以肝功能衰竭合并溶血性贫血发病的儿童肝豆状核变性13例临床特点

85《中国肝脏病杂志(电子版)》2019年第11卷第2期·病例报告·以肝功能衰竭合并溶血性贫血发病的
儿童肝豆状核变性13例临床特点
王丽旻, 张敏, 董漪, 甘雨, 陈大为, 王福川, 闫建国, 朱世殊(解放军总医院第五医学中心青少年肝病诊疗与研究中心,北京 100094)
摘要:儿童肝豆状核变性(hepatolenticular degeneration,HLD)以肝功能衰竭合并溶血性贫血发病的
临床表现不典型,需通过各项检查综合判断。肝豆状核变性进展快,预后差,需积极联系肝移植。本
文对13例以肝功能衰竭合并溶血性贫血发病的肝豆状核变性儿童患者的临床特点进行分析。
关键词:儿童;肝豆状核变性;肝功能衰竭;溶血;临床特征;预后
Clinical characteristics of 13 children of hepatolenticular degeneration with liver failure complicated
with hemolytic anemia as symptoms
WANG Li-min, ZHANG Min, DONG Yi, GAN Yu, CHEN Da-wei, WANG Fu-chuan, YAN Jian-guo, ZHU
Shi-shu (Pediatric Liver Diseases Therapy and Research Center, Fifth Medical Center of PLA General
Hospital, Beijing 100039, China)
Abstract: Hepatolenticular degeneration (HLD) in children expressed as liver failure complicated with
hemolytic anemia is often atypical in clinical manifestations. The etiological diagnosis of HLD requires comprehensive judgment of various examinations. However, the disease progresses rapidly and the prognosis
is poor. Liver transplantation should be considered actively. Clinical characteristics of 13 children with hepatolenticular degeneration expressed as liver failure complicated with hemolytic anemia were analyzed in
this article.
Key words: Children; Hepatolenticular degeneration; Liver failure; Hemolysis; Clinical characteristics;
Prognosis
肝豆状核变性(hepatolenticular degeneration,HLD)又称Wilson病(WD),是一种常染色体隐性遗传疾病,WD基因突变可引起过多的铜在肝脏和其他器官中蓄积。患者通常在5~40岁出现症状,仅少数患者表现为肝功能衰竭合并溶血性贫血,若未及时行肝移植,患者常死于肝功能衰竭[1]。本文对解放军总医院第五医学中心(原解放军302医院)青少年肝病诊疗与研究中心近12年诊断为以肝功能衰竭合并溶血性贫血发病的13例儿童肝豆状核变形患者临床特征及预后进行分析。
1临床资料
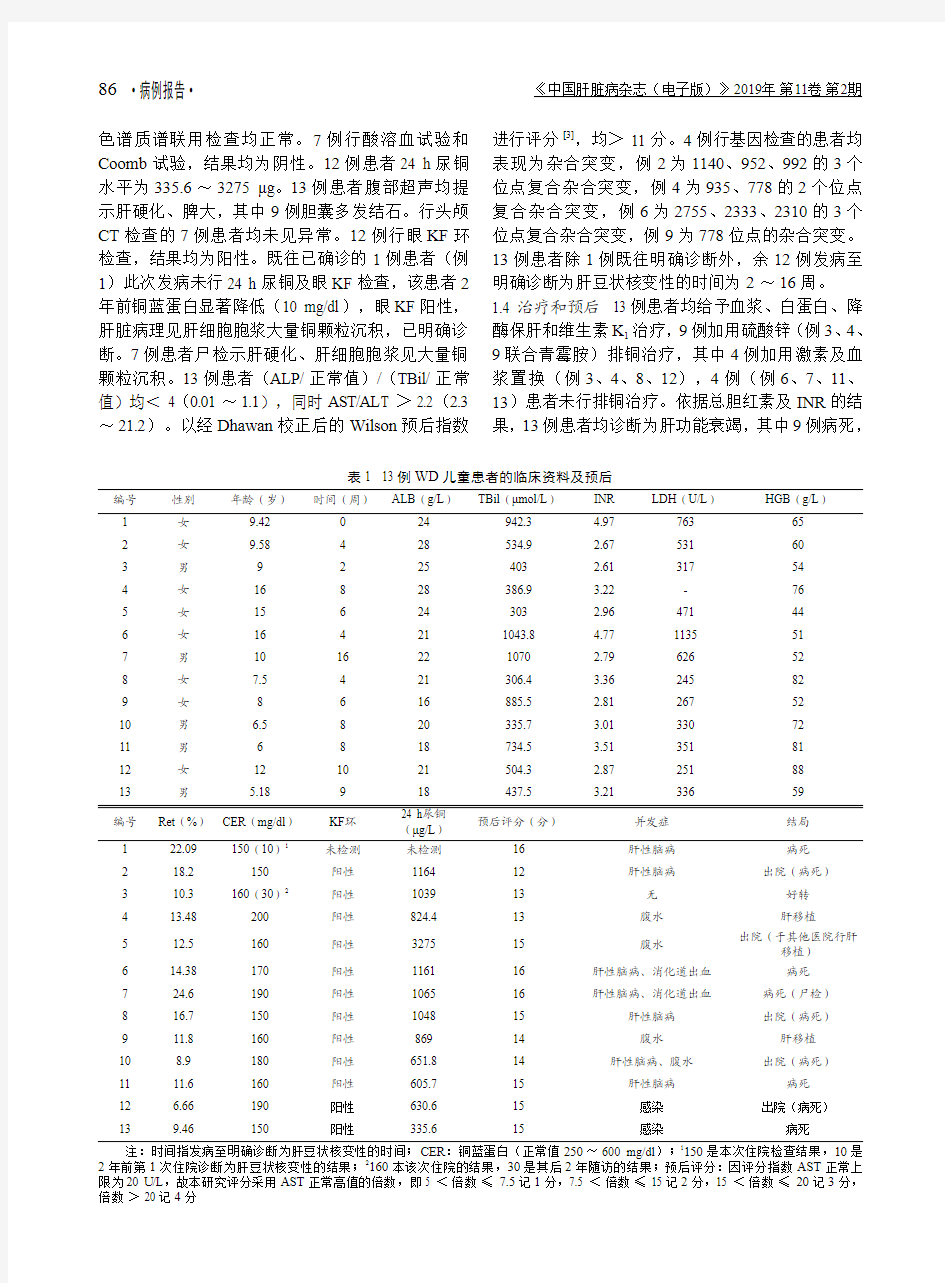
1.1 一般资料 2005年1月至2016年12月确诊的肝功能衰竭合并溶血性贫血的13例WD儿童患者中男性5例,女性8例;年龄5.18~16岁,中位年龄9.42岁。其中1例既往诊断为WD,自行停用青
DOI: 10.3969/j.issn.1674-7380.2019.02.016
基金项目:首都临床特色应用研究(Z181100001718030)
通讯作者:朱世殊 Email: 302zss@https://www.360docs.net/doc/c19387782.html, 霉胺、硫酸锌10个月后发病;1例因进食过量海产品而发病,其他起病无诱因。所有患者中仅1例患者的弟弟患病,其余12例无家族史。肝功能衰竭和肝豆状核变性的诊断符合相关标准[2,3]。
1.2 临床表现 13例患者均以眼黄、尿黄发病,病程中均有中度发热。1例患者发病同时有腹胀。患者均全身重度黄染、贫血貌;肝掌阳性者8例,皮肤黝黑者10例,脾大9例,肝大2例,移动性浊音阳性6例,下肢水肿4例。7例合并肝性脑病,其中2例还合并消化道出血;另外2例合并腹腔感染。
1.3 实验室检查所有患者总胆红素(total bilirubin,TBil)、乳酸脱氢酶(lactate dehydrogenase,LDH)、INR及网织红细胞百分比高于正常值,铜蓝蛋白和血红蛋白(hemoglobin,HGB)低于正常值(见表1)。甲型肝炎、乙型肝炎、丙型肝炎、丁型肝炎及戊型肝炎病原学检查均为阴性,自身免疫性肝病抗体阴性,CMV IgM、EBV IgM、CMV DNA、EBV DNA均阴性,血串联质谱及尿气相
小儿肝衰竭
