气相色谱谱图分析

气相色谱谱图分析
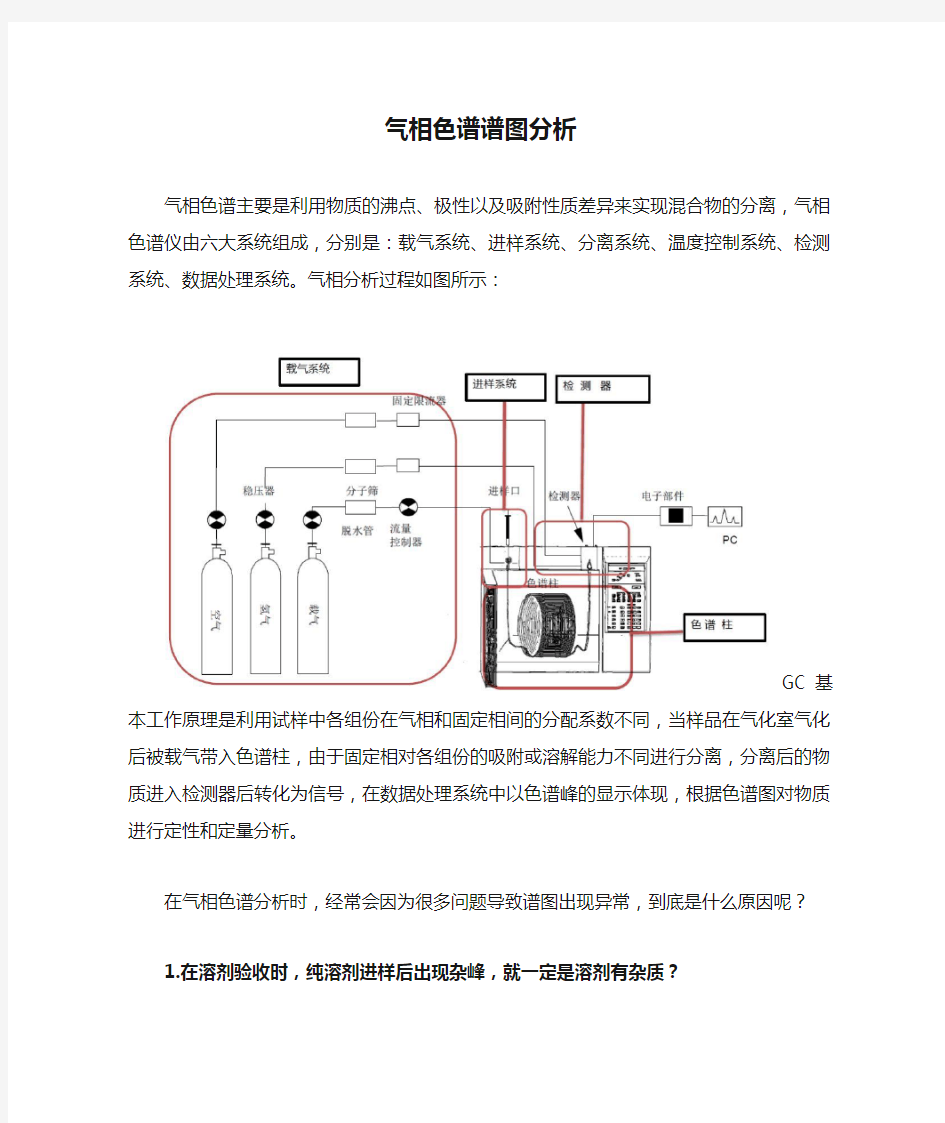
气相色谱主要是利用物质的沸点、极性以及吸附性质差异来实现混合物的分离,气相色谱仪由六大系统组成,分别是:载气系统、进样系统、分离系统、温度控制系统、检测系统、数据处理系统。气相分析过程如图所示:
GC基本工作原理是利用试样中各组份在气相和固定相间的分配系数不同,当样品在气化室气化后被载气带入色谱柱,由于固定相对各组份的吸附或溶解能力不同进行分离,分离后的物质进入检测器后转化为信号,在数据处理系统中以色谱峰的显示体现,根据色谱图对物质进行定性和定量分析。
在气相色谱分析时,经常会因为很多问题导致谱图出现异常,到底是什么原因呢?
