2 質量手冊(ISO13485第4.2.2條)
質量手冊,全稱為醫療器械質量手冊,簡稱MDQM,由S&C部參照ISO13485標準要求進行編寫和更新,系統管理部董事批准生效,管理者代表負責文本的分發、回收和處置。MDQM包括:
a. MDQMS適用的範圍(或稱之為認證部門)透過本手冊第5.5.1條的組織架構圖進行表述。
因本公司設計和製造的產品不屬於無菌醫療器械、有源植入性醫療器械和植入性醫療器
械,故本手冊刪減標準中的無菌醫療器械專用要求(7.5.1.3、7.5.2.2)、有源植入性醫療
器械和植入性醫療器械專用要求(7.5.3.2.2、8.2.4.2)
b. ISO13485:2012標準要求形成文件的程序,全稱為醫療器械操作程序,簡稱MDOP,由
S&C部董事指派相關人員編寫並由其批准生效,MDOP作為MDQM直接關聯的體系文
件,其內容可作為MDQM的引用內容。但是,如果MDOP未有詳盡的具體的操作要求,
則引用MDWI或者其他體系文件,這些文件作為本手冊的引用文件。
c. 質量管理體系各過程相互作用的表述參照QM4.1。
MDQMS有關的文件結構見本手冊第4.2.1條。MDQM的一般內容包括:
a) 封面,包括了引用文件和適用的廠名,由系統管理部董事簽署生效
b) 目錄,質量手冊的條款和要素
c) 範圍,包括應用、公司簡介和認證名稱
d) 規範性引用文件
e) 術語和定義
f) 手冊正文,包括:第四章質量管理體系、第五章管理職責、第六章資源管理、第七章產
品實現和第八章測量、分析和改進。
g) 修訂記錄表,說明修訂的內容,由系統管理部董事簽署生效。
4.2.3.文件控制(ISO13485第4.2.3條)
本手冊第4.2.1條明確的MDQMS文件應按本條款進行控制。記錄是一種特殊類型的文件,其控制要求按照本手冊第4.2.4條要求執行控制。
S&C部相關人員按照ISO13485之4.2.3的要求和顧客要求,以及結合本公司的實際情況,編制醫療器械的文件控制程序【MDOP4.2.3a MD文件控制程序】,S&C部董事批准生效。
MDOP4.2.3a的內容包括但不限於以下:
a. 文件發佈前得到評審和批准,以確保文件是充分與適宜的;
b. 必要時對文件進行評審與更新,並再次批准;
c. 確保文件的更改和現行修訂狀態得到識別;
d. 確保在使用處可獲得有關版本的適用文件;
e. 確保文件保持清晰、易於識別;
f. 確保外來文件得到識別,並控制其分發;
g. 防止作廢文件的非預期使用,若因任何原因而保留作廢文件時,對這些文件進行適當的標識。
相关文件的更改得到原审批部门或指定的其他审批部门的评审和批准,该被指定的审批部门应能获取用于作出决定的相关背景资料。
至少保存一份作废的受控文件,并确定其保存期限。这个期限应确保至少在本公司所规定的医疗器械寿命期内,可以得到此医疗器械的制造和试验的文件,但不要少于质量记录或相关法规要求所规定的保留期限。
4.2.4記錄控制(ISO13485第4.2.4條)
由S&C部相關人員按照ISO13485之4.2.4的要求和顧客要求,以及結合本公司的實際情況,編制醫療器械的文件控制程序【MDOP4.2.4a MD記錄控制程序】,S&C部董事批准生效。MDOP4.2.4a 應規定記錄的標識、貯存、保護、檢索、保存期限和處置所需的控制。
按本手冊各章節和ISO13485要求建立的記錄,各部門應按照MDOP4.2.4a要求確保記錄得到標識、貯存、保護、檢索和處置。各部門應確保記錄保持清晰、易於識別和檢索。
保留记录的期限应至少相当于公司所规定的医疗器械的寿命期,但从公司放行产品的日期起不少于2年,或按相关法规要求规定。
5.管理職責(ISO13485第5章)
5.1.管理承諾(ISO13485第5.1節)
總經理通過以下活動,對公司建立和實施的MDQMS並保持其有效性的承諾提供證據:
a. 向公司各級人員傳達滿足顧客和醫療器械適用的法律法規要求的重要性,傳達方式可透過文
件傳遞、電子郵件發放、培訓、張貼宣傳資料、專題會議、意見箱和面談等;
b. 主持制定質量方針,並透過管理評審定期對質量方針的適宜性進行評審;(見本手冊第5.3節)
c. 指派管理者代表和相關職能部門負責人制定質量目標;(見本手冊第5.4.1條)
d. 主持管理評審;(見本手冊第5.6節)
e. 對MDQMS所需的資源,對各職能部門提出的資源需求作出及時的審批,對不合理的申請專
案應要求相關人員更新的申請需求。對得到批准的申請項目,各職能部門應確保及時地採
購、安裝、使用和維護。(見本手冊第6.1~6.4節)
5.2.以顧客為關注焦點(ISO13485第5.2節)
總經理應適時地參與各項質量管理活動,包括授權質量負責人、發佈組織架構圖、審閱質量報告、參加專題會議、聽取職能部門負責人的工作彙報等。對於存在的質量問題,督促相關人員採取適宜的改進措施。具體要求見本手冊第7.2.1條和第8.2.1條要求執行。
5.3.質量方針(ISO13485第5.3節)
本節的具體要求見質量手冊QM5.3.
5.4.策劃(ISO13485第5.4節)
5.4.1質量目標(ISO13485第5.4.1條)
本條的具體要求見質量手冊QM5.4.1.
5.4.2質量管理體系策劃(ISO13485第5.4.2條)
本條的具體要求見質量手冊QM5.4.2.
5.5職責、權限與溝通(ISO13485第5.4節)
5.5.1.職責和權限(ISO13485第5.5.1條)
各職能部門負責人的主要職責見質量手冊QM3,部門負責人以外的其餘人員的職責由各部門負責人編制和批准發佈。各部門負責人應根據所形成文件的職責和權限要求與雇員取得有效溝通,並應評價雇員完全明白職責和權限要求。
新雇員、轉崗雇員在新崗位厲行職務前,各部門負責人應對雇員進行課堂培訓或/和現場實操示範教育,並採取書面考核或/和實操評價確保雇員完全明白職責要求。
總經理應適時批准發佈公司的組織架構圖,組織架構圖清晰表述各部門(主要負責人)的組織工作範圍。各部門負責人也應適時地批准發佈其部門的組織架構圖,架構圖與職責和權限要求的文件保持一致。總經理任命品管部經理作為公司的質量負責人,充分授權其對醫療器械的產品質量的監視和
測量活動、顧客溝通、質量規格發佈,确保所有从事对MDD产品质量有影响的管理、执行和验证工作的人员为完成这些任务所必须的独立性和权限。当国家或地区法规可能要求对特定人员的任命,这些人员负责的活动涉及到对从生产后阶段获取经验的监视及不良事件的报告,包括:
a. 產品質量方面與顧客溝通;(見本手冊第7.2.3條、第8.2.1條)
b. 發佈質量規格;(見本手冊第7.1節)
c. 對採購產品、外包過程進行驗證;(見本手冊第4.1節、第7.4節)
d. 對產品實現過程進行監視和測量;(見本手冊第7.1節~第7.5節)
e. 對監視和測量設備進行控制;(見本手冊第7.6節)
f. 對產品進行監視和測量;(見本手冊第8.2.4條)
g. 監督不合格品控制的有效實施;(見本手冊第8.3節)
h. 監督產品質量問題的改進措施有效實施。(見本手冊第8.5節)
5.5.2管理者代表(ISO13485第5.5.2條)
總經理授權並任命一名經理或以上級別人員為公司的管理者代表,這名管理者代表也可以由ISO9001 QMS的管理者代表兼任。根據需要,也可以任命一名副管理者代表協助管理者代表執行任務。管理者代表的職責包括:
a. 協助S&C部確保MDQMS所需的過程得到建立、實施、保持;
b. 向總經理報告MDQMS的業績和任何改進的需求;
c. 確保在公司內提高滿足醫療器械適用的法律法規和顧客要求的意識;
d. 與MDQMS有關事務的外部聯絡。
5.5.3內部溝通(ISO13485第5.5.3條)
為使MDQMS的有效運行,總經理授權和鼓勵各部門及各級人員透過以下管道就MDQMS的有關資訊進行溝通:
a. S&C部、品管部、工程部、PPC部、市場部等部門將收到的有關顧客要求、產品規格要求、
適用法律法規要求及質量管理體系要求時,通過文件、電子郵件、會議等形式傳遞給相關的部門。
b. S&C部、品管部、工程部、PPC部、廠部、生產部及其它部門均鼓勵各級人員透過信箱、電
子郵件或會議等的方式進行資訊回饋。相關部門負責人收到資訊回饋後需採取相應的措施,並應將處置結果回饋給意見(建議)提出人。
5.6管理評審(ISO13485第5.6節)
5.6.1總則(ISO13485第5.6.1條)
為了確保MDQMS持續適宜性、充分性和有效性,每間隔12個月,總經理主持對MDQMS進行管理評審。總經理委任公司管理評審成員,並明確管理評審各輸入項的負責人或跟進小組,管理者代表協助總經理收集整理管理評審的輸入資訊。
管理評審的方式可採取會議形式或報告形式進行,管理評審內容包括:
a. MDQMS改進的機會和變更需要,包括質量方針和質量目標;
b. 本手冊第5.6.2條要求的管理評審的輸入事項。
管理評審的報告(記錄)按照本手冊第4.2.4條以及MDOP4.2.4a執行管理。