小儿肝衰竭 锁定 本词条由国家卫生计生委临床医生科普项目/百科名医网提供内容并参与编辑。 声明 百度百科公开招募三甲医院副高职称医师或医学博士,加入国家卫生计生委“权威医学科普传播网络平台”,撰写医学词条。查看详情 小儿肝衰竭又称暴发型肝炎及重型肝炎,是各种原因(在我国以病毒性肝炎最为常见)导致肝细胞广泛坏死或肝功能急剧严重损害,引起的极为凶险的临床症候群,是所有肝病重症化的结局。流行病学:本症病死率极高,国外报告肝衰竭的病死率在60%~80%,病死率最高依次为慢性肝衰竭(CHF),迟发性肝衰竭(LOHF)和急性肝衰竭(AHF或暴发性肝衰竭)。 目录 1病因 2临床表现 3检查 4诊断 5治疗 1病因 1.感染 病毒性肝炎占首位近年来以乙型肝炎病毒所致者明显增多。此外,EB病毒、疱疹病毒、巨细胞病毒等均可引起。 2.中毒 包括异烟肼、利福平对乙酰氨基酚和四环素等药物中毒,毒蕈等食物中毒以及四氯化碳等化学物中毒,毒蛇咬伤等。 3.遗传性代谢缺陷 少数肝豆状核变性、半乳糖血症、果糖不耐受症、酪氨酸血症和糖原累积症Ⅳ型等也可以发生肝功能衰竭。 4.其他 (1)肝脏脑脂肪变性综合征瑞氏综合征。 (2)严重复合创伤大手术、大面积烧伤、败血症、缺血缺氧性损害、各种原因的休克等。 (3)其他侵犯肝脏的疾病,如恶性增生性组织细胞病郎汉斯细胞组织细胞增生症等。2临床表现
小儿肝衰竭的临床表现为进行性肝损害、不同程度的肝性脑病、颅内压增高、出血等,由于病因不同,尚存在原发病的症状。 1.进行性肝损害 病毒性肝炎患儿,消化道症状明显加重食欲减退、恶心、呕吐、腹胀、偶有腹泻;黄疸迅速加深,一般均为中度以上;肝脏进行性缩小,尤以肝右叶明显病情加重后肝萎缩进展极快,少数伴有脾脏增大;儿童较易出现水肿及腹水严重者呼气有肝臭味是晚期预后不良的征兆。 2.肝性脑病 根据原发病不同肝性脑病可分为内源性和外源性内源性多见暴发型肝炎,起病数天内可进入昏迷,昏迷前无前驱症状;外源性肝性脑病属门体分流性脑病多见于肝硬化,以慢性反复发作性木僵和昏迷为主,常有诱因,出现肝性脑病后存活时间因肝功能衰竭的速度和程度而异,多起病缓慢,昏迷逐步加深。 3.颅内压增高 约80%患儿伴有脑水肿,表现为颅内压增高婴儿眼神呆滞、尖叫烦躁、呕吐、前囟隆起。年长儿可有剧烈头痛、频繁喷射性呕吐血压增高、惊厥及意识障碍。伴有肢体僵直旋扭病理反射阳性。由于脑循环障碍产生高热过高热,周围血管收缩致使皮肤苍白肢端青紫、发凉。发生颞叶沟回疝时,两侧瞳孔不等大;发生枕骨大孔疝时双侧瞳孔散大,呼吸节律不齐,甚至暂停。 4.出血现象 患儿均有不同程度出血。轻者为皮肤黏膜出血或渗血,鼻出血及齿龈出血较常见。严重时内脏出血,以消化道出血发生最多可呕血或便鲜血,也可吐咖啡样物及排柏油样便,常因一次出血量很多而导致休克或加重肝性脑病;也可有其他部位出血如咯血、血尿或颅内出血等。大出血常为致死的直接原因。 3检查 1.血清学检查 (1)血清胆红素血清总胆红素一般均超过171.0μmol/L(10mg/dl),平均每天增长 17.1μmol/L(1mg/dl)或更多,以直接胆红素升高为主。 (2)酶胆分离重症肝病丙氨酸转氨酶(ALT)及谷草转氨酶(AST)显著下降,与胆红素上升呈分离现象,即“酶胆分离”。因丙氨酸转氨酶主要分布于肝细胞浆内,轻症肝炎或某些肝病患者,细胞膜通透性改变,胞浆内的酶释放入血,丙氨酸转氨酶升高;当肝细胞受到严重损伤时,线粒体也受累血中丙氨酸转氨酶则降低谷草转氨酶分布于肝细胞浆及线粒体内,人体患急性肝炎时释入血中,但失活较快,故较丙氨酸转氨酶值低;而线粒体遭破坏后,则谷草转氨酶释出进入血液循环,血中浓度增高且大于谷丙转氨酶改变了丙氨酸转氨酶与谷草转氨酶比值,故监测丙氨酸转氨酶/谷草转氨酶对判断肝细胞损伤有重要意义比值减小表示肝细胞严重坏死预后不良。 (3)血氨基酸测定支/芳氨基酸比值正常时其摩尔比为3∶1~4∶1,重症肝炎者降至1∶1~1.5∶1以下。游离色氨酸明显增高,对促进肝性脑病的发生起重要作用
肝豆状核变性
肝豆状核变性( hepatolenticular degeneration,HLD),又称为Wilson病(Wilson s disease,WD),是一种常染色体隐形遗传的铜代谢缺陷病,发病率仅0.5/ 10 万~3/10 万,其基因定位于13q14. 3,编码1 个P 型ATP 酶,此酶参与铜跨膜转运的代谢过程。目前研究多认为由于WD 基因突变使其功能降低或丧失而导致铜代谢异常,肝合成铜蓝蛋白速度减慢,胆汁排铜明显减少,铜沉积于肝、脑、肾、角膜、血细胞和关节等组织中,引起了相应脏器损害的临床症状。铜蓄积可导致肝细胞坏死、肝纤维化;从坏死的肝细胞释放的大量铜可导致溶血,并逐渐沉积在脑、肾、角膜、骨关节部位,引起多器官受累。不同程度的肝细胞损害,脑退行性病变和角膜边缘有铜盐沉着(K-F环)为其临床特征。早期诊断和治疗可避免严重的不可逆的组织器官损害,常可获得与健康人一样的生活和寿命。 1921年Hall首先分析68例有血缘关系的HLD病人,其中34例至少与另一名患者有血缘关系,且绝大多数是表(堂)亲。因此Hall确信HLD属遗传性疾病,并且指出是由2个不同的隐匿性代谢缺陷的基因传递所致,其传递形式与常染色体隐性遗传相一致。我国调查167例HLD 先证者的家系,615位先证者及其同胞中,278人证实为HLD,按Hardy-Weinberg公式计算;(278-167)/(615-167)=0.248。此值与常染色体隐性遗传的预期值0.25极接近,统计学无显著差异(P>0.05),符合常染色体隐性遗传。 肝豆状核变性的临床表现 肝豆状核变性是一种先天代谢缺陷病。其病理生理基础是铜代谢呈正平衡。全身组织内有铜的异常沉积。本病散见于世界各地不同的民族。从遗传学说研究估计本病发病率约为人口的16万分之一。我国(包括台湾)的不完全统计有220例。大多数在少年或青年期发病,以10~2 5 岁最多,男女发病率相等。幼儿发病多呈急性,在数月或数年内死亡,30岁以后发病多属慢性型。 (1)早期表现为一般消化道症状:消化不良、嗳气、食欲不振等,脾肿大、黄疸、肝功能异常、类似肝炎,迁延不愈。以后肝脏缩小、质硬、表面有结节,发展为坏死性肝硬化。 (2)精神症状常表现为性格异常、忧郁、癔病样发作,以及智力、记忆力减退、言语等表达能力障碍。 (3)神经方面症状以锥体外系运动障碍为主:不自主运动、震颤、共济失调、甚至肌强直、全身痉挛。 (4)眼部出现Kayser-Fleischer角膜色素环。铜浓缩在溶酶体中,肝铜含量显著增加。血铜蓝蛋白浓度降低。铜在红细胞内沉着引起溶血性贫血和黄疸。白细胞的细胞色素氧化酶明显减少。 (5)肾脏损害主要表现为氨基酸尿、尿中铜、尿胆素原、钙、磷酸和尿酸的排泄量增加。