蛋白-小分子对接(含同源建模)

蛋白-小分子对接(含同源建模)
1.项目说明
采用同源模建方法构建单链抗体(以下简称“抗体”)的三维结构,通过分子对接方法预测化合物的结合模式(图 1)。
图1.化合物两种构型的化学结构
2.计算方法
本研究采用的计算方法简述如下(详见《计算方法》文档):
A.采用在线工具PIGSPro预测抗体的三维结构,通过分子动力学模拟优化结构;
B.采用在线工具POCASA 1.1预测优化的抗体结构上潜在的结合位点;
C.采用DOCK 6.7将化合物对接到各个预测位点中,根据打分和结合模式,挑选最佳的结合模式进行分析。
3.结果分析
A.同源模建
采用在线工具PIGSPro
(http://cassandra.med.uniroma1.it/AbPrediction/web/)对抗体进行同源模建。首先进行序列比对,采用单序列模式,从已知三维结构的数据库中分别针对L链和H链搜索序列相似的蛋白质结构。L链的模板为XXX,H链的模板为XXX和XXX。序列比对如下(保密需要,部分数据不公开):
Light Chain Target - Template alignment:
Heavy Chain Target - Template alignment:
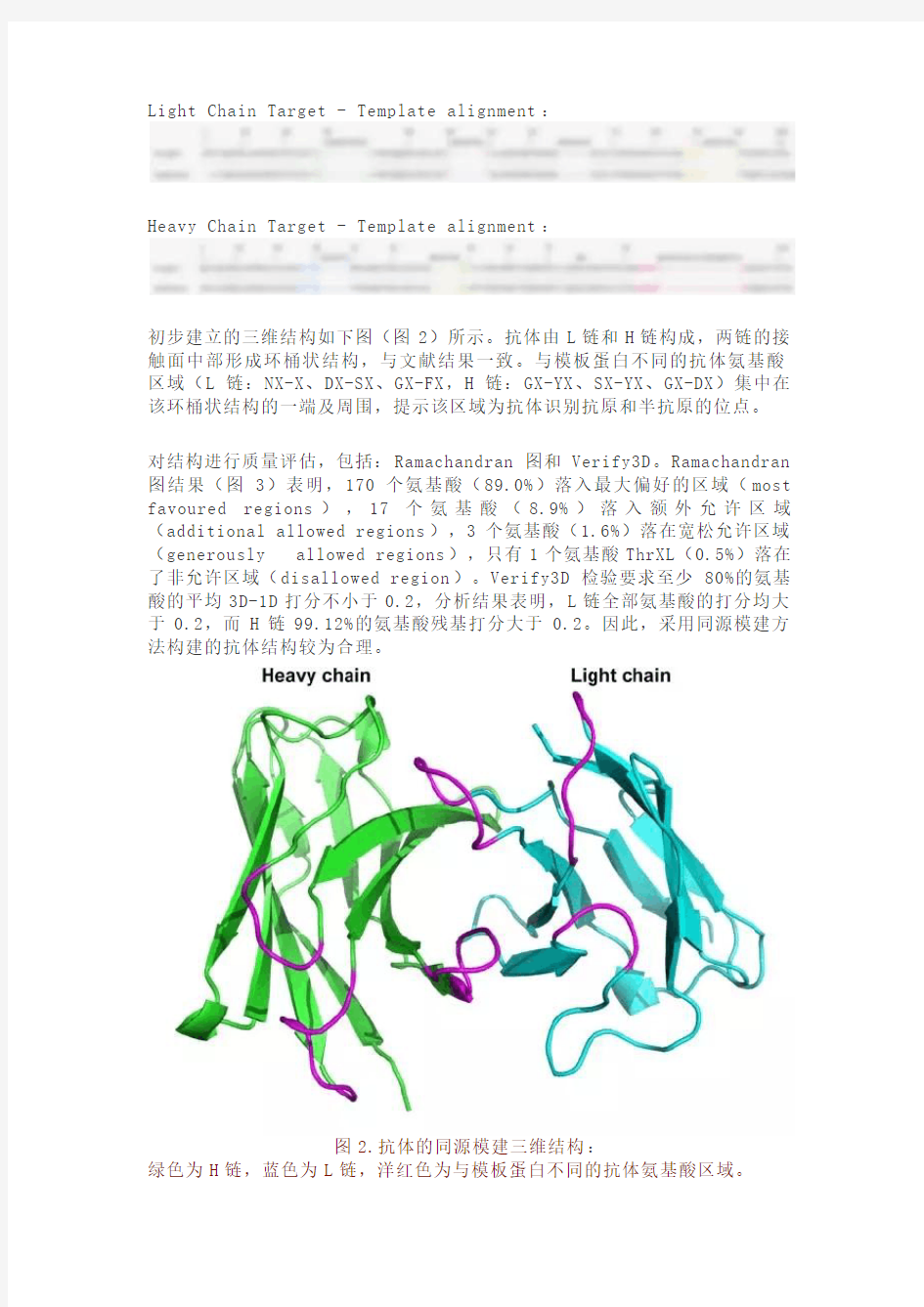
初步建立的三维结构如下图(图2)所示。抗体由L链和H链构成,两链的接触面中部形成环桶状结构,与文献结果一致。与模板蛋白不同的抗体氨基酸区域(L链:NX-X、DX-SX、GX-FX,H链:GX-YX、SX-YX、GX-DX)集中在该环桶状结构的一端及周围,提示该区域为抗体识别抗原和半抗原的位点。
对结构进行质量评估,包括:Ramachandran图和Verify3D。Ramachandran 图结果(图3)表明,170个氨基酸(89.0%)落入最大偏好的区域(most favoured regions),17个氨基酸(8.9%)落入额外允许区域(additional allowed regions),3个氨基酸(1.6%)落在宽松允许区域(generously allowed regions),只有1个氨基酸ThrXL(0.5%)落在了非允许区域(disallowed region)。Verify3D检验要求至少80%的氨基酸的平均3D-1D打分不小于0.2,分析结果表明,L链全部氨基酸的打分均大于0.2,而H链99.12%的氨基酸残基打分大于0.2。因此,采用同源模建方法构建的抗体结构较为合理。
图2.抗体的同源模建三维结构:
绿色为H链,蓝色为L链,洋红色为与模板蛋白不同的抗体氨基酸区域。
图3.抗体结构优化前后的Ramachandran图
B.动力学优化
采用同源模建方法构建的抗体三维结构仍然存在一些质量问题,比如L链的ThrX落在非允许区域内,H链的ArgX、LysX和GluX落在宽松允许区域。其中,LysX和GluX位于环桶状区域内,GluX可能是抗体的识别位点之一。因此,我们采用分子动力学模拟方法对结构进行优化。
我们采用Gromacs 2016在水溶液中进行了10 ns的动力学模拟。骨架C原子的RMSD曲线表明(图4),体系已经达到平衡状态。基于RMSD的聚类分析表明,蛋白质各帧构象非常相似(RMSD < 0.15nm),全部帧落入同一簇中,因此,采用最后一帧的构象作为抗体的最终优化结构。
Ramchandran质量评估结果表明(图3),原出于非允许区域和宽松允许区域的氨基酸残基均得到优化。Verify3D结果也表明,L链全部氨基酸残基合格,H链99.12%的氨基酸残基合格。因此,该结构已得到优化,可用作分子对接。
图4.分子动力学模拟RMSD图
C.结合位点预测
在优化的抗体结构基础上,采用POCASA
(http://altair.sci.hokudai.ac.jp/g6/service/pocasa/)在线工具预测结合位点。结果表明(图 5),抗体存在4个可能的结合位点(Site 1-4),其中Site 1-3位于环桶状结构区域开口同一侧,Site 4位于另一侧。Site 4体积最小,不足以容纳小分子,我们认为该区域为伪位点。因此,我们选取Site 1-3作为分子对接的3个潜在的结合位点。
图5.抗体的预测结合位点