5.6.2.評審輸入(ISO13485第5.6.2條)
MDQMS管理評審的輸入應包括但不限於以下事項:
a. 審核結果,包括內部審核、客戶審核、第三方審核的審核結果。
b. 顧客回饋,包括顧客的書面投訴、口頭投訴、滿意度調查、海外退貨。
c. 過程的業績和產品的符合性,包括採購產品驗證、前工序驗證、成品驗證以及顧客QC驗貨
結果等。
d. 預防和糾正措施的狀況,包括管理體系和產品方面的預防和糾正措施。
e. 以往管理評審的跟蹤措施。
f. 可能影響MDQMS的變更,主要指公司的架構變更、經營場所和產品類型變更。
g. 改進建議,各級人員對MDQMS的一些改進建議或意見。
h. 醫療器械適用的法規的變更,包括新發佈和修訂的法規,這些法規包括我國大陸的法律法規、
產品銷售地(含國家和地區)的法規。
由S&C部相關人員按照ISO13485之5.6.2的要求以及結合本公司的實際情況,編制【MDOP5.6.2 a MD法規收集、識別、分發和監督程序】,S&C部董事批准生效。相關部門按照此程序的要求執行醫療器械適用的法規的收集、整理、識別、分發、儲存、監督實施,必要時,S&C部及相關部門應把法規的適用內容納入內部文件。MDQMS適用的法規包括我國大陸地區、香港地區、澳門地區、台灣地區的與醫療器械有關法律法規和標準,產品銷售目的地與醫療器械有關的法規和標準,與醫療器械有關的國際標準。
5.6.3評審輸出(ISO13485第5.6.3條)
管理評審輸出包括但不限於以下方面有關的任何決定和措施:
a. 保持MDQMS及其過程有效性所需的改進。
b. 與顧客要求有關的產品的改進。
c. 資源需求,包括新增加資源以及資源重新分配。
管理評審輸出應形成書面記錄,並得到總經理的簽批,按照本手冊第4.2.4條以及MDOP4.2.4a執行管理。
6.資源管理(ISO13485第6章)
6.1.資源提供(ISO13485第6.1節)
各部門負責人及相關人員應確定並提供以下方面所需的資源:
a. 實施MDQMS並保持其有效性;
b. 滿足醫療器械適用的法規要求和顧客要求。
關於資源提供的具體執行要求按照QM6.1。
6.2.能力、意識和培訓(ISO13485第6.2節)
6.2.1.總則(ISO13485第6.2.1條)
公司明確規定從事影響產質量量管理工作的人員應是能夠勝任的。為了確保和實現這些人員能夠滿足要求,應確保這些人員基於適當的教育、培訓、技能和經驗。相關人員的能力要求按本手冊第6.2.2條執行。
6.2.2.能力、意識和培訓(ISO13485第6.2.2條)
為了確保從事影響產質量量管理工作的人員能夠勝任其工作,應滿足以下能力、意識和培訓要求:
a. 總經理和各部門負責人應分別確定相關人員應具有的工作能力要求:
a) 總經理應確定各部門負責人應有的能力,這些能力包括學歷及所學專業、崗位所需的
培訓專案、崗位所需的技能要求和工作經驗年限要求,應把相關要求形成書面文件以
及得到實施保持;
b) 各部門負責人應確定其屬下的各級人員應有的能力,這些能力包括學歷及所學專業、
崗位所需的培訓專案、崗位所需的技能要求和工作經驗年限要求,應把相關要求形成
書面文件以及得到實施保持。
c) 關於部門負責人及各級員工的能力要求之文件可結合本手冊第5.5.1條同時制定和執
行。
b. 提供培訓或採取其他措施以滿足相關的能力要求
a) 總經理應評估和安排部門負責人的培訓需求,給予相關人員適當的新入職培訓和在職
培訓安排,可按照QM6.2執行;
b) 各部門負責人應評估和安排其屬下的各級人員的培訓需求,給予相關人員適當的新入
職培訓和在職培訓安排,可按照QM6.2執行;
c) 與醫療器械有關的生產車間、貨倉部、車間QC應接受醫療器械的儲存、生產過程環
境、設備設施的清潔和防止污染的知識培訓。
d) 除了培訓外,也可以安排相關人員外出參觀交流學習、參加研討會、知識分享等措施。
c. 各管理層成員應評價所採取措施的有效性,評價方式包括(但不限於)實操考核、書面考試;
d. 各管理層人員可通過培訓、宣傳等方確保屬下各級人員清晰到個人的崗位技能要求和職責要
求,瞭解個人從事崗位對組織影響的相關性和重要性。
e. 各級人員應聘時提交的教育、技能和經驗的相關記錄由人事部負責人儲存,儲存期限按照本
手冊第4.2.4條執行。
f. 国家和地区法规规定对MDD相关人员的培训要求
6.3基礎設施(ISO13485第6.3節)
為達到產品符合性要求所需的基礎設施的確定、提供和維護按照QM6.3執行。本公司的基礎設施包括:
a. 寫字樓、車間、貨倉等建築物和相關的設施;
b. 生產設備及其配套軟體、發電機及其配套設施等過程設備的硬體和軟體;
c. 電腦及電腦系統、電話及電話系統、車輛、電梯、消防系統和消防器材等支援性服務的設備
設施。
負責設備設施維護保養的部門應建立維護保養制度和計畫,計畫應明確維護保養責任部門、責任人、保養週期、保養專案等內容。負責維護保養的人員應按相關制度和計畫對設備設施進行維護保養工作,並把保養結果形成書面記錄,記錄應按照本手冊第4.2.4條執行管理。
6.4工作環境
為達到產品符合要求的工作環境的確定和一般管理按照MDOP6.4a、MDOP6.4b、MDOP6.4c 執行。與醫療器械有關的工作環境及相關設施應符合以下要求:
a. 生產工人、車間管理人員、車間QC及其它與生產過程直接接觸的人員的身體健康、進入工
作場所的消毒清潔、著裝等方面的要求按照【MDOP6.4a MD雇員健康、清潔和服裝管理程序】要求執行。
b. 生產車間、貨倉部、實驗室等工作場所的工作環境條件按照【MDOP6.4b MD工作環境條件
和作業指導書管理程序】要求執行,相關部門按照該程序進行日常的監視和控制,日常監視和控制過程出現異常,相關人員應採取以下措施
a) 採取與異常情況相適應的糾正行動,確保環境條件按照所策劃的要求;
b) 品管部應對受影響的產品重新驗證和評估,確保產品符合放行準則;
c) 相關人員應記錄環境條件的監視和控制結果,以及出現異常情況所採取的措施也應記
錄。
c. 因產品特性和客人要求,對清潔和防止污染有特別要求的產品,工程師或/和QAE需針對這
類產品制定適用的產品環境作業要求。
d. 在特殊工作場所的臨時工作人員,應接受清潔和防止污染的培訓,這些人員也可以在已經接
受過培訓的人員的監督下工作。無論採用哪種方式,相關車間(部門)應將這些培訓或監督結果形成書面記錄。
e. 對受污染或易於污染的產品的控制,按照【MDOP6.4c MD受污染或易於污染的產品控制的
特殊安排程序】要求執行。
7.產品實現(ISO13485第7章)
7.1產品實現的策劃(ISO13485第7.1節)
接到新產品時,工程部或產品小組應策劃和開發產品的實現過程。產品實現的策劃應與本手冊第4.1條所確定的其他過程一致。
產品實現的策劃的內容,包括但不限於以下項目:
a. 產品的質量目標和要求,應結合適用的法律法規、客人要求策劃產品的質量要求,質量目
標的策劃按照本手冊第5.4.2條;
b. 針對產品確定生產過程、生產工藝要求、人力資源、生產設備設施、檢驗設備、工作環境
的需求,這些方面的策劃按照本手冊第7.3節;
c. 根據客人的產品規格及產品特性策劃採購物料、生產過程、成品的驗證、核對總和試驗活
動,以及制定內部QC檢驗規格,這部份的策劃由品管部負責;
d. 為實現過程及其產品滿足要求提供證據所需的記錄,相關的記錄在本手冊的第4章、第5
章、第6章、第7章第7.2?7.6節以及第8章相關章節所表述的記錄,部份記錄可能引
用QM(ISO9001的質量手冊)所對應的章節。
產品實現策劃的輸出形式應適合於公司的運作方式。上述這些策劃內容可從本手冊第7.2條?第7.6條,以及本手冊第8.2條作為產品實現過程的開發。
在產品實現全過程中的風險管理按照程序【MDOP7.1a MD風險管理程序】執行,風險管理引起的記錄應按照本手冊第4.2.4a條執行管理。
7.2與顧客有關的過程(ISO13485第7.2節)
7.2.1與顧客有關的要求的確定(ISO13485第7.2.1條)
公司接收新產品訂單時和產品實現過程中,相關部門應在產品的設計和開發階段初期確定以下要求的執行:
a. 顧客規定的要求,包括對交付及交付後活動的要求。
工程部或/和品管部或/和客戶服務部或/和PPC部等部門接收到顧客的訂單合同、產品規格、樣辦、設計文件等資訊時,應按照本手冊第7.2.2條對這些要求評審,並把適用的要求轉化為公司內部的生產工藝要求、產品檢驗規格、內部程序文件、設計文件等,這些要求應發放給相關職能部門和操作者。在操作人員對這些要求容易出現理解和操作上的偏差的情況下,應對其作出適當的培訓教育,並確保操作者能正確運用這些知識。
b. 顧客雖然沒有明示,但規定的用途或已知的預期用途所必需的要求
工程部或/和品管部應對此類要求在內部的生產工藝要求、產品檢驗規格、內部程序文件、設計文件上作出規定。
c. 與產品有關的法律法規要求