1.在溶剂验收时,纯溶剂进样后出现杂峰,就一定是溶剂有杂质?
如果进空白针后也存在杂峰,连续进针后,峰面积逐渐减少,优先考虑仪器系统流路问题出现的杂峰。可以从以下几个方面逐一排查:
1)气源是否有问题;
2)进样针,洗针瓶,隔垫,衬管,分流平板是否有污染;
3)色谱柱是否有污染;
4)检测器是否有污染等。
2.出现前沿峰
1)样品过载,需稀释样品,减少进样量;
2)载气流速过高;
3)柱温太低,升高柱温;
4)气化室温度太低;
5)可能存在干扰峰,需要优化色谱条件;
6)色谱柱选型错误,老化程度不够等。
3.出现拖尾峰
1)衬管、分流平板或色谱柱被污染,或色谱柱安装不当,存在死体积;
2)柱温或进样器温度低,升高温度;
3)载气流量偏低;
4)进样量大,减少进样量货增大分流比;
5)进样器或气化室被高沸点杂质或残留污染等。
4.出现鬼峰
1)色谱柱有残留,未完全老化;
2)气化室、注射针等被污染或载气纯度不够;
3)气化温度过高使样品某些组分分解;
4)样品中有空气或TCD、ECD等密封性差(有漏气)等。
5.操作条件不变,原来可以分离的峰不见了?
1)色谱柱被污染或者失效;
2)载气系统被污染(载气纯度低或过滤器失效);
3)注射垫或注射针漏气等。
6.进样后不出峰或者峰很小?
1)检查检测器的信号值,信号值正常时,优先考虑进样口问题;
2)进样针漏气或者堵塞;
3)进样温度太低导致样品不能气化或柱温太低,导致样品在柱中冷凝;
4)如果是FID,需要检查FID火焰是否点燃等。
7.连续进样时,重复性差
1)进样技术差;
2)载气泄露或者流速不稳定;
3)检测器、色谱柱或衬管等被污染;
4)注射针有泄漏等。
8.基线向上漂移
1)检测器温度达到设定温度并稳定2h以上,基线不稳定,检测器受污染,需要进行清洗;2)色谱柱未完全老化,有残留;
3)载气流速下降,重新调整载气压力;
4)色谱柱固定相可能被破坏等。
9.基线向下漂移
1)进样口隔垫漏气;
2)气路系统漏气或载气流量波动;
3)色谱柱未老化完全等。
10.样品分离度下降
1)色谱柱被样品或其他杂质污染;
2)色谱柱固定相流失严重导致无法达到所需要的柱效或分离度等。
气相色谱仪原理(图文详解)
气相色谱仪原理(图文详解) 什么是气相色谱 本章介绍气相色谱的功能和用途,以及色谱仪的基本结构。 气相色谱(GC)是一种把混合物分离成单个组分的实验技术。它被用来对样品组分进行鉴定和定量测定: 基子时间的差别进行分离 和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC)是基于时间差别的分离技术。 将气化的混合物或气体通过含有某种物质的管,基于管中物质对不同化合物的保留性能不同而得到分离。这样,就是基于时间的差别对化合物进行分离。样品经过检测器以后,被记录的就是色谱图(图1),每一个峰代表最初混合样品中不同的组分。 峰出现的时间称为保留时间,可以用来对每个组分进行定性,而峰的大小(峰高或峰面积)则是组分含量大小的度量。 图1典型色谱图
系统 一个气相色谱系统包括 可控而纯净的载气源.它能将样品带入GC系统进样口,它同时还作为液体样品的气化室色谱柱,实现随时间的分离 检测器,当组分通过时,检测器电信号的输出值改变,从而对组分做出响应 某种数据处理装置图2是对此作出的一个总结。 样品 载气源一^ 进样口一^ 色谱柱一^ 检测器一_ 数据处理」 图2色谱系统 气源 载气必须是纯净的。污染物可能与样品或色谱柱反应,产生假峰进入检测器使基线噪音增大等。推荐使用配备有水分、烃类化合物和氧气捕集阱的高纯载气。见图