综上所述,抗体通过氢键、π-π堆积、π-阳离子作用、卤键及疏水作用等相互作用特异性地识别化合物,而针对不同构型的化合物,识别模式有一些区别。
图6.化合物R构型(A)和S构型(B)在抗体Site 1的结合模式:
抗体以泥绿色(smudge)卡通(cartoon)形式表示,关键残基以麦色(wheat)棍状(stick)表示,化合物则以绿色(green)棍状表示;蓝色虚线表示氢键,绿色虚线表示卤键,橙色虚线表示π-π堆积,黄色虚线表示π-阳离子相互作用,灰色虚线表示疏水作用。
SWISSMODEL蛋白质结构预测教程
SWISS-MODEL 蛋白质结构预测 SWISS-MODEL是一项预测蛋白质三级结构的服务,它利用同源建模的方法实现对一段未知序列的三级结构的预测。该服务创建于1993年,开创了自动建模的先河,并且它是讫今为止应用最广泛的免费服务之一。 同源建模法预测蛋白质三级结构一般由四步完成: 1.从待测蛋白质序列出发,搜索蛋白质结构数据库(如PDB,SWISS-PROT等),得到许多相似序 列(同源序列),选定其中一个(或几个)作为待测蛋白质序列的模板; 2.待测蛋白质序列与选定的模板进行再次比对,插入各种可能的空位使两者的保守位置尽量对齐; 3.建模:调整待测蛋白序列中主链各个原子的位置,产生与模板相同或相似的空间结构——待测 蛋白质空间结构模型; 4.利用能量最小化原理,使待测蛋白质侧链基团处于能量最小的位置。 最后提供给用户的是经过如上四步(或重复其中某几步)后得到的蛋白质三级结构。 SWISS-MODEL工作模式 SWISS-MODEL服务器是以用户输入信息的最小化为目的设计的,即在最简单的情况下,用户仅提供一条目标蛋白的氨基酸序列。由于比较建模程序可以具有不同的复杂性,用户输入一些额外信息对建模程序的运行有时是有必要的,比如,选择不同的模板或者调整目标模板序列比对。该服务主要有以下三种方式: ?First Approach mode(简捷模式):这种模式提供一个简捷的用户介面:用户只需要输入一条氨基酸序列,服务器就会自动选择合适的模板。或者,用户也可以自己指定模板(最多5条),这些模板可以来自ExPDB模板数据库(也可以是用户选择的含坐标参数的模板文件)。如果一条模板与提交的目标序列相似度大于25%,建模程序就会自动开始运行。但是,模板的可靠性会随着模板与目标序列之间的相似度的降低而降低,如果相似度不到50%往往就需要用手工来调整序列比对。这种模式只能进行大于25个残基的单链蛋白三维结构预测。 ?Alignment Interface(比对界面):这种模式要求用户提供两条已经比对好的序列,并指定哪一条是目标序列,哪一条是模板序列(模板序列应该对应于ExPDB模板数据库中一条已经知道其空间结构的蛋白序列)。服务器会依据用户提供的信息进行建模预测。
蛋白-小分子对接(含同源建模)
蛋白-小分子对接(含同源建模) 1.项目说明 采用同源模建方法构建单链抗体(以下简称“抗体”)的三维结构,通过分子对接方法预测化合物的结合模式(图 1)。 图1.化合物两种构型的化学结构 2.计算方法 本研究采用的计算方法简述如下(详见《计算方法》文档): A.采用在线工具PIGSPro预测抗体的三维结构,通过分子动力学模拟优化结构; B.采用在线工具POCASA 1.1预测优化的抗体结构上潜在的结合位点; C.采用DOCK 6.7将化合物对接到各个预测位点中,根据打分和结合模式,挑选最佳的结合模式进行分析。 3.结果分析 A.同源模建 采用在线工具PIGSPro (http://cassandra.med.uniroma1.it/AbPrediction/web/)对抗体进行同源模建。首先进行序列比对,采用单序列模式,从已知三维结构的数据库中分别针对L链和H链搜索序列相似的蛋白质结构。L链的模板为XXX,H链的模板为XXX和XXX。序列比对如下(保密需要,部分数据不公开):