磷、钙的丢失可引起骨、关节异常。 肝豆状核变性的病因和发病机理 肝豆状核变性的一系列病变是铜代谢障碍引起的。好发于10~25岁。正常人从食物中获取铜,95%的铜在肝脏内合成铜蓝蛋白,极少以直接反应铜存在。80%的铜从胆汁、粪便中排泄。Wilson病人肠道吸收铜增强,肝脏内有一种特异蛋白,对铜的亲和力强,可阻止铜蓝蛋白合成,血液中铜蓝减少。这种蛋白和铜结合松散,铜释放入血,铜盐沉积在肝脏、脑、肾、角膜等。有研究证实,肝细胞中异常蛋白称作金属硫团(Metallothionien),与铜有极高亲和力,与铜形成不可溶聚集物,不能被酶分解,也不能从胆汁排出。这些聚集物沉积在肝脏、脑、肾、角膜,引起一系列毒性反应。 关于肝豆状核变性发生机制有以下几个学说:①该类患者胆汁中与铜结合的正常物质缺陷,可能是鹅脱氧胆酸与牛磺酸结合缺陷,导致胆汁分泌铜功能障碍。不支持这一学说的证据是:该类患者胆汁铜结合蛋白没有质的改变,而且没有证据表明该类患者存在胆酸代谢异常。②肝脏铜结合蛋白合成异常,导致蛋白对铜的亲合力增加,支持这一学说的证据有,Wilson病患者铜结合蛋
【精品】肝豆状核变性
肝豆状核变性(hepatolenticulardegeneration,HLD),又称为Wilson病(Wilsonsdisease,WD),是一种常染色体隐形遗传的铜代谢缺陷病,发病率仅0。5/10万~3/10万,其基因定位于13q14.3,编码1个P型ATP酶,此酶参与铜跨膜转运的代谢过程.目前研究多认为由于WD基因突变使其功能降低或丧失而导致铜代谢异常,肝合成铜蓝蛋白速度减慢,胆汁排铜明显减少,铜沉积于肝、脑、肾、角膜、血细胞和关节等组织中,引起了相应脏器损害的临床症状。铜蓄积可导致肝细胞坏死、肝纤维化;从坏死的肝细胞释放的大量铜可导致溶血,并逐渐沉积在脑、肾、角膜、骨关节部位,引起多器官受累。不同程度的肝细胞损害,脑退行性病变和角膜边缘有铜盐沉着(K-F环)为其临床特征。早期诊断和治疗可避免严重的不可逆的组织器官损害,常可获得与健康人一样的生活和寿命。 1921年Hall首先分析68例有血缘关系的HLD病人,其中34例至少与另一名患者有血缘关系,且绝大多数是表(堂)亲。因此Hall确信HLD属遗传性疾病,并且指出是由2个不同的隐匿性代谢缺陷的基因传递所致,其传递形式与常染色体隐性遗传相一致.我国调查167例HLD先证者的家系,615位先证者及其同胞中,278人证实为HLD,按Hardy-Weinberg公式计算;(278-167)/(615-167)=0.248。此值与常染色体隐性遗传的预期值0。25极接近,统计学无显著差异(P>0。05),符合常染色体隐性遗传。 肝豆状核变性的临床表现 肝豆状核变性是一种先天代谢缺陷病.其病理生理基础是铜代谢呈正平衡。全身
病毒感染与肝功能衰竭(文档版)
1.1甲型肝炎病毒(HA V)感染.HA V是小RNA病毒.主要 经粪——口途径传播。人感染HAV,可表现为临床型和亚临床型感染。6岁以下儿童感染HAV后,70%为亚临床型感染;大龄儿童和成人感染HAV后,70%以上为临床型感染。病死率为0.3%~0.6%;50岁以上患者的病死率为1 .8%;慢性肝病者感染HAV后,发生急性肝衰竭的危险性升高.。HAv感染大多表现为自限性、急性临床过程,单独HAV感染进展为急性肝衰竭的几率低(约0.1%~0.35%),但一印度医院报道的70例HAV感资的儿童中,l4例发展为急性肝衰竭.几率为20%。调查解放军302医院1977例肝衰竭患者的病因未发砚单独HAV感染急性肝衰竭,发现5例HAV感染引起的肝衰竭皆为在慢性HBV感染基础上发生。有研究显示HAV感染进展为急性肝衰竭与HAV受体TIM1的基因多态性有关1.2乙型肝炎病毒(HBV)感染目前我国慢性HBV感染者 达9300万,HBV感染是我国引起肝衰竭的最主要病因。有研究显示,HBV感染在1977例急性、亚急性、慢加急性肝衰竭患者病因中占87.25%。 1.3 丙型肝炎病毒(HCV}感染HCV感染是欧美国家终末期肝病的最主要原因。目前世界范围内确诊为HCV感染患者的人数达1.7亿,我国抗-HCV阳性率为3.2%,有近4千万感染者。HCV感染后一般临床症状较轻,引起急性肝衰竭少见,仅20%感染者表现为急性并呈自限性发病过程,而
15%~80%发展为慢性感染,肝硬化、肝愛发生率也较高,约25%的慢性HCV感染者发展为肝硬化,大多发展为以严重腹水、腹膜炎、肝性脑病、消化道出血等严重并发症为特征的慢性肝衰竭。慢性HCV感染基础上发生的慢加急性肝衰竭少见。 1.4 丁型肝炎病毒(HDV)感染HDV是一个小型的、有缺陷的RNA病毒,只感染有HBV感染基础患者,全球超过1500万人感染。HDV虽一定程度上抑制HBV复制,但会加速病程进展为肝硬化和慢性肝衰竭。有研究报道在HBV感染相关的急性、亚急性肝衰竭患者中合并HDV感染者约占30% ,远远高于普通型乙型肝炎患者中HDV感染的阳性率,说明HDV/HBV感染发生急性、亚急性肝衰竭的几率远远高于单独H BV感染者。 1.5戊型肝炎病毒(H EV)感染同HAV感染相似,HEV经消化道传播,大多呈自限性,孕妇、年老体弱者及慢性H BV 感染者感染HEV后易发展为肝衰竭,病死率也相对较高。孕妇感染HEV的预后比感染其他嗜肝病毒的预后差,我国上海地区孕妇感染HEV后的病死率为31%,印度北方地区病死率为39.1%。范振平等报道424例戊型肝炎患者中,青年、中年、老年组重型肝炎病例分别占单纯性戊型肝炎的3.0%、5. 99%和14.0%,汪茂荣等报道45例戊型重型肝炎平均年龄58岁,混合或重叠其它肝炎病毒感染19例(4 2. 2%),
肝豆状核变性患者饮食手册范本
肝豆状核变性患者饮食手册 常用食物含铜量表(mg/100g) 食物名称含铜量建议 粮食(干) 标准粉 0.42 少食 富强粉 0.26 可食 麦胚粉 0.83 禁食 麦麸皮 2.03 禁食 挂面(平均) 0.39 少食 面条(平均) 0.17 可食 玉米面(黄) 0.35 少食 玉米面(白) 0.23 可食 梗米(标一) 0.19 可食 籼米 0.23 可食 糯米(平均) 0.11 可食 大麦 0.63 禁食 青稞 5.13 禁食 小米 0.54 禁食 黄米 0.90 禁食 高粱米 0.53 禁食 荞麦面 0.89 禁食 薏米 0.29 可食 豆类(干) 黄豆(平均) 1.4 禁食 豆奶粉(平均 5.57 禁食 豆腐皮 1.86 禁食 腐竹 1.31 禁食 绿豆 1.08 禁食