钢瓶阀 若使用气体发生器而不是气体钢瓶时,应对每一台GC都装配净化器,并且使气源尽可能靠近仪器的背面。 进样口 进样口就是将挥发后的样品引入载气流。最常用的进样装置是注射进样口和进样阀。注射进样口 用于气体和液体样品进样。常用来加热使液体样品蒸发。用气体或液体注射器穿透隔垫将样品注入载气流。其原理(非实际设计尺寸)如图4所示。
热分析考试考试)20121210)
热分析习题 一、填空(10分,共10题,每题1分)。 1、差热分析是在程序控温条件下,测量样品坩埚与坩埚间的温度差与温 度的关系的方法。(参比) 2、同步热分析技术可以通过一次测试分别同时提供-TG或 -TG两组信号。(DTA-TG ,DSD-TG) 3、差示扫描量热分析是在程序控温条件下,测量输入到物质与参比物的功率差与温度的关 系的方法,其纵坐标单位为。(mw或mw/mg) 4、硅酸盐类样品在进行热分析时,不能选用材质的样品坩埚。(刚玉) 5、差示扫描量热分析根据所用测量方法的不同,可以分类为热流型DSC 与 型DSC。(功率补偿) 6、与差热分析(DTA)的不同,差示扫描量热分析(DSC)既可以用于定性分析,又可以 用于分析。(定量) 7、差热分析(DTA)需要校正,但不需要灵敏度校正。(温度) 8、TG热失重曲线的标注常常需要参照DTG曲线,DTG曲线上一个谷代表一个失重阶段, 而拐点温度显示的是最快的温度。(失重) 9、物质的膨胀系数可以分为线膨胀系数与膨胀系数。(体) 10、热膨胀系数是材料的主要物理性质之一,它是衡量材料的好坏的一个重要指 标。(热稳定性) 二、名词解释 1.热重分析答案:在程序控温条件下,测量物质的质量与温度的关系的方法。 2.差热分析答案:在程序控温条件下,测量物质与参比物的温度差与温度的关系的方法。 3.差示扫描量热分析答案:在程序控温条件下,测量输入到物质与参比物的功率差与温度的关系的方法。 4.热膨胀分析答案:在程序控温条件下,测定试样尺寸变化与温度或时间的关系的方法。 三、简答题 1.DSC与DTA测定原理的不同 答案:DSC是在控制温度变化情况下,以温度(或时间)为横坐标,以样品与参比物间温差为零所需供给的热量为纵坐标所得的扫描曲线。DTA是测量T-T 的关系,而DSC是保持T = 0,测定H-T 的关系。两者最大的差别是DTA只能定性或半定量,而DSC的结果可用于定量分析。DTA在试样发生热效应时,试样的实际温度已不是程序升温时所控制的温度(如
怎样分析气相色谱图
在实际工作中,当我们拿到一个样品,我们该怎样定性和定量,建立一套完整的分析方法是关键,下面介绍一些常规的步骤: 1、样品的来源和预处理方法 GC能直接分析的样品通常是气体或液体,固体样品在分析前应当溶解在适当的溶剂中,而且还要保证样品中不含GC不能分析的组分(如无机盐),可能会损坏色谱柱的组分。这样,我们在接到一个未知样品时,就必须了解的来源,从而估计样品可能含有的组分,以及样品的沸点范围。如果样品体系简单,试样组分可汽化则可直接分析。如果样品中有不能用GC直接分析的组分,或样品浓度太低,就必须进行必要的预处理,如采用吸附、解析、萃取、浓缩、稀释、提纯、衍生化等方法处理样品。 2、确定仪器配置 所谓仪器配置就是用于分析样品的方法采用什么进样装置、什么载气、什么色谱柱以及什么检测器。 一般应首先确定检测器类型。碳氢化合物常选择FID检测器,含电负性基团(F、Cl等)较多且碳氢含量较少的物质易选择ECD检测器;对检测灵敏度要求不高,或含有非碳氢化合物组分时,可选择TCD检测器;对于含硫、磷的样品可选择FPD检测器。 