品管部或/和工程部或/和S&C部應收集與醫療器械適用的法律法規、國家標準,以及產品銷售地(國家或地區)適用的法規,並將此類要求在內部的生產工藝要求、產品檢驗規格、內部程序文件、設計文件上作出規定。
d. 組織確定的任何附加要求
7.2.2與產品有關的要求的評審(ISO13485第7.2.2條)
工程部在收到新產品資料時,應按照QM7.2.1/7.2.2對新產品作出合約評審。合約評審應確保:
a.產品要求已經轉化為內部文件;
b.沒有產品規格、合同或訂單的要求不一致的地方;
c.公司的生產設備、人力資源、生產工藝、生產車間、設計和開發、物流管理等方面的能力能滿足產品的設計和開發、生產管理、質量管理、物流管理等方面。
《合約評審表》和相關後續跟進記錄按照本手冊第4.2.4 條執行管理。
顧客用非正式的產品規格提出產品要求時,工程部或品管部應對這些要求進行確認。非正式的產品要求包括口頭通知、會議通報或電子郵件等。
在產品實現任何階段,產品要求發生變更時,工程部或品管部應在一個月內修改相應的文件,並確保相關人員知道已變更的要求。修改文件應符合以下規定:
a. 修改處應與原稿文件有明顯的識別;臨時手改形式應確保正本和所有副本的修改內容一致,
修改人員和批准人員須在修改處簽名和注明修改日期。
b. 修改版應標明修改版次、日期、修改內容,並由原批准人簽名批准生效。當原批准人離職或
不再負責該項目時,由同等職位的人員簽名批准。
c. 修改版生效後應確保舊版收回註銷,如需保留作廢文件時應標識清楚和採取適宜的措施確保
不會被誤用。
7.2.3顧客溝通(ISO13485第7.2.3條)
在產品實現適宜階段,品管部或/和工程部或/和PPC部按照QM7.2.3流程圖要求與顧客進行溝通,溝通內容包括但不限於以下內容:
a.產品信息;
b.問詢、合同或訂單的處理,包括對其的修改;
c.顧客反饋,包括顧客投訴,關於顧客反饋方面的信息收集、整理、分析和改進見本手冊第8.2.1條。
d.忠告性通知,具體的發佈、實施見本手冊第8.5.1條和程序MDOP8.5.1a。
7.3設計和開發(ISO13485第7.3節)
本公司現有MDD產品設計開發由客戶負責,本手冊保留7.3為對應標準和公司應對以後產品設計開發所需要求。如現有產品99900/99901/99902/氧氣面罩等。
7.3.1設計和開發策劃(ISO13485第7.3.1條)
醫療器械的設計和開發的策劃、輸入、輸出、評審、驗證、確認和設計更改的控制按照程序【MDOP7.3 MD設計和開發程序】執行。
接收新產品訂單時,工程部按照MDOP7.3a對醫療器械的設計和開發進行策劃和控制。策劃的內容包括但不限於以下內容:
a 設計和開發階段;
b適合於每個設計和開發階段的評審、驗證、確認和設計轉換活動;
c設計和開發的職責和權限。
接到新產品的設計和開發任務後,工程師應確定與產品要求有關的設計輸入,這些設計輸入應儲存在產品文件案中,並按本手冊4.2.4執行管理。輸入內容包括但不限於以下內容:
a.客人要求和產品規格,以及根據產品預期用途規定產品的功能、性能和安全要求;
b.醫療器械適用的我國的法律法規和標準要求,以及銷售目的地的法規要求;
c.適用時,以前類似產品的設計資訊;
d.設計和開發所必需的其他要求;
e.在產品實現策劃進行風險管理時的輸出;
工程師在實施設計和開發前,應對設計和開發的輸入內容進行評審,以確保輸入內容是充分與適宜的,且完整、清楚和不自相矛盾,並經工程部經理簽名批准。
7.3.3 設計和開發輸出(ISO13485第7.3.3條)
在實施設計和開發的任何過程,工程師應確保設計和開發的是根據輸入內容進行設計和開發工作,設計和開發的輸出應以能夠針對設計和開發的輸入進行驗證的方式提出,並應在放行前得到工程部經理批准。設計和開發輸出應符合以下要求:
a.滿足設計和開發輸入的要求;
b.產品相片、圖紙、產品規格、樣板、MOQ Checklist、TOOL PLAN、DECO PLAN、BOM、工夾模具表、工位指導書、工序質量規格、流程圖、裝配排拉表等產品生產要求;
c.包含或引用產品檢驗規格;
d.規定對產品的安全和正常使用所必需的產品特性。
工程部應按照本手冊第4.2.4條建立產品文件檔案,並把上述輸出形成書面記錄和按照本手冊第4.2.4條要求進行管理。
7.3.4 設計和開發評審(ISO13485第7.3.4條)
在實施設計和開發的任何過程,工程部(也可稱為機械或產品工程部)的工程師或其上級領導應對依據所策劃的安排(見7.3.1)設計和開發進行系統的設計評審,設計評審可透過會議形式或評審報告形式進行。無論採用哪種方式進行設計評審,設計評審小組成員包括與所評審的設計和開發階段有關的職能的代表,通常包括機械工程部、電子工程部、車縫工程部、品管部、PPC部、生產部等部門的代表。
設計評審結果應形成書面記錄,並且得到小組成員的簽名確認。設計評審的各項目均應明確處置結果,需要後續跟進的項目應明確具體執行措施、項目負責人、計劃完成日期等信息,並且將這些必要措施記錄,設計評審記錄及任何後續措施應予保持(見4.2.4)。
設計評審應滿足以下要求:
a) 評價設計和開發的結果滿足要求的能力;
b) 識別任何問題並提出必要的措施。
為確保設計和開發輸出滿足輸入的要求,在產品的設計和開發的適宜階段,工程師依據所策劃的安排(見7.3.1)設計和開發進行驗證。驗證的方法和要求具體流程按照MDOP7.3的要求執行。
設計驗證的結果及任何必要措施的記錄應予保持(見4.2.4)。
7.3.6 設計和開發確認(ISO13485第7.3.6條)
為確保產品能夠滿足規定的使用要求或已知預期用途的要求,工程師依據所策劃的安排(見7.3.1)對設計和開發進行確認。設計確認分EP、FEP和PP等三個階段進行,工程師按照公司要求或/和客戶要求在適宜的設計和開發階段將EP樣板、FEP樣板、PP樣板提交XXX實驗室做測試,客戶要求提交給其實驗室或第三方實驗室進行測試的,則按其要求執行。EP、FEP和PP等階段的實驗室測試是設計和開發確認的主要方法,每個階段的測試所發現的質量問題應採取充分適宜的措施并得到解決,除非得到客戶的書面批准,否則不能進入下一個設計和開發階段。
EP或/和FEP階段出現的質量問題得到妥善的解決后,由工程部董事/副董事批准放行后才能進入PP試產階段。
完成PP階段的試產后,由QAE召集產品小組全體成員對產品進入PS階段的放行進行核准,產品小組成員一致同意放行和簽署【新產品投產核准報告】后,才能進入PS量產階段。
當國家或地區法規要求實施醫療器械臨床評價和/或性能評價的醫療器械產品,工程師和/或QAE 應按照相關法規執行設計和開發確認。
與設計確認有關的記錄按照MDOP4.2.4a要求執行儲存管理。
7.3.7 設計和開發更改的控制(ISO13485第7.3.7條)
產品在設計和開發階段、PP試產階段、PS生產階段因客戶要求或產品要求需要對設計進行更改,工程師按照MDOP7.3a要求對設計更改進行識別、控制、文件記錄發放。在適當時,應對設計和開發的更改進行評審、驗證和確認,並在實施前得到批准,設計和開發更改的評審應包括評價更改對產品組成部分和已交付產品的影響,具體流程按照MDOP7.3a的要求執行。
與設計更改有關的記錄按照MDOP4.2.4a執行儲存管理。
7.4 採購(ISO13485第7.4節)
7.4.1 採購過程(ISO13485第7.4.1條)
採購部、品管部/貨倉部按照按照QM7.4.1/7.4.2對供方及採購的產品以及採購過程進行控制。
供方選擇、評價和重新評價準則按照指引QA027和QA208執行管理。
7.4.2 採購信息(ISO13485第7.4.2條)
採購部或PPC部按照QM7.4.1/7.4.2執行採購的管理,採購信息包括但不限於以下内容:
a) 產品、程序、過程和設備批准的要求;
b) 人員資格的要求;
c) 質量管理體系的要求。
在與供方溝通前,組織應確保規定的採購要求是充分與適宜的。
當採購物料有追溯性要求時,應按照本標準規定的可追溯性要求的範圍和程度,並保持相關的採購資訊,如採購檔、採購記錄等。
與採購過程和採購信息有關的文件按照MDOP4.2.3a執行管理,相關記錄則按MDOP4.2.4a執行管理。
7.4.3 採購產品的驗證(ISO13485第7.4.3條)
品管部按照按照QM7.4.1/7.4.2、QA700確定採購品和外包加工品的檢驗、試驗和測量活動,並實施檢驗或其他必要的活動,以確保採購的產品滿足規定的採購要求。
採購品或加工品的檢驗、試驗和測量活動,在本公司範圍内進行。品管部經理可根據產品的特性安排品檢員在供方的現場進行檢驗、試驗和測量活動,如果在供方現場驗證採購品或加工品,品管部應規定驗證后的產品的運輸和追蹤要求。
保持與採購品或加工品有關的檢驗、試驗和測量等驗證記錄,並應按MDOP4.2.4a執行儲存管理。
7.5 生產和服務提供(ISO13485第7.5條)