蚕豆 0.99 禁食 扁豆 1.27 禁食 豇豆 2.10 禁食 豌豆 0.57 禁食 鲜根菜类 萝卜(平均) 0.04 可食胡萝卜(红) 0.08 少食胡萝卜(黄) 0.03 可食丕蓝 0.02 可食 甜菜根 0.15 禁食 地瓜 0.07 少食 山药 0.24 禁食 芋头 0.37 禁食 马铃薯 0.12 少食 红薯 0.17 禁食 鲜豆类 扁豆 0.12 少食 蚕豆 0.39 禁食 豆角 0.15 禁食 荷兰豆 0.06 少食 龙豆 0.35 禁食 四季豆 0.11 少食 豇豆 0.14 禁食 黄豆芽 0.14 禁食 绿豆芽 0.10 少食 豌豆苗 0.20 禁食 鲜茄瓜类 茄子(平均) 0.10 少食西红柿 0.06 少食
生瓜 0.03 可食 冬瓜 0.07 少食 佛手瓜 0.02 可食 葫芦(长) 0.04 可食黄瓜 0.05 可食 苦瓜 0.06 少食 南瓜 0.03 可食 丝瓜 0.06 少食 鲜葱蒜韭类 大蒜头 0.22 禁食 蒜苗 0.05 可食 青蒜 0.05 可食 蒜黄 0.09 少食 大葱 0.08 少食 大葱(红皮) 0.34 禁食洋蒜头 0.05 可食 韭菜 0.08 少食 韭黄 0.10 少食 韭苔 0.05 可食 鲜茎、叶、花 大白菜(平均 0.05 可食黄芽白 0.03 可食 青白菜 0.04 可食 小白菜 0.07 少食 乌菜 0.13 少食 油菜 0.06 少食 白菜苔 0.18 禁食 油菜苔 0.18 禁食 卷心菜 0.04 可食
爆发性肝衰竭急救预案
起草人:刘全忠 暴发性肝衰竭急救预案SOP Ⅰ目的:建立暴发性肝衰竭急救预案的标准操作规程。 Ⅱ范围:本规程适用于暴发性肝衰竭急救处理。 Ⅲ规程 1. 定义 暴发性肝衰竭是指出现首发症状8周内发生Ⅱ度以上肝性脑病、血浆凝血酶原活动度低于40%、肝活检或尸解证明有大块肝坏死的急性肝损害。 2. 临床表现 2.1 基本临床表现 健康状况全面衰退、显著乏力、消化道症状严重(食欲极度减退、上腹闷胀不适、腹部明显胀气、肠鸣音减少或消失)、黄疸进行性加深、出血倾向明显、焦虑和烦躁、低热、出现肝臭等。 2.2 肝性脑病 最早出现的是性格的改变,表现为欣快、抑郁或孤僻,昼夜睡眠颠倒,可有扑翼样震颤。随着病情的进展,患者的智能发生改变,表现为对时间和空间概念不清,人物概念模糊,吐词不清,书写困难,计算力、定向力下降。继而出现较明显的意识障碍,开始处于昏睡状态,以后进入全昏迷状态。 2.3 腹水 主要根据腹部叩诊法加以识别。中等量腹水可出现显著的移动性浊音,大量腹水时两侧胁腹膨出如蛙腹。 2.4 凝血功能障碍 肝功能衰竭时,可出现血浆凝固的异常,包括内源性和外源性凝血系统的异常,血小板质和量以及形态的改变,DIC及抗凝系统的异常等,导致凝血功能障碍而致出血。 3 急救措施 3.1 基础支持治疗 原则上应绝对卧床休息,给予适量的水分、电解质,补充足够的能量、维生
素和微量元素,阻抑肝细胞进行性坏死,促进肝细胞修复和再生,调节肝细胞的代谢,纠正体内的各种代谢失衡,维持内环境稳定等。对急性肝衰竭的各种并发症,特别是上消化道出血、肝肾综合征、感染等要充分警惕。对某些并发症可给予预防性治疗,如适当输注新鲜血浆以补充凝血因子、给予抑酸剂以防止消化道出血、减少侵入性操作等以防止外源性继发感染。 ①肝细胞生长因子20-100mg/日溶于葡萄糖液内静脉滴注; ②前列腺素E1(凯时)10μg溶于20ml生理盐水中静推,没日一次; ③门冬氨酸钾镁20 mg溶于10%葡萄糖液内静脉滴注; ④阿拓莫兰1200 mg加入10%葡萄糖液250ml中静滴,15-60天为一疗程; ⑤易善复注射液(肝得健)10-20ml/日缓慢静推,每日一次; ⑥丹参注射液10-20 ml溶于10%葡萄糖液250ml中静滴,每日1-2次。 3.2 人工肝支持系统 人工肝脏是借助体外机械、化学或生物性装置,暂时替代或部分替代肝脏功能,从而协助治疗肝功能不全或相关疾病的方法。常用方法有血浆置换、血液灌流、血液透析、血液滤过、分子吸附循环系统以及连续性血液净化技术。其临床意义在于: ①遏制病情进展,促进肝脏自发恢复;暴发性肝衰竭时,尽管采取多种内科治疗,其死亡率仍在70%-80%以上,其中出现Ⅳ期肝性脑病患者的病死率高达90%-95%。在这种情况下,人工肝是迅速改善机体内环境,部分解除或缓解毒性物质对肝脏及全身的毒害作用,促使病情稳定甚至恢复的有效手段。 ②部分代偿衰竭肝脏的基本功能;通过血浆置换治疗,可补充体内急需的白蛋白、凝血因子及调理素等,部分代偿肝脏的合成功能。 ③减低内毒素和促炎性细胞因子的水平,防止和改善多脏器功能衰竭; ④改善肝移植患者的术前条件,顺利渡过术中的无肝期以及术后的肝脏无功能期。 3.3 肝脏移植 原则上,一切终末期肝病和未播散的恶性肿瘤,经现有的治疗方法不能治愈,预计在短期内无法避免死亡者,均是肝移植的适应症。 4.监测要点:
儿童肝功能衰竭