Light Chain Target - Template alignment: Heavy Chain Target - Template alignment: 初步建立的三维结构如下图(图2)所示。抗体由L链和H链构成,两链的接触面中部形成环桶状结构,与文献结果一致。与模板蛋白不同的抗体氨基酸区域(L链:NX-X、DX-SX、GX-FX,H链:GX-YX、SX-YX、GX-DX)集中在该环桶状结构的一端及周围,提示该区域为抗体识别抗原和半抗原的位点。 对结构进行质量评估,包括:Ramachandran图和Verify3D。Ramachandran 图结果(图3)表明,170个氨基酸(89.0%)落入最大偏好的区域(most favoured regions),17个氨基酸(8.9%)落入额外允许区域(additional allowed regions),3个氨基酸(1.6%)落在宽松允许区域(generously allowed regions),只有1个氨基酸ThrXL(0.5%)落在了非允许区域(disallowed region)。Verify3D检验要求至少80%的氨基酸的平均3D-1D打分不小于0.2,分析结果表明,L链全部氨基酸的打分均大于0.2,而H链99.12%的氨基酸残基打分大于0.2。因此,采用同源模建方法构建的抗体结构较为合理。 图2.抗体的同源模建三维结构: 绿色为H链,蓝色为L链,洋红色为与模板蛋白不同的抗体氨基酸区域。
蛋白质三级结构预测(swiss-model同源建模)
利用同源建模预测蛋白质的三级结构 首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。 我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行! 对于一个未知结构的蛋白质, 白质建立结构模型。 那么,我们首先要做的就是找到和我们 空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。 打开SWISS-MODEL网站:https://www.360docs.net/doc/a46732737.html,/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。 这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。 接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。找到以后,以FASTA格式显示。 接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我 大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~ SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择: Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (https://www.360docs.net/doc/a46732737.html,/sprot )编目号(accession)可以直接通过web界面提交。服务器会完全自动地为目标序列建立模型。 用户可以选择指定模板结构,模板可以来自由PDB数据库(https://www.360docs.net/doc/a46732737.html, )抽取得到的内建模板库,也可以上传PDB格式的坐标文件。 Alignment mode(联配模式): 这个模式需要多序列联配的结果,序列中至少包括目标序列和模板(最多可输入5条序列)。服务器会基于比对结果建模。 与模板的联配结果。这个结果也要上传到服务器。这种方式提供对建模过程中细
生物信息学课程报告 几种蛋白质二级结构预测方法评价
几种蛋白质二级结构预测方法的评价 摘要蛋白质二级结构的预测是了解蛋白质空间结构及其作用机理的重要步骤,二级结构的预测方法也越来越多,为便于广大研究者选择合适的预测方法,本文利用SARS 数据集,采用统一的评价标准,对蛋白质二级结构预测的三种典型方法PSIPRED V3.0、APSSP2、GOR4进行评测。结果显示,PSIPRED预测效果较好,可以作为相关研究的首选,而GOR4表现最差,对β折叠的预测能力最缺乏。 