7.5.1 生產和服務提供的控制(ISO13485第7.5.1條)
7.5.1.1總要求(ISO13485第7.5.1.1款)
由S&C部相關人員按照ISO13485之7.5.1的要求和顧客要求,以及結合本公司的實際情況,編制醫療器械的【MDOP7.5.1a 生産和服務提供的控制程序】,S&C部董事批准生效。該程序對策劃生産和服務提供的受控條件作出規定。生產和服務提供受控條件應包括:
a.獲得表述產品特性的信息;
b.獲得與產品有關的程序文件、產品要求、工藝要求、作業指導書、檢驗規格;
c.使用適宜的設備;
d.獲得和使用監視和測量裝置;
e.實施監視和測量;
f.放行、交付和交付後活動的實施;
g.規定的標簽和包裝操作的實施。
程序還對要求記錄醫療器械的每批次的產品編號、產品名稱、生産數量、批准銷售的數量、生産日期、生産日期碼(Date Code)等信息,每一份記錄應得到授權人員批准簽署。這些記錄按MDOP4.2.4a執行儲存管理。
7.5.1.2生產和服務提供的控制--- 規定要求(ISO13485第7.5.1.2款)
7.5.1.2.1產品的清潔和污染的控制(ISO13485第7.5.1.2.1項)
每一款產品正式投產前,工程部和/或品管部按【MDOP7.5.1b產品清潔和污染的控制程序】要求和顧客要求對產品的清潔和防止污染作出文件規定,生產部、制程QC按工程部和/或品管部的文件要求進行日常運行控制。應編制產品的清潔和防止污染的文件的範圍包括但不限於:
a. 在滅菌和/或使用前由組織進行清潔的產品;或
b. 以非無菌形式提供的而需在滅菌和/或使用前先進行清潔處理的產品;或
c. 作為非無菌使用提供的而使用時清潔是至關重要的產品;或
d. 在生產中,可能需要應用一些輔助的生產物料,但這些輔助生產物料不是產品的組成部分
應,因此在完成生產的每一個階段應有效清潔,以確保產品不受污染。例如,注塑工序使
用的脫模劑。
如產品按照本條第7.5.1.2.1a款和第7.5.1.2.1b款的要求進行產品清潔的,則在清潔處理前不必滿足本手冊第6.4條的a款和b款的要求。
7.5.1.2.2安裝活動(ISO13485第7.5.1.2.1項)
本公司現有MDD產品暂不适用此条款。保留本条款為對應標準和公司應對以後產品所需要求。如指99900/99901/99902/氧氣面罩等。
每一款產品正式投產前,工程部按【MDOP7.5.1c MD安裝活動管理程序】和顧客要求編制醫療器械的制程要求,生產部及其他有關部門按工程部發出的醫療器械的制程要求進行日常運行控制;品管部按【MDOP7.5.1c MD安裝活動管理程序】和顧客要求編制醫療器械的制程和成品的檢驗、測量和試驗要求,生產車間和QC檢驗員按照這些要求對制程巡查和製品(含成品)的檢驗、測量和試驗。檢驗、測量和試驗結果應得到適宜的處置,適用時符合本手冊第8.2.4節、第8.3條、第8.5.2節、第8.5.3節的要求。
經顧客同意,醫療器械的部份制程可交由加工商完成,如果把部份制程交由加工商時,PPC部(VO)應把工程部發出的“制程要求”和品管部發出的“制程和成品的檢驗、測量和試驗要求”轉發給加工商,並且應確保加工商理解和遵照執行這些要求。PPC部、品管部應參照本手冊第7.4條要求對加工商進行管理。
與安裝活動有關的記錄按照本手冊第4.2.4節要求進行儲存管理。
7.5.1.2.3服務活動(ISO13485第7.5.1.2.1項)
本公司現有MDD產品暂不适用此条款。保留本条款為對應標準和公司應對以後產品所需要求。如指99900/99901/99902/氧氣面罩等。
公司生產的醫療器械屬非自主品牌,售後服務的安排按照顧客要求執行。涉及顧客的退貨、換貨、維修活動按照QM8.2.1執行,涉及產品召回則按照工作指引QA017執行。與服務活動有關的記錄按照本手冊第4.2.4節要求進行儲存管理。
7.5.1.3無菌醫療器械的專用要求(ISO13485第7.5.1.3款)
公司生產的醫療器械屬於非無菌醫療器械,因此,本條款不適用。
7.5.2 生產和服務提供過程的確認(ISO13485第7.5.2條)
7.5.2.1總要求(ISO13485第7.5.2.1款)
經過評估,公司生產的醫療器械類型不能由後續的監視或測量加以驗證的情況不存在,這類過程
有源医疗器械质量手册-模板
WUYY 北京XXXX医疗科技有限公司 质量手册 编制:年月日 审核:年月日 批准:年月日 版本号: 分发号: XXXX-XX-X发布 XXXX-XX-XX实施北京XXXX医疗科技有限公司发布
目录 0.1 颁布令 (2) 0.2 管理者代表任命书 (3) 0.3 公司概况 (4) 0.4 修订情况 (5) 1.0 质量手册说明 (6) 2.0公司质量管理体系结构图 (9) 4.0 质量管理体系 (13) 4.1质量管理体系的总要求: (14) 4.2文件的要求 (18) 5.0 管理职责 (22) 5.1管理承诺: (22) 5.2以顾客为关注焦点 (23) 5.3质量方针 (24) 5.4策划 (25) 5.5职责和权限 (27) 5.6 管理评审 (32) 6.0 资源管理 (35) 6.1资源提供 (35) 6.2 人力资源管理 (35) 6.3基础设施 (36) 6.4工作环境和污染控制(ISO9001 标准7.1.4 过程运行环境): (36) 7.0 产品实现 (37) 7.1产品实现的策划 (38) 7.2与顾客有关的过程 (38) 7.3设计和开发 (40) 7.4采购控制 (43) 7.5生产和服务的提供 (45) 7.6监视和测量设备的控制 (49) 8.0 测量、分析和改进 (51) 8.1总则 (51) 8.2 监视和测量 (51) 8.3 不合格品控制 (54) 8.4 数据分析 (55) 8.5 改进 (56) 附录一程序文件清单 (58)
0.1 颁布令 本《质量手册》是依据ISO 9001:2015《质量管理体系要求》、YY/T 0287-2017/ISO 13485:2016《医疗器械质量体系用于法规的要求》和《医疗器械生产质量管理规范》,结合XXXXXX公司生产产品的实际和特点编制而成的。它阐述了公司的质量方针、质量目标和质量承诺。它包括或引用上述标准要求的和质量管理体系所要求的形成文件的程序及对经识别和建立的质量管理体系的过程之间的相互作用给予描述,并对公司的质量体系提出了具体要求。 本《质量手册》是公司质量管理的基本法规,是公司全体员工贯彻质量方针、实现质量目标、履行质量职责的行为准则和行动纲领,也是公司向社会和所有顾客提供质量保证的展示性文件。 本《质量手册》自XX年XX月XX日起正式实施,公司全体人员自本手册实施之日起,必须遵照执行。 特此发布! 总经理(签字): (公司盖章) 日期:
医疗器械质量手册
质量手册 引用标准: YY/T 0287-2017 、ISO 13485:2016 医疗器械生产质量管理规范 文件编号: 版本: 编制/日期:年月日 审核/日期:年月日 批准/日期:年月日 受控状态: 发放号: 发布日期XXXX年XX月0XX日实施日期XXXX年XX月XX日 XXXXXXXXXX有限公司
目录 《质量手册》颁布令 (3) 修订记录 (4) 公司简介 (5) 管理者代表任命书 (6) 第一章质量体系范围 (8) 1.1发行目的 (8) 1.2手册适用范围 (8) 第二章质量手册有关说明 (9) 2.1质量手册编制说明 (9) 2.2手册的编制、审核及发布 (9) 2.3手册的构成 (9) 2.4质量管理体系覆盖场所 (9) 2.5手册的不适用、删减条款及理由 (9) 2.6手册的更改和换版 (10) 2.7质量手册的管理 (10) 第三章术语、定义和引用标准 (13) 3.1公司的质量手册采用标准中术语和定义 (13) 3.2公司内部术语 (13) 3.3手册引用标准和法规 (13) 第四章质量管理体系 (14) 4.1总要求 (14) 4.2文件要求 (15) 第五章管理职责 (19) 5.1管理承诺 (19) 5.2以顾客为关注焦点 (19) 5.3质量方针 (20) 5.4策划 (20) 5.5职责、权限与沟通 (21) 5.6管理评审 (23) 第六章资源管理 (25) 6.1资源提供 (25) 6.2人力资源 (25) 6.3基础设施 (26) 6.4工作环境和污染控制 (26)
第七章产品实现 (28) 7.1产品实现的策划 (28) 7.2与顾客有关的过程 (28) 7.3设计开发 (30) 7.4采购 (34) 7.5生产和服务提供 (36) 7.6监视和测量设备的控制 (39) 第八章测量、分析和改进 (40) 8.1总则 (40) 8.2监视和测量 (40) 8.3不合格品控制 (42) 8.4数据分析 (43) 8.5改进 (44) 第九章相关附件 (46) 附件一:公司组织机构图 (47) 附件二:质量管理体系职能分配表 (48) 附件三:程序文件清单 (50)