儿童急性肝衰竭 急性肝衰竭(ALF)是严重的肝病症候群。临床上虽属罕见或少见,但因为一旦发生病死率极高,因此必须予以足够重视。多年来,各国学者对肝衰竭的定义、分类、诊断和治疗等问题不断进行探索,2005年美国肝病学会发布了对急性肝衰竭处理的建议[1]。为适应临床工作的需要,规范我国肝衰竭的诊断和治疗,中华医学会感染病分会和中华医学会肝病学分会组织国内有关专家于2006年制定了我国第一部《肝衰竭诊疗指南》[2]。这些建议和指南针对成人肝衰竭而制定,儿童肝衰竭的病因及临床表现等与成人都有较大差别。2008年8月28-30日在上海召开的第九次全国儿科肝病学术会议由中华医学会感染病学分会主办,上海市肝病学会和上海市感染病学会协办,中华医学会感染病学分会小儿肝病及感染学组和复旦大学附属儿科医院承办。为提高对儿童急性肝功能衰竭的诊断水平和救治能力,会议特邀中华医学会感染病学分会主任委员李兰娟院士和英国国王学院医院儿童肝病科主任、欧洲儿童肝病专委会主席Anil Dhawan教授等就有关问题作了专题报告。特将其中的新进展进行总结。 一、儿童肝衰竭的定义 急性肝衰竭最初的定义为无慢性肝病的患者,在起病8周内出现大块的肝坏死伴肝性脑病。以后许多学者认为一些先前无症状的慢性肝病,包括肝豆状核变性、垂直获得性HBV 或自身免疫性肝炎患者可能已存在肝硬化,但呈急性起病者,仍应纳入ALF 范畴。还有学者不以最早出现症状的时间,而以出现黄疸到进展为肝性脑病的时间进行分类。2005年美国肝病学会(AASLD)定义为原来没有肝硬化的患者,在发病26周内出现凝血障碍( INR ≥1. 5)和不同程度神智障碍(肝性脑病),包括急性起病表现的肝豆状核变性等。既往国内对于肝衰竭多称为重型肝炎,但命名、分类和定义等和国际上所称的肝衰竭并不一致。我国《肝衰竭诊疗指南》将肝衰竭定义为由多种因素引起肝细胞严重损害,导致其合成、解毒和生物转化等功能障碍,出现以黄疸、凝血功能障碍、肝性脑病和腹水等为主要临床表现的一种临床综合征。临床上可将其分为急性肝衰竭、亚急性肝衰竭、慢加急性肝衰竭和慢性肝衰竭四种。急性肝衰竭指起病2周以内出现肝衰竭表现,亚急性肝衰竭指起病15天至24周出现肝衰竭,分别相当于既往的急性或亚急性重症肝炎。慢加急性肝衰竭之在慢性肝病基础上出现急性或亚急性肝衰竭,是本次肝衰竭标准中新提出的。慢性肝衰竭指在慢性肝病基础上出现肝功能进行性减退或造成失代偿,是慢性肝硬化的结果,和国际上的定义相同。
儿童暴发性心肌炎诊治进展
儿童暴发性心肌炎诊治进展 儿童暴发性心肌炎(FM)为起病2周以内,迅速发展为心力衰竭和循环功能障碍,或阿斯综合征的心肌炎患者,是儿童重症医学和心脏病学的严重危重疾病。儿童FM通常由病毒感染引起,常见有柯萨奇病毒和腺病毒,由微小病毒B19和甲型H1N1流感病毒导致的也有报道。近年来认为肠道病毒EV71也是FM的重要病因。 一、儿童FM的早期发现与诊断 1.主要临床表现:(1)泵衰竭型:突发心力衰竭和(或)心源性休克。(2)脑缺血缺氧发作型:也称阿斯综合征发作型,突然起病,迅速出现晕厥。(3)心动过速型:表现为室上性心动过速及室性心动过速。有报道FM症状最多依次为发热、疲乏、咳嗽,以呼吸困难为主要表现者仅占39%左右,而以休克为首发症状者约占29%。 2.早期诊断:儿科门急诊医师提高对FM的认识和警惕,是早期诊断FM的关键。参考指标如下:(1)有急性发作或急性加重的心脏疾病患;(2)收缩压降至同年龄正常血压低限以下;(3)有周围循环不足表现:如苍白、发绀、心率快、少尿或无尿、足底毛细血管再充盈时间延长;(4)有心功能不全体征:如心音低钝、奔马律、肝脏肿大、双肺湿罗音或血性分泌物、中心静脉压>6cmH2O;(5)心超EF<0.55, FS <0.3;(6)除外其他类型休克。1、2、 5、6为必备指标,加3、4中任意2个症状体征即纳入。 二、FM血流动力学监测 对FM进行血流动力学测定对循环功能维护,特别是正性肌力药物保用和液体管理很有帮助。 1.无创血流动力监测:超声心脏功能测定及连续无创超声心输出量监测。FM患儿抢救时尽量维持CI3.5-5.5L/(min.m2),LVEF40%以上,LVEF低于40%时,提示左心功能障碍,及时上调正性肌力药物。 2.脉搏指示连续心排血量技术(PiCCO):通过大动脉内测量温度时间变化曲线,测量全心血流动力学参数。 三、治疗 儿童FM一旦诊断,就就立即开始救治,纠正严重的血流动力学障碍(包括心力衰竭)和严重心律失常,保障有效组织循环。 1.机械循环支持治疗: FM最严重的情况是心源性休克、阿斯综合征和严重心律失常,单纯药物治疗往往是不够的,经常需要进行有效的生命支持措施。 (1)心脏临时起搏器:安装临时起搏器是严重心律失常,特别是心室率过慢(通常<40次/分)的FM最直接的救治手段,可帮助患儿渡过最危险的时期。设置起搏器电压一般为4-6mV,起搏后心率设置为基础心率上浮5-10次/分,一般婴幼儿100次/分,儿童70-80次/分。 (2)体外膜氧合器(ECMO):ECMO将血液从体内引流到体外,经膜式氧合器氧合后再用泵将血液注入体内,可以暂时代替以及的泵功能和肺的氧合功能,保证机体有充分的循环灌注与氧供,为心源性休克患儿短期内提供心肺功能支持,早期应用可尽快达到血流动力学的稳定。 2.严重心律失常的处理 (1)心动过缓或完全房室传导阻滞:心率低于50次/分(婴儿低于70-80次/分)时,静脉注射阿托品0.01-0.03mg/(kg.次),或异丙肾上腺素0.05-0.2mg/(kg.min)。无效装临时起搏器。 (2)室性心动过速或室颤:合并血流动力学障碍可道选电击复律,电击能量为0.5-1J/
肝豆状核变性护理常规
肝豆状核变性护理常规 肝豆状核变性(hepatolenticular degeneration,HLD)由Wilson在1912年首先描述,故又称为Wilson 病(Wilson Disease,WD)。是一种常染色体隐性遗传的铜代谢障碍性疾病,以铜代谢障碍引起的肝硬化、基底节损害为主的脑变性疾病为特点,对肝豆状核变性发病机制的认识已深入到分子水平。WD的世界范围发病率为1/30 000~1/100 000,致病基因携带者约为1/90。本病在中国较多见。WD好发于青少年,男性比女性稍多,如不恰当治疗将会致残甚至死亡。WD也是至今少数几种可治的神经遗传病之一,关键是早发现、早诊断、早治疗。 发病机制: 正常人每日自肠道摄取少量的铜,铜在血中先与白蛋白疏松结合,在肝细胞中铜与α2-球蛋白牢固结合成具有氧化酶活性的铜蓝蛋白。循环中90%的铜与
铜蓝蛋白结合,铜作为辅基参与多种重要生物酶的合成。铜在各脏器中形成各种特异的铜-蛋白组合体,剩余的铜通过胆汁、尿和汗液排出。 疾病状态时,血清中过多的游离铜大量沉积于肝脏内,造成小叶性肝硬化。当肝细胞溶酶体无法容纳时,铜即通过血液向各个器官散布和沉积。基底节的神经元和其正常酶的转运对无机铜的毒性特别敏感,大脑皮质和小脑齿状核对铜的沉积也产生症状。铜对肾脏近端小管的损害可引起氨基酸、蛋白以及钙和磷酸盐的丢失。铜在眼角膜弹力层的沉积产生K-F环。与此同时,肝硬化可产生门静脉高压的一系列变化。 临床表现: 1.神经和精神症状 神经症状以锥体外系损害为突出表现,以舞蹈样动作、手足徐动和肌张力障碍为主,并有面部怪容、张口流涎、吞咽困难、构音障碍、运动迟缓、震颤、肌强直等。震颤可以表现为静止或姿势性的,但不像帕金森