关键词二级结构PSIPRED SARS数据集 Evaluation on three prediction methods of protein secondary structure Abstract Protein secondary structure prediction plays a key role in recognizing the protein’s three-dimensional structure and mechanism. With more and more prediction methods developed, three prediction methods of protein secondary structure including PSIPRED V3.3,APSSP2,GOR4 had been utilized in SARS to evaluate their applicability. The findings suggested that PSIPRED performed best, and GOR4 had poor effect in the protocol, especially in the prediction of β strands. Keywords protein secondary structure ;PSIPRED ; SARS 由于蛋白质的生物学功能在很大程度上依赖于其空间结构,因而进行蛋白质的结构预测对了解未知蛋白生物学功能具有重要意义[1]。 通常,蛋白质结构包括4个层次[2]:一级结构即氨基酸的排列顺序;二级结构主要是由氢键维持的α-螺旋和β-折叠;三级结构是完全折叠的蛋白质的空间结构残基的立体排列模式;四级结构是多个蛋白质亚基组成的蛋白质复合体的结构(即蛋白质之间的交互作用)。用计算机对蛋白质二级结构的预测对认识蛋白质结构与功能的有重要意义。一方面,蛋白质二级结构预测为三级结构模型构建的起点,是三、四级结构预测的基础[3]。另一方面,由于利用X光绕射、核磁共振等实验方法对蛋白质二级结构预测受到一
蛋白质3D建模,酶与底物分子模拟对接 autodock
摘要 多环芳烃(polycylic aromatic hydrocarbons,PAHs)是一类典型的芳香烃类有机污染物,其种类繁多,常见的共有16种。近年来多环芳烃的污染已经引起人们的高度重视,随着对PAHs 微生物降解研究的深入,已经发现大量在耗氧条件下对四环以下PAHs 有降解能力的细菌,但微生物对五环及五环以上PAHs的降解能力较低,为了提高菌群的PAHs底物范围,对其降解途径中的关键酶进行分子改造具有非常重要的意义。萘双加氧酶(Naphthalene dioxygenase,NDO)是多环芳烃降解途径中的关键酶,。本论文通过计算机模拟的方式研究不同来源的萘双加氧酶与多环芳烃的相互作用规律,考察影响其活性中心口袋大小的关键氨基酸,为使用定点突变等基因工程技术提高萘双加氧酶的降解效率提供参考。本实验从数据库下载了9种来源不同的萘双加氧酶的α亚基氨基酸序列,采用3种方式进行同源建模,经过3种方法对模型进行评价,选取质量最好的一组模型与16个PAHs分子进行对接。通过比较这些不同菌种来源的NDO与PAHs的对接结果,寻找影响其相互作用的关键氨基酸。实验结论如下:通过同源模建及模型评价,发现工具Phyre2获得的模型质量相对较好;使用Autodock Tools(ADT)将模型与PAHs进行对接后获得了不同来源NDO与PAHs相互作用的特征曲线,PAHs环数的多少会显著影响NDO与PAHs的结合能力;通过对对接结果的统计,发现来自Rhodococcus sp.的萘双加氧酶(Q9X3R9)和PAHs的结合能最低,结合能力最强。通过统计9种不同来源的NDO活性中心18个氨基酸的突变情况和偏移量发现,相对于实验室的JM-2序列,比较保守的氨基酸包括N205、F206、D209、H212、H217、G255、V264、D368、G208。而这些不同来源的BDO活性中心氨基酸组成差异主要发生于V213、L257、H301、N303、T316、L364、A412七个位置,其变异性较强,结构位置不稳定,对七个氨基酸进行改造,增大NDO的活性口袋,能增强酶对高环PAHs的结合能力,为NDO的分子改造提供参考。。 关键词:萘双加氧酶;同源建模;分子对接;活性中心;蛋白设计