2017最新医疗器械质量手册
控制状态:受控□ 非受控□质量手册 依据:YY/T0287-2017 文件编号: 编制: 审核: 批准: 2017-08-28发布2017-08-28实施**********有限公司发布
目录 1. 质量手册发布令 (5) 2. 企业概况 (6) 2.1管理者代表任命书 (7) 2.2质量方针与质量目标 (8) 3. 图表 (9) 3.1组织结构图 (9) 3.2质量管理体系机构图 (10) 3.3质量管理体系职能分配图 (11) 4. 质量管理体系 (12) 4.1总要求 (12) 4.2文件要求 (13) 4.2.1总则 (13) 4.2.2质量手册 (14) 4.2.3医疗器械文档 (17) 4.2.4文件控制 (19) 4.2.5记录控制 (24) 5. 管理职责 (26) 5.1管理承诺 (26) 5.2以客户为关注焦点 (26) 5.3质量方针 (27) 5.4策划 (28)
5.4.2质量管理体系策划 (28) 5.5职责、职权与沟通 (29) 5.5.1职责与权限 (29) 5.5.2管理者代表 (36) 5.5.3内部沟通 (37) 5.6管理评审 (38) 5.6.1总则 (38) 5.6.2评审输入和输出 (39) 6.资源管理 (42) 6.1资源提供 (42) 6.2人力资源 (42) 6.3基础设施 (44) 6.4工作环境和污染的控制 (44) 6.4.1工作环境 (44) 6.4.2污染控制 (45) 7.产品实现 (45) 7.1产品实现的策划 (45) 7.2与顾客有关的过程 (47) 7.2.1产品要求的确定、评审、沟通 (47) 7.3设计和开发 (48) 7.3.1总则 (48) 7.3.2设计和开发策划 (49) 7.3.3设计和开发输入 (50) 7.3.4设计和开发输出 (51)
医疗器械生产企业质量手册(V)
质量手册 审核: 批准: 日期: 陕西三八妇乐科技股份有限公司 前言 本《质量手册》是我公司质量管理体系的纲领性文件。本文件由公司总经理组织各有关部门根据医疗器械生产质量管理规范及ISO9001:2000,结合本公司的实际情况编制而成,经过认真审核。现预发布实施。 《质量手册》内容包括: 1)本公司质量管理体系的范围。 2)本公司质量管理体系所形成文件的程序; 3)本公司质量管理体系过程之间的顺序和相互作用的表述。 本《质量手册》从颁发之日起,要求公司各部门、全体员工严格。贯彻执行。 任命书 为了贯彻执行医疗器械生产质量管理规范,旨在本公司有效建立、实施和保持一个完善的质量管理体系,特任命为本公司管理者代表,并担当如下职责和权限: 1)确保质量管理体系所需的过程得到建立,实施和保持; 2)向最高管理者报告质量管理体系的业绩和任何改进的需求; 3)确保在整个组织内提高满足法规要求和顾客要求的意识; 4)负责就质量管理体系有关事宜的外部联络。 质量方针 人民健康至上,产品质量第一 。 质量目标 一. 严格执行医疗器械生产质量管理规范及ISO9001:2000。
二. 产品的性能指标达到医疗器械企业标准。 三. 产品出厂合格率达到100%。 四. 不断开发系列化新产品,以填补国内空白。SB-WJ-01-01 目录 0.1 目录 0.2 质量手册说明 0.3 企业概况质量 0.4 公司组织机构图 05 公司质量管理体系结构图 06 质量管理体系过程职责分配表 07 质量管理体系 08 文件控制程序 09 质量记录控制程序 10 管理职责 11 管理评审控制程序 12 人力资源控制程序 13 工作环境控制程序职责和权限 14 与顾客有关的过程控制程序 15 评定供方控制程序资源管理 16 采购控制程序 17 生产和服务提供控制程序 18 内部审核程序产品实现 19 过程和产品的测量和监控程序 20 不合格控制程序 21 纠正、预防、改进措施控制程序 22 顾客信息反馈控制程序 质量记录清单: SB-JL-01-01- SB-JL-01-055
医疗器械企业如何建立有效的质量管理体系
医疗器械企业如何建立有效的质量管理体系医疗器械企业如何建立有效的质量管理体系建立的总体流程如下:识别要求(4.1)→实施培训(4.2)→策划建立体系(4.3)→运行体系(4.4) 1识别医疗器械企业质量管理体系的特殊要求 医疗器械是一种特殊的商品,是救死扶伤的工具,其质量好坏直接关系到人民的身体健康,所以医疗器械企业必须坚持"质量第一"的方针,加强质量管理,建立有效的质量管理体系,从根本上保证产品质量,提高社会效益和经济效益。 1.1医疗器械必须遵循法律法规的要求 每个国家都对医疗器械规定了一些法律法规,满足法律法规的要求是其企业生产的首要条件,法律法规将是医疗器械企业质量管理体系的基础。 1.2出口的医疗器械产品要遵循到岸国家的法律法规 出口的医疗器械,就必须遵循到岸国家的医疗器械指令,否则产品将不能在当地上市,例如欧盟的三个医疗器械指令是: a)有源植入性医疗器械指令(90/385/EEC,AIMDD) b)医疗器械指令(93/42/EEC,MDD) c)实验室用诊断医疗器械指令(98/79/EC,IVD) 1.3在建立质量管理体系时,以ISO13485为标准 ISO13485 是基于ISO9001基础上的对医疗器械的专用标准,从2003年开始成为一个独立的标准,名为《医疗器械质量管理体系用于法规的要求》,此标准的主要目的是便于实施经协调的质量管理体系的法规要求,此标准包含了一些医疗器械的专用要求,删减了ISO9001中不适用于作为法规要求的某些要求。ISO13485的所有要求是针对提供医疗器械的组织,不论组织的类型或规模。我公司在咨询过程中是以 ISO13485为标准的。 1.4医疗器械企业质量管理体系中要渗入GMP GMP是英文名Good Manufacturing Practices的缩写,我国一般称其为"良好的生产管理规范"。GMP是人类社会科学技术进步和管理科学发展的必然产物,它是适应保证药品或医疗器械生产管理的需要而产生的。医疗器械最终质量的保证必须依靠整个生产过程中的良好管理,才能降低最终产品出现不合格的风险,使医疗器械的安全性加强。所以企业在建立质量管理体系时要立足ISO13485,引入GMP,提高产品质量,保护消费者的利益。 2医疗器械企业质量管理体系的建立 2.1优先培训决策层——导入ISO13485质量管理体系的前奏现代的质量管理观念强调:"质量从头头开始,从头开始。"也就是强调质量观念的更新、根植,质量策划的运筹,都需要从领导做起。 2.2决策层的关键作用
精选医疗器械企业质量管理体系的建立
医疗器械企业质量管理体系的建立 2.1、优先培训决策层一一导入ISO13485质量管理体系的前奏 现代的质量管理观念强调:"质量从头头开始,从头开始。"也就是强调质量观念的更新、 根植,质量策划的运筹,都需要从领导做起。 2.2、决策层的关键作用 1994版ISO9000标准中曾将一个企业选用质量管理体系标准的驱动动机分为两类:管理者驱动和受益者推动。而实际上,无论管理者(此为决策领导)自主推动,亦或是来自于受益者的推动压力而被动选用,最终都要经过决策领导的导入决定。最高管理者是企业成功推 行ISO13485标准的关键,,应在企业内形成一种重视质量、关注顾客的氛围,并提供充足的资源,为推行ISO13485标准做好领导作用。 2.3、决策层的培训 决策领导是企业的核心,其决策及表现对整个企业具有决定性影响和放大效应。 (1)选择适宜的培训方式。我公司选择有经验的咨询老师到企业进行培训,确保企业在正常生产的同进完成培训工作。 (2)确保重点培训内容。决策领导需要掌握的ISO13485知识至少应包括:ISO13485标准 的产生背景,发展形式和趋势,成功运作ISO13485质量管理体系的成功组织的案例,质量 方针和目标的设定,质量意识的强化、管理职责,质量策划,管理评审,质量成本管理、质 量管理体系与企业管理其他部分的关系等等。 3、医疗器械企业质量管理体系文件的建立 3.1、根据ISO13485标准的要求策划质量管理体系。 3.2、识别ISO13485,确定标准中适用的条款和不适用的条款。 3.3、根据标准的要求确定文件的等级,一般文件的等级如下: a)第一层次文件:质量手册 b)第二层次文件:程序文件 c)第三层次文件:作业指导书类,即是操作类文件 3.4、起草企业的《质量手册》。
医疗器械生产质量管理制度
医疗器械生产质量 管理制度 1 2020年4月19日