【疾病名】肝豆状核变性
【疾病名】肝豆状核变性 【英文名】hepatolenticular degeneration 【缩写】 【别名】Wilson病;威尔逊变性;威尔逊氏变性;威尔逊氏病;威尔逊氏综合征;威尔逊病 【ICD号】K76.8 【概述】 肝豆状核变性(hepatolenticular degeneration)又称Wilson病,本病于1911年首先由Wilson报道,此为一种常染色体隐性遗传性疾病,是先天性铜代谢障碍性疾病。临床上以肝损害、锥体外系症状与角膜色素环等为主要表现。 【流行病学】 本病人群发病率为0.5/10万~3/10万,病人同胞患病风险为1/4,人群中杂合子或病变基因携带者频率为1/100~1/200,阳性家族史达25%~50%。患病率约为1/3万活婴,杂合子频率约1%。绝大多数限于一代同胞发病或隔代遗传,连续两代发病罕见。Wilson病在大多数欧美国家罕见,某些国家和地区,如意大利南部撒丁岛及西西里岛、以色列及东欧犹太人和罗马尼亚发病率较高,日本发病率高达1/2万。我国尚无准确统计资料,但临床报告病例相当多见,据不完全统计,1950~1998年国内有关WD文献报告就有791篇,报告病例4501例。 【病因】 肝豆状核变性系常染色体隐性遗传性疾病,受累基因与铜代谢紊乱有关,与位于染色体的酯酶D基因与视网膜母细胞瘤基因紧密连锁。 【发病机制】 WD的发病机制有胆道排泄减少、铜蓝蛋白合成障碍、溶酶体缺陷、金属巯蛋白基因异常及调节基因异常等学说,目前以前二种学说获得多数学者赞同。 1.铜代谢合成障碍 多数实验室用64Cu对体内铜代谢研究证明,血清铜蓝蛋白减少是WD体内铜积蓄的主要原因。但铜蓝蛋白为何缺乏,尚未完全阐明。B ichtrrich根据铜蓝蛋白电泳发现,正常成人是由先构成的未分化的铜蓝蛋白D在肝脏内经肽酶
儿童肝功能衰竭
儿童肝功能衰竭
————————————————————————————————作者: ————————————————————————————————日期: ?
儿童急性肝衰竭 急性肝衰竭(ALF)是严重的肝病症候群。临床上虽属罕见或少见,但因为一旦发生病死率极高,因此必须予以足够重视。多年来,各国学者对肝衰竭的定义、分类、诊断和治疗等问题不断进行探索,2005年美国肝病学会发布了对急性肝衰竭处理的建议[1]。为适应临床工作的需要,规范我国肝衰竭的诊断和治疗,中华医学会感染病分会和中华医学会肝病学分会组织国内有关专家于2006年制定了我国第一部《肝衰竭诊疗指南》[2]。这些建议和指南针对成人肝衰竭而制定,儿童肝衰竭的病因及临床表现等与成人都有较大差别。2008年8月28-30日在上海召开的第九次全国儿科肝病学术会议由中华医学会感染病学分会主办,上海市肝病学会和上海市感染病学会协办,中华医学会感染病学分会小儿肝病及感染学组和复旦大学附属儿科医院承办。为提高对儿童急性肝功能衰竭的诊断水平和救治能力,会议特邀中华医学会感染病学分会主任委员李兰娟院士和英国国王学院医院儿童肝病科主任、欧洲儿童肝病专委会主席Anil Dhawan教授等就有关问题作了专题报告。特将其中的新进展进行总结。 一、儿童肝衰竭的定义 急性肝衰竭最初的定义为无慢性肝病的患者,在起病8周内出现大块的肝坏死伴肝性脑病。以后许多学者认为一些先前无症状的慢性肝病,包括肝豆状核变性、垂直获得性HBV 或自身免疫性肝炎患者可能已存在肝硬化,但呈急性起病者,仍应纳入ALF 范畴。还有学者不以最早出现症状的时间,而以出现黄疸到进展为肝性脑病的时间进行分类。2005年美国肝病学会(AASLD)定义为原来没有肝硬化的患者,在发病26周内出现凝血障碍(INR≥1. 5)和不同程度神智障碍(肝性脑病),包括急性起病表现的肝豆状核变性等。既往国内对于肝衰竭多称为重型肝炎,但命名、分类和定义等和国际上所称的肝衰竭并不一致。我国《肝衰竭诊疗指南》将肝衰竭定义为由多种因素引起肝细胞严重损害,导致其合成、解毒和生物转化等功能障碍,出现以黄疸、凝血功能障碍、肝性脑病和腹水等为主要临床表现的一种临床综合征。临床上可将其分为急性肝衰竭、亚急性肝衰竭、慢加急性肝衰竭和慢性肝衰竭四种。急性肝衰竭指起病2周以内出现肝衰竭表现,亚急性肝衰竭指起病15天至24周出现肝衰竭,分别相当于既往的急性或亚急性重症肝炎。慢加急性肝衰竭之在慢性肝病基础上出现急性或亚急性肝衰竭,是本次肝衰竭标准中新提出的。慢性肝衰竭指在慢性肝病基础上出现肝功能进行性减退或造成失代偿,是慢性肝硬化的结果,和国际上的定义相同。
肝豆状核变性的护理体会
肝豆状核变性的护理体会 作者:邱祖燕章冬梅来源: 肝豆状核变性又称Willson病(WD病),是常染色体隐性性疾病,铜蓝蛋白(CP)合成障碍是本病最基本的遗传缺陷。WD是由于铜代谢障碍,导致体内大量的铜沉积在肝、脑、角膜、肾脏、骨骼等部位,而出现相应的表现,如、锥体外系症状、角膜色素环(K-F 环)和肾功能损害等。发病年龄4~50岁,平均18岁[1]。现将我院2003~2004年共收治的3例总结如下。 1 临床资料 本组3例,男1例,女2例,年龄12~20岁,平均17岁。3例均以神经系统症状为首发症状,尚无肝功能损害。有锥体外系症状的2例,有吐词不清的1例,3例均有角膜色素环。头颅MRI均显示豆状核部位异常信号改变,血清铜蓝蛋白显著降低而确诊。 2 护理 心理护理少年型患者应保持与学校、父母和医生间的联系,让学校、老师、同学了解其病情,帮助其克服心理上的障碍和学习上的困难,增强自我保护意识,避免某些危险的活动、游戏和区域。 饮食护理减少铜的摄入,防止铜盐蓄积。向患者及家属介绍有关疾病的知识,懂得长期限制饮食的重要性,以取得患者及家属完全合作。 2.2.1 尽量避免食用含铜量高的食物,严禁食用贝类、坚果类、蘑菇、动物脂肪、海产品及其他含铜高的食品。注意勿使用铜制的炊具和餐具。 2.2.2 宜长期饮用低铜高蛋白的牛奶,长期多量食用还有排铜功效。 用药护理告知药物的作用与用法,注意观察药物的疗效与,发现异常情况及时报告医生处理。 2.3.1 D-青霉胺使用前需做青霉素皮试,根据病型、病程、严重程度及体差异等因素,从小剂量开始,逐渐加量至症状缓解后减为维持量的个体化用药;长期应用可致维生素B 6 缺乏,应予及时补充。