对于液体样品可选择隔膜垫进样方式,气体样品可采用六通阀或吸附热解析进样方法,一般色谱仅配置隔膜垫进样方式,所以气体样品可采用吸附-溶剂解析-隔膜垫进样的方式进行分析。 根据待测组分性质选择适合的色谱柱,一般遵循相似相容规律。分离非极性物质时选择非极性色谱柱,分离极性物质时选择极性色谱柱。色谱柱确定后,根据样本中待测组分的分配系数的差值情况,确定色谱柱工作温度,简单体系采用等温方式,分配系数相差较大的复杂体系采用程序升温方式进行分析。 常用的载气有氢气、氮气、氦气等。氢气、氦气的分子量较小常作为填充柱色谱的载气;氮气的分子量较大,常作为毛细管气相色谱的载气;气相色谱质谱用氦气作为载气。 3、确定初始操作条件 当样品准备好,且仪器配置确定之后,就可开始进行尝试性分离。这时要确定初始分离条件,主要包括进样量、进样口温度、检测器温度、色谱柱温度和载气流速。进样量要根据样品浓度、色谱柱容量和检测器灵敏度来确定。样品浓度不超过10mg/mL时填充柱的进样量通常为1-5uL,而对于毛细管柱,若分流比为50:1时,进样量一般不超过2uL。进样口温度主要由样品的沸点范围决定,还要考虑色谱柱的使用温度。原则上讲,进样口温度高一些有利,一般要接近样品中沸点最高的组分的沸点,但要低于易分解温度。
有机波谱综合谱图解析
综合谱图解析 1.某未知物分子式为C5H12O,它的质谱、红外光谱以及核磁共振谱如图,它的紫外吸收光谱在200 nm以上没有吸收,试确定该化合物结构。并解释质谱中m/z 57和31的来源。
2?待鉴定的化合物(I )和(II )它们的分子式均为C 8H 12O 4。它们的质谱、红外 光谱和核磁共振谱见图。也测定了它们的紫外吸收光谱数据:(I )入max 223nm , S 4100; (II )入max 219nm 2300,试确定这两个化合物。 未之物(I )的谱图 127 100-1 - 10 10 曲 凹 M 亠亲) ? 册 -J P 科 J S W
未之物(II)的谱图
3、某未知物的分子式为C 9H 10O 2,紫外光谱数据表明:该物入max 在26 4、262 I? 257、252nm (&maxIOI 、158、147、194、153);红外、核磁数据如图所示,试 0 LOtMio. sopoiggg 翌g 嚴效 却31卿]卿丄电00 uyo iw mo 推断其结构,并说明理 由。 ! \ \ 「 1 CCh 1 I J —' 1 1 _■ ____ __ _ ,B . _ ,- T J.亠」亠亠」亠 | * --------------- U 5>0 4. 0 d/ppm
4.某未知物C ii H i6的UV 、IR 、中NMR 、MS 谱图及13C NMR 数据如下,推导 未知物结构。 序号 S c ( ppm ) 碳原子个数 序号 S c ( ppm ) 碳原子个数 1 143.0 1 6 32.0 1 2 128.5 2 7 31.5 1 3 128.0 2 8 22.5 1 4 125.5 1 9 10.0 1 5 36.0 1 MS(E[] 100 so 30D A/tnn 350 血 >0624*68<)2 4 內 OS n 2 2 98765^43211 0SU 'H bMRfCDCI^
四大图谱综合解析