医疗器械生产质量管理制度 第一章总则 第一条为了加强医疗器械生产监督管理,规范医疗器械生产质量管理体系,根据《医疗器械生产质量管理规范》,制定本细则。 第二条医疗器械生产企业(以下简称生产企业)应当根据产品的特点,按照医疗器械生产质量管理规范和本细则的要求,建立质量管理体系,并保持有效运行。 作为质量体系的一个组成部分,医疗器械生产企业应当在产品实现全过程中实施风险管理。 第三条本细则适用于除无菌医疗器械、植入性医疗器械、体外诊断试剂等已发布分类实施细则以外的其它医疗器械的设计开发、生产、销售和服务的全过程。 第二章管理职责 第四条生产企业应当建立相应的组织机构,规定各机构的职责、权限,明确质量管理职能。生产管理部门和质量管理部门负责人不得互相兼任。 第五条生产企业负责人应当具有以下职责: 1.组织制定生产企业的质量方针和质量目标; 2.组织策划并确定产品实现过程,确保满足顾客要求; 3.确保质量管理体系有效运行所需的人力资源、基础设施和工 作环境; 4.组织实施管理评审并保持记录; 5.指定人员负责相关法律法规的收集,确保相应法律法规在企 业内部贯彻和执行。 第六条生产企业负责人应当确定一名管理者代表,负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高企业员工满足法规和顾客要求的意识。 第三章资源管理 第七条生产、技术和质量管理部门的负责人应当掌握医疗器械的法规、具有质量管理的实践经验和相应的专业知识,有能力对生产和质量管理中的实际问题做出正确的判断和处理。
第八条生产企业应当确定影响医疗器械质量的岗位,规定这些岗位人员所必须具备的专业知识水平、工作技能、工作经验,并对从事这些岗位工作人员的能力进行评价,对未满足规定要求的,要采取相应的措施,以满足要求。医疗器械生产操作及质量检验人员,应当经过相应技术培训,具有相关理论知识和实际操作技能。 第九条生产企业应当具备并维护产品生产所需的生产场地、生产设备、监视和测量装置、仓储场地等基础设施以及工作环境,生产环境应当符合相关法规和技术标准的要求。 第十条若工作环境条件可能对产品质量产生不利影响,生产企业应当建立对工作环境条件要求的控制程序并形成文件或作业指导书,以监视和控制工作环境条件。 第十一条生产企业应当有整洁的生产环境。厂区周边环境不应对生产过程和产品质量造成影响,行政区、生活区和辅助区的总体布局合理,不得对生产区有不良影响。 第十二条生产企业应当确定产品生产中须避免污染、在相应级别洁净室(区)进行生产的过程。空气洁净级别不同的洁净室(区)之间的静压差应大于5帕,洁净室(区)与室外大气的静压差应大于10帕,并应有指示压差的装置。洁净室(区)级别设置应当符合《医疗器械生产质量管理规范无菌医疗器械实施细则》附录要求。 第十三条洁净室(区)应当按照生产工艺流程及所要求的空气洁净度级别进行合理布局。同一洁净室(区)内或相邻洁净室(区)间的生产操作不得互相交叉污染。 洁净室(区)的温度和相对湿度应当与产品生产工艺要求相适应。在无特殊要求时,温度应控制在18~28℃,相对湿度控制在45%~65%。 第十四条生产厂房应当设置防尘、防止昆虫和其它动物进入的设施,门、窗及安全门应当密闭。生产操作间的内表面应当便于清洁,能耐受清洗和消毒。 第十五条生产企业应当制定卫生管理文件,按照规定进行清洁、清洗和消毒,并做好记录。所用的消毒剂或消毒方法不得对设备、工艺装备、物料和产品造成污染。 第十六条生产企业应当建立对人员健康的要求,并形成文件。建立人员健康档案,对患有传染性和感染性疾病的人员不得从事直接接触产品的工作。 第十七条生产企业应当建立对人员服装和人员清洁的要求,并形成文件。 第十八条生产企业应当确定所需要的工艺用水,当生产过程中使用工艺用水时,应当配备相应的制水设备,并有防止污染的措施。工艺用水应当满足产品质量的要求。
(完整版)医疗器械质量手册
******公司 质量手册 版号:A版 分发号: 受控状态: 发布日期:年月日实施日期:年月日
编制:日期:年月日批准:日期:年月日
本质量手册由管理者代表组织编写,经审查符合GB/T19001—2000idt ISO9001:2000《质量管理体系要求》和YY/T0287—2003 idtISO13485:2003《医疗器械质量管理体系用于法规的要求》标准,现予以批准、发布。 质量手册作为全公司职工质量工作的法规和纲领性文件,用以统一协调全公司的质量管理活动,各级人员都要在本职工作中认真贯彻执行。 总经理: 年月日
D 企业概况 ******公司,现有员工5人,中专以上学历5名。公司总面积318平方米,其中经营50面积平方米,仓储面积880平方米。相应的检测仪器、仓储设备齐全,基本符合医疗器械验收、贮存要求。 公司拟经营一、二、三类医疗器,包括植、介入医疗器械和一次性使用无菌医疗器械产品。公司严格按照IS09001、YY/0287标准质量管理体系的要求,建立质量管理网络,从医疗器械的进货、储存、体系的要求,从产品的产品检验、售后服务以及各个环节建立了控制程序,严把进货关、质量关。并按照质量管理备了计算机管理系统,能满足产品经营管理全过程及质量控制要求。确保了公司销出使顾客满意的医疗器械。公司按照《一次性使用无菌医疗器械监督管理办法(暂行)》(国家药品监督管理局局令第24号)以及《山东省〈医疗器械经营企业许可证管理办法〉实施细则补充规定》的要求,完善质量管理。 地址: 电话: 邮编: 法定代表人:
E 管理者代表任命书 为确保本公司质量体系的建立、实施和保持有效运行,兹任命同志担任本公司管理者代表,代表本公司管理层负责本公司质量体系的建立、实施和保持,向公司管理层报告质量体系的运行情况,及时处理影响质量体系运行的有关问题。 总经理: 年月日
医疗器械生产厂家质量手册2018
XXX医疗器械有限公司 第A/1版文件编号:MMM/A-A/1 质量手册 依据YY/T0287-2017&ISO13485:2016标准 医疗器械生产质量管理规范 编制: 审核: 批准: 2018-1-1发布2018-1-1实施 XXX医疗器械有限公司发布
质量手册修改控制页
0.1 发布令 质量手册发布令 为了满足市场的需求和顾客的要求,适应市场的竞争,并使公司的质量管理与国际接轨,不断提高公司的质量管理水平,依据最新颁布的《医疗器械管理条例》、《医疗器械质量管理规范》等医疗器械行业有关法规及YY/T0287-2017&ISO13485:2016《医疗器械——质量管理体系用于法规的要求》,依据标准要求,结合本公司产品特性,在管理者代表的组织下,通过对企业质量管理体系的策划和建立,对原质量手册(ZLQM/A-A/0)进行修订,编制本质量手册(包括程序文件),现予以发布实施。 本《质量手册》描述了公司的质量管理体系,对公司的质量管理体系提出了具体要求。阐明了质量方针和质量目标,是公司法规性文件,也是向顾客和认证机构提供信任的依据.本手册主要目的是为实施经协调的质量管理体系的法规要求,并以此作为本公司医疗器械设计、开发、生产和服务的依据,证实本公司有能力提供持续满足顾客要求和适用于医疗器械和相关服务的法规要求。本手册所规定的质量管理体系要求是对产品技术要求的补充。 本质量手册自发布之日起生效实施,要求全体员工认真学习,充分理解,正确执行公司的质量方针,严格按照“质量手册”的要求落实质量责任,开展质量管理活动,满足法律法规要求和顾客期望,达到顾客满意。本公司现有文件体系中与本手册相悖或不一致处,应按本手册为准。 原质量手册同日起废止。 总经理:阿林 2018年1月1日
医疗器械管理体系手册
广州健福医疗科技有限公司 质量手册 文件编号: 版本:0 页数:31 受控状态: 2012-05-01发布 2012-05-01 实施编制:小组审核:吴少乐批准:张晓东
颁布令 本公司按 9001:2008《质量管理体系要求》和《化妆品生产许可工作规范》,结合本公司的实际情况,遵循国家、地区和行业有关法规编制本手册。手册中各页由质量负责人组织编写,经审核后由董事长批准,现予发布执行。 本手册是用来阐明本公司质量方针和描述实施质量管理活动的纲领性文件,对内进行质量管理,对外是本公司品质保证能力的证实文件,目的在于帮助我们清楚品质管理体系及其运作,为内部质量管理体系管理和外部相关方满意提供工作指南和依据。 本质量手册发布后,要求“广州健福医疗科技有限公司”职员应落实贯彻执行,并就手册中相关程序或作业文件做定期研讨、修正,以保证公司整体经营效益与产品品质不断提升,本着“持续进步”的目标不懈努力。 现行的质量手册是本公司质量管理的证据及第三方认证的依据。 现予批准发行并于2012年05月01日开始实施。 签署: 日期:2012-05-01
任命书 兹正式任命苏为广州健福医疗科技有限公司的企业负责人,贯彻执行国家法律法规、方针政策和强制性标准、组织监督相关人员建立和完善各项规章制度并执行。 董事长签署:张 日期:2012-05-01 任命书 兹正式任命由吴担任本公司质量负责人,其职责和权限如下: 1.《化妆品生产许可检查要点》的组织实施; 2.质量管理制度体系的建立和运行; 3.产品质量问题的决策。 董事长签署:张 日期:2012-05-01