儿童肝功能异常原因待查首次病程
肝功能异常原因待查 小儿8月余以来外院多次查肝功能均异常,故可明确诊断肝功能异常,外院做血、骨髓、肝穿刺病理等检查均未确诊,分析可能病因如下: 感染因素:8月余以来反复间断发热,应注意感染因素。 (1)细菌感染:各种细菌感染,以及全身及局部严重感染,均可损害肝功能,导致肝功能异常。但此类异常多为一过性,随感染控制肝功能应恢复正常。患儿病程中感染控制后,肝功能持续异常,与此不符。 (2)病毒感染:各种病毒感染,以及全身及局部感染,亦均可损害肝功能,特别如HBV、CMV、EBV等感染。外院曾查EBV抗体阳性,需考虑本病毒感染致肝功能持续异常,入院后进一步查乙肝五项、EBV抗体四项、四病毒抗体等协诊。 (3)其它病原体如结核、支原体、真菌、阿米巴原虫等感染,但患儿目前无相关感染表现,需进一步观察。 非感染因素:无感染征象时患儿肝功能已有异常,故需注意非感染因素 (1)营养因素:患儿家庭条件好,营养供应充足,虽患儿食欲一般,且有挑食,但患儿无营养不良表现,营养及生长发育中等,可除外本病。 (2)代谢性疾病:如糖原累积病、粘多糖病、尼曼-匹克综合征、戈谢病等,患儿常有体格发育畸形,肝脾肿大等表现,不支持本病。
(3)肿瘤:原发或转移性肝脏肿瘤,可致肝功能异常,但同时多伴肝脏肿大。患儿肝脏不肿大,曾做腹部彩超未见腹部脏器异常,肝脏活检亦未见肿瘤细胞,不支持。 (4)非炎症性肌病:肝功能可异常,多同时伴有CK增高。患儿无CK增高等肌损害表现,不支持。 (5)风湿性疾病:患儿无皮疹、关节痛等表现,曾查ANA、ds-DNA、ANCA系列等均阴性,不支持。 1.入院后予以清热合剂口服清热解毒。 2.积极完善辅助检查如血尿便常规,血沉,CRP,血生化,心电图,胸片,结核菌素试验、四病毒抗体、肺炎支原体抗体、ASO、腹部彩超等。 3.向家长交待病情,患儿目前诊断肝功能异常原因待查,病因不明确,入院后可完善辅助检查以明确诊断。入院后可能病情加重,院内交叉感染,费用较高,家长表示理解。 4. 请示上级医师指导诊治。
2017年主管护师儿科护理学试题
2017年主管护师儿科护理学试题 考前的备考工作是很重要的,高效率的复习方法能让考生们更快地进入备考状态,下面是为大家准备的2017年主管护师儿科护理学试题,供大家参考借鉴! A1型题 1.为诊断21-三体综合征最重要的检查是 A.骨穿 B.腰穿 C.血常规 D.甲状腺功能 E.染色体核型分析 2.小儿最常见的染色体病是 A.苯丙酮尿症 B.肝豆状核变性 C.21-三体综合征 D.系统性红斑狼疮 E.先天性甲状腺功能减低症状 3.21-三体综合征标准型的核型是 A.47,XY(或XX),+21 B.47,XY(或XX),+22 C.46,XY(或XX),-14,+t(14q21q) D.46,XY(或XX),-15,+t(15q21q) E.46,XY(或XX),-21,+t(21q21q) 4.21-三体综合征最常见的伴发器官畸形是 A.肾脏 B.心脏 C.消化道 D.呼吸道 E.生殖器 5.21-三体综合征核型分析最常见的是 A.标准型 B.易位型 C.嵌合体型 D.D/G易位型 E.G/G易位型 6.21-三体综合征患儿手皮纹征特点不包括 A.通贯手 B.atd角增大 C.atd角减小 D.第4、5指桡箕增多 E.第5指只有一条指褶纹 7.苯丙酮尿症最突出的临床特点是 A.皮肤白皙 B.智力低下 C.伴有惊厥
D.肌张力减低 E.头发黄褐色 8.典型苯丙酮尿症的病因是缺乏 A.多巴胺 B.5-羟色胺 C.四氢生物蝶呤 D.酪氨酸羟化酶 E.苯丙氨酸羟化酶 9.典型苯丙酮尿症最主要的治疗方法是给予 A.酪氨酸 B.5-羟色胺 C.左旋多巴 D.四氢生物蝶呤 E.低苯丙氨酸饮食 10.苯丙酮尿症患儿一般在生后何时初现症状 A.1个月~2个月 B.3个月~6个月 C.7个月~12个月 D.13个月~18个月 E.19个月~24个月 11.苯丙酮尿症新生儿筛查应采用的试验是 A.尿三氯化铁试验 B.苯丙氨酸耐量试验 C.血清苯丙氨酸浓度测定 D.尿2,4-二硝基苯肼试验 E.GuthriE细菌生长抑制试验 12.苯丙酮尿症患儿的外观特点是 A.眼裂小 B.头发黑色 C.皮肤白皙 D.皮肤粗糙 E.眼外角上斜 13.苯丙酮尿症患儿的尿液特点是 A.血尿 B.甜味尿 C.蛋白尿 D.管型尿 E.鼠尿臭味 14.苯丙酮尿症患儿,低苯丙氨酸饮食需服用至 A.3岁 B.5岁 C.10岁 D.终生 E.青春期后 15.苯丙酮尿症属于 A.内分泌病 B.染色体畸变 C.免疫性疾病
人工肝与肝衰竭
人工肝支持系统治疗的适应证 .重型病毒性肝炎:包括急性重型、亚急性重型和慢性重型肝炎,原则上以早、中期为好,凝血酶原活动度控制在,血小板>×109/L者为宜,晚期重型肝炎和凝血酶原活动度<者也可进行治疗,但并发症多见,应慎重。 .其他原因引起的肝功能衰竭(包括药物、毒物、手术、创伤、过敏等)。 .晚期肝病肝移植围手术期治疗。 .各种原因引起的高胆红素血症(肝内胆汁淤积、术后高胆红素血症等),内科治疗无效者。.临床医师认为适合人工肝支持系统治疗的其他疾病。 二、人工肝支持系统治疗的相对禁忌证 .疾病晚期,出现难以逆转的呼吸衰竭、重度脑水肿伴有脑疝等频危症状者禁用。 .有严重全身循环功能衰竭者禁用。 .伴有弥散性血管内凝血状态者禁用。 .有较重的活动性出血者应慎用。 .对治疗过程中所用药品如血浆、肝素、鱼精蛋白等高过敏者,应慎用。 .临床医师认为不能耐受治疗的其他情况患者。 三、人工肝支持系统治疗的疗效判断 人工肝支持系统治疗临床用近期疗效和远期疗效来进行疗效判断。 .近期疗效 ()治疗前后有效率 临床治疗前后有效率是以患者乏力、纳差、腹胀、尿少、出血倾向和肝性脑病等临床症状、体征的改善,血胆红素下降,胆碱脂酶活力增高;凝血酶原活动度改善;血内毒素下降及血芳香氨基酸和支链氨基酸比值的好转等指标来评价。 ()患者出院时的治愈好转率 ①急性、亚急性重型肝炎以临床治愈率作为判断标准。临床治愈标准:乏力、纳差、腹胀、尿少、出血倾向和肝性脑病等临床症状消失。黄疸消退,肝脏恢复正常大小。肝功能检查基本恢复正常。恢复正常。 ②慢性重型肝炎以临床好转率为判断标准。临床好转标准:乏力、纳差、腹胀、出血倾向等临床症状明显好转,肝性脑病消失。黄疸、腹水等体征明显好转。肝功能:检查明显好转总胆红素降至正常的以下,凝血酶原活动度在以上。 .远期疗效 存活率:分治疗后半年存活率和年后存活率两种。 本篇文章来源于中华肝病网|https://www.360docs.net/doc/c19387782.html, 原文链接:https://www.360docs.net/doc/c19387782.html,/ganbingyanjiu/2608.html 。转载时请以链接的形式注明原始出处、原文链接及本声明。 人工肝治疗的适应证 2.1 肝脏衰竭各种原因引起的急性、亚急性和慢性肝衰竭进展期均可考虑,以早、中期应用为佳。 2.2 肝功能不全综合判断有明显向肝衰竭发展者,一旦内科治疗效果不明显,应考虑配合人工肝治疗。 2.3 肝移植围术期治疗可暂时改善机体状态,为移植手术争取时间或改善术前条件。
病毒感染与肝功能衰竭(文档版)
1.1甲型肝炎病毒(HAV)感染.HAV是小RNA病毒.主要 经粪——口途径传播。人感染HAV,可表现为临床型和亚临 床型感染。6岁以下儿童感染HAV后,70%为亚临床型感染; 大龄儿童和成人感染HAV后,70%以上为临床型感染。病死率为0.3%~0.6%;50岁以上患者的病死率为1 .8%;慢性肝病 者感染HAV后,发生急性肝衰竭的危险性升高.。HAv感染大多表现为自限性、急性临床过程,单独HAV感染进展为急性肝衰竭的几率低(约0.1%~0.35%),但一印度医院报道的70例HAV感资的儿童中,l4例发展为急性肝衰竭.几率为 20%。调查解放军302医院1977例肝衰竭患者的病因未发砚单独HAV感染急性肝衰竭,发现5例HAV感染引起的肝衰竭皆为在慢性HBV感染基础上发生。有研究显示HAV感染进展为急性肝衰竭与HAV受体TIM1的基因多态性有关1.2乙型肝炎病毒(HBV)感染目前我国慢性HBV感染者 达9300万,HBV感染是我国引起肝衰竭的最主要病因。有研究显示,HBV感染在1977例急性、亚急性、慢加急性肝衰竭患者病因中占87.25%。 1.3 丙型肝炎病毒(HCV}感染HCV感染是欧美国家终末期肝病的最主要原因。目前世界范围内确诊为HCV感染
患者的人数达1.7亿,我国抗-HCV阳性率为3.2%,有近4千万感染者。HCV感染后一般临床症状较轻,引起急性肝衰竭少见,仅20%感染者表现为急性并呈自限性发病过程,而15%~80%发展为慢性感染,肝硬化、肝愛发生率也较高,约25%的慢性HCV感染者发展为肝硬化,大多发展为以严重腹水、腹膜炎、肝性脑病、消化道出血等严重并发症为特征的慢性肝衰竭。慢性HCV感染基础上发生的慢加急性肝衰竭少见。 1.4 丁型肝炎病毒(HDV)感染HDV是一个小型的、有缺陷的RNA病毒,只感染有HBV感染基础患者,全球超过1500万人感染。HDV虽一定程度上抑制HBV复制,但会加速病程进展为肝硬化和慢性肝衰竭。有研究报道在HBV 感染相关的急性、亚急性肝衰竭患者中合并HDV感染者约占30% ,远远高于普通型乙型肝炎患者中HDV感染的阳性率,说明HDV/HBV感染发生急性、亚急性肝衰竭的几率远远高于单独H BV感染者。 1.5戊型肝炎病毒(H EV)感染同HAV感染相似,HEV经消化道传播,大多呈自限性,孕妇、年老体弱者及慢性H BV 感染者感染HEV后易发展为肝衰竭,病死率也相对较高。孕妇感染HEV的预后比感染其他嗜肝病毒的预后差,我国上海地区孕妇感染HEV后的病死率为31%,印度北方地区病死率为39.1%。范振平等报道424例戊型肝炎患者中,青
住院医师综合练习题 儿科练习 (5)
儿科练习 一 . 单选题(共90题,每题1分) 1 .不同先心病的X线表现女孩,6岁,发育稍差,乏力、活动后气促、心悸,平时易发生反复呼吸道感染,胸骨左缘第2、3肋间闻及Ⅱ-Ⅲ级吹风样杂音,P2亢进伴固定分裂.心电图示电轴右偏,不完全性右束支传导阻滞,其X线改变是 A . 心脏向两侧扩大,呈蛋形,肺野充血 B . 心脏稍扩大,以右室增大为主呈靴形心,肺纹理少 C . 肺纹理少,肺动脉段突出,右房、右室增大 D . 肺门血管影增强,肺纹理增多,主动脉弓增大 E . 右房、右室增大,肺纹理增多,肺门舞蹈,主动脉影缩小 F . 心外形增大以右、左室增大为主,肺野充血,肺动脉段突出 2 .某婴孕42+周分娩,出生体重4100g,脐带绕颈。Apgar评分1分钟6分,该患儿不应诊断为 A . 新生儿窒息 B . 高危儿 C . 巨大儿 D . 正常新生儿 E . 过期产儿 3 .女孩,12岁。患Turner综合征。下列核型中,哪种最常见 A . 45,Xo B . 45,x/46xx C . 46,xdel(xP) D . 46,xdel(xq) E . 46,xL(xp) 4 . 2岁女孩,因皮肤淤点4天住院,淤点以颜面及双下肢稍多;肝脏肋下2cm,脾脏不大。血常规:血小板20×109/L,红细胞和白细胞数量正常,形态无异常;骨髓检查见骨髓巨核细胞增多,该患儿初步的诊断为: A . 血友病甲 B . 急性ITP C . 急性白血病 D . 弥散性血管内凝血 E . 缺铁性贫血 5 . 10个月女婴,腹泻、呕吐2d,大便每天10余次,稀水样。呕吐每天6~7次,12h无尿。体格检查精神委靡,呼吸较深长,皮肤弹性差,四肢凉。紫绀,脉细弱。血钠125mmol/L,CO2-CP10.68mmol/L该患儿符合哪种诊断 A . 等渗重度脱水,重度酸中毒 B . 低渗重度脱水,重度酸中毒