2013/12/2四大图谱综合解析[解] 从分子式CHO,求得不饱和度为零,故未知物应为512饱和脂肪族化合物。 1 某未知物分子式为CHO,它的质谱、红外光谱以及核磁共振谱如图,512未知物的红外光谱是在CCl溶液中测定的,样品的CCl稀溶液它的紫外吸收光谱在200 nm以上没有吸收,试确定该化合物结构。44-1的红外光谱在3640cm处有1尖峰,这是游离O H基的特征吸收峰。样品的CCl4浓溶液在3360cm-1处有1宽峰,但当溶液稀释后复又消失,说明存在着分子间氢键。未知物核磁共振谱中δ4. 1处的宽峰,经重水交换后消失。上述事实确定,未知物分子中存在着羟基。未知物核磁共振谱中δ0.9处的单峰,积分值相当3个质子,可看成是连在同一碳原子上的3个甲基。δ3.2处的单峰,积分值相当2个质子,对应1个亚甲基,看来该次甲基在分子中位于特丁基和羟基之间。质谱中从分子离子峰失去质量31(-CHOH)部分而形成基2峰m/e57的事实为上述看法提供了证据,因此,未知物的结构CH是3CCl稀溶液的红外光谱, CCl浓溶液44 CHOH C HC在3360cm-1处有1宽峰23 CH3 2. 某未知物,它的质谱、红外光谱以及核磁共振谱如图,它的根据这一结构式,未知物质谱中的主要碎片离子得到了如下紫外吸收光谱在210nm以上没有吸收,确定此未知物。解释。CH CH3+3.+ +C CH HCOH CHOH C HC3223 m/e31CH CH33 m/e88m/e57-2H -CH-H-CH33m/e29 CH m/e73CHC23+ m/e41 [解] 在未知物的质谱图中最高质荷比131处有1个丰度很小的峰,应从分子量减去这一部分,剩下的质量数是44,仅足以组为分子离子峰,即未知物的分子量为131。由于分子量为奇数,所以未成1个最简单的叔胺基。知物分子含奇数个氮原子。根据未知物的光谱数据中无伯或仲胺、腈、CH3N酞胺、硝基化合物或杂芳环化合物的特征,可假定氮原子以叔胺形式存CH3在。红外光谱中在1748 cm-1处有一强羰基吸收带,在1235 cm-1附近有1典型正好核磁共振谱中δ2. 20处的单峰(6H ),相当于2个连到氮原子上的宽强C-O-C伸缩振动吸收带,可见未知物分子中含有酯基。1040 的甲基。因此,未知物的结构为:-1cm处的吸收带则进一步指出未知物可能是伯醇乙酸酯。O核磁共振谱中δ1.95处的单峰(3H),相当1个甲基。从它的化学位移来CH3N看,很可能与羰基相邻。对于这一点,质谱中,m/e43的碎片离子CHCHCHOC223CH(CHC=O)提供了有力的证据。在核磁共振谱中有2个等面积(2H)的三重33峰,并且它们的裂距相等,相当于AA’XX'系统。有理由认为它们是2个此外,质谱中的基峰m /e 58是胺的特征碎片离子峰,它是由氮原子相连的亚甲-CH-CH,其中去屏蔽较大的亚甲基与酯基上的氧原子22的β位上的碳碳键断裂而生成的。结合其它光谱信息,可定出这个相连。碎片为至此,可知未知物具有下述的部分结构:CHO3NCH2CHCHCHOCCH32231 2013/12/23.某未知物CH的UV、IR、1H NMR、MS谱图及13C NMR数据如下,推[解] 1. 从分子式CH,计算不饱和度Ω=4;11161116导未知物结构。 2. 结构式推导未知物碳谱数据UV:240~275 nm 吸收带具有精细结构,表明化合物为芳烃;序号δc序号δc碳原子碳原子IR ::695、740 cm-1 表明分子中含有单取代苯环;(ppm)个数(ppm)个数MS :m/z 148为分子离子峰,其合理丢失一个碎片,得到m/z 91的苄基离子;1143.01632.01 313C NMR:在(40~10)ppm 的高场区有5个sp杂化碳原子;2128.52731.51 1H NMR:积分高度比表明分子中有1个CH和4个-CH-,其中(1.4~1.2)3128.02822.5132 ppm为2个CH的重叠峰;4125.51910.012因此,此化合物应含有一个苯环和一个CH的烷基。511536.01 1H NMR 谱中各峰裂分情况分析,取代基为正戊基,即化合物的结构为:23