任命书 兹正式任命由吴担任本公司质量管理部门负责人,其职责和权限如下: 1.负责内部检查及产品召回等质量管理活动; 2.确保质量标准、检验方法、验证和其他质量管理规程有效实施; 3.确保原料、包装材料、中间产品和成品符合质量标准; 4.评价物料供应商; 5.负责产品的放行; 6.负责不合格品的管理; 7.负责其他与产品质量有关的活动。 董事长签署:张 日期:2012-05-01 任命书 兹正式任命由吴担任本公司生产负责人,其职责和权限如下: 1.确保产品按照批准的工艺规程生产、储存; 2.确保生产相关人员经过必要和持续的培训; 3.确保生产环境、设施设备满足生产质量需求。 董事长签署:张
医疗器械质量手册
医疗器械质量手册 质量手册 文件编号:×××××/QH-01 编制:××× 2005 年×月×日 审核:××× 2004 年×月×日 批准:××× 2004年×月×日 版号: , 分发号:2005第××号 受控状态: 持有者: 2005-××-××发布 2005-××-××实施 ×××××××××××有限公司发布 质量手册颁发令 质量是企业的生命和希望,全体员工务必牢记本公司的质量方针,并以此为己任,在质量管理体系活动中贡献力量。 2000《质量管理体为实施本公司的质量方针,有效地开展质量活动,依据 GB/T 19001-系要求》(idt ISO 9001: 2000)及YY/T 0287-2003《医疗器械质量管理体系用于法规的要求》(idt ISO13485:2003)、国家药品监督管理局令第22号《医疗器械生产公司质量体系考核办法》的要求,结合本公司实际编制了《质量手册》A版,本《质量手册》描述了公司的质量管理体系,对公司的质量管理体系提出了具体要求。阐明了质量方针和质量目标,是公司法规性文件,也是向顾客和认证机构提供信任的依据,本手册与公司的其它管理体系相一致。现予以批准发布。
本手册主要目的是为实施经协调的质量管理体系的法规要求,并以此作为医疗器械设计、开发、生产和服务的依据,证实本公司有能力提供持续满足顾客要求和适用于医疗器械和相关服务的法规要求。本手册所规定的质量管理体系要求是对产品技术要求的补充。 在不影响提供满足顾客和适用法律法规要求的产品和不影响公司相应责任的那些质量管理要求前提下,依据公司提供产品的性质、法律法规要求,对质量管理体系要求进行合理删减,删减后质量管理体系符合YY/T 0287-2003《医疗器械质量管理体系用于法规的要求》、国家药品监督管理局令第22号《医疗器械生产公司质量体系考核办法》。 本手册覆盖的产品为: 本手册是公司质量管理的基本法规,质量体系的纲领性文件,质量管理体系运行的准则,质量活动应遵循的基本规则,也是对所有顾客的承诺,所有员工自本手册实施之日起,必须遵照执行,以保证公司质量管理体系的有效运行和质量方针、目标的实施。 本手册自×××× 年××月×× 日起生效实施,全体员工必须理解、贯彻并效力。本公司现有文件体系中与本手册相悖或不一致处,应按本手册为准。 为保持质量管理体系的持续有效,特任命副总经理×××先生为管理者代表,其主要职责和权限为: a)确保公司质量管理体系的过程得到建立和保持; b)向总经理报告质量管理体系的业绩,包括改进的需求; c)在整个公司内促进顾客要求意识的形成; d)就质量管理体系的有关事宜与外部联系。 总经理: 2005 年××月××日
医疗器械02 质量手册
质量手册 发布日期2016-03-28 1.公司简介: XXX医疗器械制造有限公司,主要从事设计和制造本企业商标和客户各类医疗器械产品。公司总占地面积200余亩,主要产品是:骨科的各类医疗器械产品,前期生产高分子夹板、绷带,产品主要销售到国内外市场。 2.公司地址:XXX; 电话:0769-XXXXXXX。 2.1术语; a.本品质手册使用的有关术语和定义参照并采用GB/T19000:2000(IDT ISO9000:2000)/YY/T0287(IDT ISO13485:2003)。 b.本品质手册所采用的供应链为:供方组织客户 c.本品质手册中,有时“组织”也被称为“本公司”。 2.2代号说明: 品质系统所有系统文件,指南,工程、生产所使用受控文件的代码,在《文件格式程序》中规定。 3.1职责 3.1.1《品质手册》由管理者代表组织文件编写小组编写,副总经理审核后,呈总经 理批准颁布实施。 3.1.2文件控制中心负责体系文件的统一发放、回收、销毁、记录、保存的管理。3.2管理: 3.2.1《品质手册》由文件控制中心统一编号、发放、记录,受控副本则于副本文件 封面及每一内页盖上“受控文件”印章,具体依QPMD003《文件控制程序》 QPMD002《工程变更管理程序》执行。 3.2.2文件控制组根据管理者代表指定的发放范围进行发放复印副本。 3.3修订: 3.3.1《品质手册》在执行过程中出现下列情况时需进行修订和再版: a.GB/T19001:2000(IDT ISO9001:2000)/YY/T0287:2003(IDTISO13485:2003)发生修改; b.组织结构发生变动; c.产品结构发生变动; d.品质管理体系审核及管理评审提出改进要求; e.市场要求变化以及本公司持续改进需要; f.品质方针和目标调整。 3.3.2《品质手册》的修订版本号及文件编号的原则,依照QPMD006《文件格式程序》 执行。
医疗器械生产企业质量管理体系质量手册(DOC 52页)
医疗器械生产企业质量管理体系质量手册(DOC 52页)
医疗器械生产企业质量管理体系质量手册
目录 章节号标题页 码 章节号标题页码 0.1 前言 3 附录4 适用法规明细表51 0.2 组织结构图 4 附录5 适用法律法规明细表52 0.3 发布令 5 附录6 工艺流程图53 0.4 质量方针和质量目标 6 0.5 过程指责分配表7 0.6 管理者代表任命书9 0.7 企业概况10 1.0 范围11 2.0 引用标准12 3.0 术语和定义13 4.0 质量管理体系15 5.0 管理职责19 6.0 资源管理25 7.0 产品实现30 8.0 测量分析和改进42 附录1 手册的管理48 附录2 更改控制记录49 附录3 引用程序文件明细表50
0.1 前言 医疗器械涉及到人身安全和健康,相对于其他产品,其安全性和有效性具有较高的要求。而产品的安全性和有效性集中反映了产品的质量。产品的质量与生产企业的质量管理体系密切相关。为保证产品的质量,提高企业在国内外的市场竞争能力,根据本企业的产品特点、实际管理情况以及国家有关法律、法规的要求,按照YY/T 0287—2003 IDT ISO13485:2003《医疗器械质量管理体系用于法规的要求》的模式,建立质量管理体系文件—《质量手册》。 本手册是按照03版YY/T 0287的要求,编制而成。手册阐明了企业的质量方针和质量目标,对企业的质量管理体系进行了描述,包括组织结构、职责权限等。是企业从事质量活动的纲领法规性文件和准则,全体员工必须认真遵照执行,以确保能向顾客提供满意的、满足法规要求的产品,从而提高企业的管理水平和经济效益。 本手册强调以过程为基础的质量管理体系模式。 质量管理体系的改进 顾 客法规 顾 客管理职责 资源管理 测量、分 析和改进
医疗器械经营企业质量手册
绩溪同帆医疗器械销售有限公司质量手册 版号:A版 分发号:2-01 受控状态:受控 发布日期:2011年12月20日 实施日期:2011年12月20日
编制:日期:2011年12月20日批准:日期:2011年12月20日
本质量手册由管理者代表组织编写,经审查符合GB/T19001—2000idt ISO9001:2000《质量管理体系要求》和YY/T0287—2003 idtISO13485:2003《医疗器械质量管理体系用于法规的要求》标准,现予以批准、发布。 质量手册作为全公司职工质量工作的法规和纲领性文件,用以统一协调全公司的质量管理活动,各级人员都要在本职工作中认真贯彻执行。 总经理: 2011年12月20日
D企业概况 绩溪同帆医疗器械销售有限公司,现有员工12人,中专以上学历12名。公司总面积396平方米,其中经营186平方米,仓储面积210平方米。相应的检测仪器、仓储设备齐全,基本符合医疗器械验收、贮存要求。 公司拟经营二、三类医疗器械,包括植、介入医疗器械产品。公司严格按照IS09001、YY/0287标准质量管理体系的要求,建立质量管理网络,从医疗器械的进货、储存、体系的要求,从产品的产品检验、售后服务以及各个环节建立了控制程序,严把进货关、质量关。并按照质量管理备了计算机管理系统,能满足产品经营管理全过程及质量控制要求。确保了公司销出使顾客满意的医疗器械。公司按照《一次性使用无菌医疗器械监督管理办法(暂行)》(国家药品监督管理局局令第24号)以及《河南省〈医疗器械经营企业许可证管理办法〉实施细则补充规定》的要求,完善质量管理。 地址:郑州市金水区南阳路301号附11号楼 电话: 邮编:450000 法定代表人:
医疗器械公司质量手册
医疗器械公司质量手册 This manuscript was revised by the office on December 10, 2020.
受控编号: XXXX医疗器械有限公司 第A/0版文件编号:XX/QB(PS)质 量手册 (包括:程序文件) 依据:YY/T0287-2003 编制: 审核: 批准: 20XX-05-28发布 20XX-05-28实施XXXX医疗器械有限公司发布 质量手册发布令
依据YY/T0287-2003标准要求,结合本公司产品特性,在管理者代表的组织下,通过对企业质量管理体系的策划和建立,编制了本质量手册(包括程序文件),现予以发布实施。 本手册是本企业质量管理体系的法规性文件,具体阐述了企业质量方针和质量目标,描述了质量管理体系的范围、过程和相互作用。是指导企业建立实施和保持质量管理体系的纲领和行动准则。企业全体员工应严格遵守执行。 总经理: 20XX年5月28日 任命书
为贯彻执行YY/T0287-2003标准,加强企业质量管理体系运行的监督、检查和考核,特任命同志为本公司管理者代表,全面负责质量管理体系的建立、实施,保持和改进。 总经理: 20XX年5月28日
1 XXXX医疗器械有限公司
主题内容 第A/0版 XX/ 本质量手册阐述了本公司的质量方针和质量目标,依据并引用了YY/T0287-2003及YY/T0316-2003标准的核心内容,对质量管理体系的范围、过程和相互作用进行了识别和描述,是企业开展质量方针、质量控制、质量保证和质量改进活动的纲领性文件。 编制:批准: 20XX-05-28 2
医疗器械质量手册新建
质量手册
前言 本《质量手册》是我公司质量管理体系的纲领性文件。本文件由公司总经理组织各有关部门根据医疗器械生产质量管理规范及ISO9001:2000,结合本公司的实际情况编制而成,经过认真审核。现预发布实施。 《质量手册》内容包括: 1)本公司质量管理体系的范围。 2)本公司质量管理体系所形成文件的程序; 3)本公司质量管理体系过程之间的顺序和相互作用的表述。 本《质量手册》从颁发之日起,要求公司各部门、全体员工严格。贯彻执行。
任命书 为了贯彻执行医疗器械生产质量管理规范,旨在本公司有效建立、实施和保持一个完善的质量管理体系,特任命为本公司管理者代表,并担当如下职责和权限: 1)确保质量管理体系所需的过程得到建立,实施和保持; 2)向最高管理者报告质量管理体系的业绩和任何改进的需求; 3)确保在整个组织内提高满足法规要求和顾客要求的意识; 4)负责就质量管理体系有关事宜的外部联络。
质量方针 人民健康至上,产品质量第一 。 质量目标 一. 严格执行医疗器械生产质量管理规范及ISO9001:2000。 二. 产品的性能指标达到医疗器械企业标准。 三. 产品出厂合格率达到100%。 四. 不断开发系列化新产品,以填补国内空白。
目录0.1 目录 0.2 质量手册说明 0.3 企业概况质量 0.4 公司组织机构图 05 公司质量管理体系结构图 06 质量管理体系过程职责分配表 07 质量管理体系 08 文件控制程序 09 质量记录控制程序 10 管理职责 11 管理评审控制程序 12 人力资源控制程序 13 工作环境控制程序职责和权限 14 与顾客有关的过程控制程序 15 评定供方控制程序资源管理 16 采购控制程序 17 生产和服务提供控制程序 18 内部审核程序产品实现 19 过程和产品的测量和监控程序 20 不合格控制程序 21 纠正、预防、改进措施控制程序 22 顾客信息反馈控制程序 质量记录清单: SB-JL-01-01- SB-JL-01-055 SB-WJ-01-02
医疗器械质量手册2017
质量手册 文件类别:□受控本□非受控本文件持有部门: 文件发放编号:
0.0 目录 0.0 质量手册目录┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄1 0.1 修改页┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 5 0.2 批准页┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄6 0.3 公司简介┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 7 0.4 管理者代表任命书┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 8 0.5 质量方针、质量目标┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 9 0.6 各部门质量目标分解┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 10 0.7 公司组织机构图┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 11 0.8 质量体系组织结构图┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 12 0.9 质量管理职能分配表┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄ 13 1 范围┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄16 2 引用标准┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄┄17
(完整word版)医疗器械生产质量管理制度
医疗器械生产质量管理制度 第一章总则 第一条为了加强医疗器械生产监督管理,规范医疗器械生产质量管理体系,根据《医疗器械生产质量管理规范》,制定本细则。 第二条医疗器械生产企业(以下简称生产企业)应当根据产品的特点,按照医疗器械生产质量管理规范和本细则的要求,建立质量管理体系,并保持有效运行。 作为质量体系的一个组成部分,医疗器械生产企业应当在产品实现全过程中实施风险管理。 第三条本细则适用于除无菌医疗器械、植入性医疗器械、体外诊断试剂等已发布分类实施细则以外的其他医疗器械的设计开发、生产、销售和服务的全过程。 第二章管理职责 第四条生产企业应当建立相应的组织机构,规定各机构的职责、权限,明确质量管理职能。生产管理部门和质量管理部门负责人不得互相兼任。 第五条生产企业负责人应当具有以下职责: 1.组织制定生产企业的质量方针和质量目标; 2.组织策划并确定产品实现过程,确保满足顾客要求; 3.确保质量管理体系有效运行所需的人力资源、基础设施和工 作环境; 4.组织实施管理评审并保持记录; 5.指定人员负责相关法律法规的收集,确保相应法律法规在企 业内部贯彻和执行。 第六条生产企业负责人应当确定一名管理者代表,负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高企业员工满足法规和顾客要求的意识。 第三章资源管理 第七条生产、技术和质量管理部门的负责人应当掌握医疗器械的法规、具有质量管理的实践经验和相应的专业知识,有能力对生产和质量管理中的实际问题做出正确的判断和处理。 第八条生产企业应当确定影响医疗器械质量的岗位,规定这些岗位人员所必须具备的专业知识水平、工作技能、工作经验,并对从事这些岗位工作人员的能力进行评价,对未满足规定要求的,要采取相应的措施,以满足要求。医疗器械生产操作及质量检验人员,应当经过相应技术培训,具有相关理论知识和实际操作技能。 第九条生产企业应当具备并维护产品生产所需的生产场地、生产设备、监视和测量装置、仓储场地等基础设施以及工作环境,生产环境应当符合相关法规和技术标准的要求。 第十条若工作环境条件可能对产品质量产生不利影响,生产企业应当建立对工作环境条件要求的控制程序并形成文件或作业指导书,以监视和控制工作环境条件。 第十一条生产企业应当有整洁的生产环境。厂区周边环境不应对生产过程和产品质量造成影响,行政区、生活区和辅助区的总体布局合理,不得对生产区有不良影响。 第十二条生产企业应当确定产品生产中须避免污染、在相应级别洁净室(区)进行生产的过程。空气洁净级别不同的洁净室(区)之间的静压差应大于5帕,洁净室(区)与室外大气的静压差应大于10帕,并应有指示压差的装置。洁净室(区)级别设置应当符合《医疗器械生产质量管理规范无菌医疗器械实施细则》附录要求。 第十三条洁净室(区)应当按照生产工艺流程及所要求的空气洁净度级别进行合理布局。同一洁净室(区)内或相邻洁净室(区)间的生产操作不得互相交叉污染。
医疗器械质量管理体系文件之质量手册
受控编号:XXXX医疗器械有限公司 第A/0版文件编号:XX/QB(PS)01-0.1-8.5 质量手册 (包括:程序文件) 依据:YY/T0287-2003 编制: 审核: 批准: 20XX-05-28发布20XX-05-28实施 XXXX医疗器械有限公司发布 质量手册发布令 依据YY/T0287-2003标准要求,结合本公司产品特性,在管理者代表的组织下,通过对企业质量管理体系的策划和建立,编制了本质量手册(包括程序文件),现予以发布实施。 本手册是本企业质量管理体系的法规性文件,具体阐述了企业质量方针和质量目标,描述了质量管理体系的范围、过程和相互作用。是指导企业建立实施和保持质量管理体系的纲领和行动准则。企业全体员工应严格遵守执行。 总经理:
20XX年5月28日 任命书 为贯彻执行YY/T0287-2003标准,加强企业质量管理体系运行的监督、检查和考核,特任命同志为本公司管理者代表,全面负责质量管理体系的建立、实施,保持和改进。 总经理: 2008年5月28日 目录
1 XXXX医疗器械有限公司 主题内容 第A/0版XX/QB01-0.2 本质量手册阐述了本公司的质量方针和质量目标,依据并引用了YY/T0287-2003及YY/T0316-2003标准的核心内容,对质量管理体系的范围、过程和相互作用进行了识别 和描述,是企业开展质量方针、质量控制、质量保证和质量改进活动的纲领性文件。编制:批准:20XX-05-28 2 XXXX医疗器械有限公司 企业概况 第A/0版XX/QB01-0.2 公司简介 3 XXXX医疗器械有限公司 目的范围 第A/0版XX/QB01-1.0 1 目的 为依据YY/T0287-2003、YY0033-2000及YY/T0316-2003标准建立、实施和保持质
医疗器械13485质量手册
质量手册 [依据:医疗器械-质量管理体系-用于法规的要求] 编制/日期: 审核/日期: 批准/日期: 受控状态:□受控□非受控 持有者: 2010年09月01日发布2010年10月01日实施 XXXXXXXX
颁布令 总经理: 2010年9月1日
管理者代表任命书 为确保我企业质量管理体系的建立、实施和有效运行,并持续改进其有效性,现任命 XXX同志为我公司质量管理体系管理者代表,全权负责质量管理体系的相关事宜。 管理者代表职责: 1.确保质量管理体系公司需的过程得到建立、实施和保持; 2.向总经理报告质量管理体系的业绩和任何改进的需求; 3.确保在全公司范围内提高满足法规要求和顾客要求的意识; 4.负责本公司质量管理体系有关事宜的对外联络。 希望各部门和全体员工给予积极的支持和配合。 2010年9月1日
目录 0.1 企业简介 (7) 0.2 公司组织机构图 (8) 1质量体系职能分配表 (9) 2引用标准和有关定义 (10) 2.1 引用标准: (10) 2.2 文献 (10) 2.3 定义术语 (10) 3质量手册说明 (11) 4质量管理体系 (12) 4.1 质量管理体系总要求 (13) 4.2 质量管理体系文件要求 (14) 4.2.1总则 (14) 4.2.2质量手册 (14) 4.2.3文件控制 (14) 4.3 文件控制程序 (15) 4.3.1目的 (15) 4.3.2范围 (15) 4.3.3职责 (15) 4.3.4程序 (15) 4.3.4.1 文件编号 (15) 4.3.4.2 文件编写、审核、批准 (16) 4.3.4.3 文件登记 (16) 4.3.4.4 文件发放 (16) 4.3.4.5 文件受控状态 (16) 4.3.4.6 文件更改和换版 (16) 4.3.4.7 文件保存、作废与销毁 (16) 4.3.4.8 外来文件控制 (17) 4.3.5相关文件 (17) 4.3.6质量记录 (17) 4.4 质量记录控制程序 (18) 4.4.1目的 (18) 4.4.2范围 (18) 4.4.3职责 (18) 4.4.4程序 (18) 4.4.4.1 记录标识编号 (18) 4.4.4.2 记录填写 (18) 4.4.4.3 记录的保存、保护 (18) 4.4.4.4 记录发放、借阅和复制 (19) 4.4.4.5 记录销毁处理 (19)