仪器分析气相色谱分析习题+答案.doc
气相色谱习题 一 . 选择题 ( ) 1.色谱图上一个色谱峰的正确描述是( ) A. 仅代表一种组分 ; B. 代表所有未分离组分 ; C. 可能代表一种或一种以上组分; D. 仅代表检测信号变化( )2.下列保留参数中完全体现色谱柱固定相对组分滞留作用的是( ) A. 死时间 ; B. 保留时间 ; C.调整保留时间; D.相对保留时间 ( )3.气-液色谱系统中,待分离组分的k值越大,则其保留值: A. 越大; B. 越小; C.不受影响; D.与载气流量成反比 ( )4.关于范第姆特方程式,正确的说法是: A. 最佳线速这一点,塔板高度最大; B. 最佳线速这一点,塔板高度最小; C. 塔板高度最小时,线速最小; D.塔板高度最小时,线速最大 ( )5.根据范第姆特方程式H=A+B/u+Cu,下列说法正确的是: A.H 越大,则柱效越高,色谱峰越窄,对分离有利; B. 固定相颗粒填充越均匀,则柱效越高; C. 载气线速越高,柱效越高; D. 载气线速越低,柱效越高 ( )6.在范第姆特方程式中,涡流扩散项主要受下列哪个因素影响 A. 载体填充的均匀程度 ; B.载气的流速大小; C.载气的摩尔质量; D.固定液的液膜厚度
( )7.用气相色谱法定量分析试样组分时,要求分离达98%,分离度至少为: ( )8.在气相色谱中,当两组分未能完全分离时,我们说: A. 柱效太低; B. 柱的选择性差; C.柱的分离度低; D. 柱的容量因子大 ( )9.分离非极性组分和极性组分混合物时,一般选用极性固定液,这是利用极性固定液的: A. 氢键作用; B. 诱导效应; C.色散作用; D.共轭效应 ( )10.苯和环已烷的沸点分别是80.10 °C 和 80.81 ° C,都是非极性分子。气相色谱分析中,若采用极性固定 液,则保留时间关系是: A. 苯比环已烷长; B. 环已烷比苯长; C. 二者相同; D. 无法确定 ( )11. 已知苯的沸点为80.10 ° C,环已烷的沸点为80.81 °C。当用邻苯二甲酸二壬酯作固定液分析这二种组 分时,环已烷比苯先出峰,其原因是固定液与被测组分间的: A. 静电力; B. 诱导力; C.色散力; D.氢键力 ( )12.使用热导池检测器时,一般选用H 2或He作载气,这是因为它们: A. 扩散系数大; B. 热导系数大; C.电阻小; D. 流量大 ( )13.氢火焰离子化检测器优于热导检测器的主要原因是: A. 装置简单; B. 更灵敏; C.可以检出许多有机化合物; D.较短的柱能够完成同样的分离
四大图谱综合解析
2013/12/2
四大图谱综合解析
1 某未知物分子式为C5 H12 O,它的质谱、红外光谱以及核磁共振谱如图,
它的紫外吸收光谱在200 nm以上没有吸收,试确定该化合物结构。
CCl4稀溶液的红外光谱, CCl4浓溶液 在3360cm-1处有1宽峰
[解] 从分子式C5H12O,求得不饱和度为零,故未知物应为 饱和脂肪族化合物。 未知物的红外光谱是在CCl4溶液中测定的,样品的CCl4稀溶液 的红外光谱在3640cm-1处有 1尖峰,这是游离 O H基的特征吸收 峰。样品的CCl4浓溶液在 3360cm-1处有 1宽峰,但当溶液稀释 后复又消失,说明存在着分子间氢键。未知物核磁共振谱中δ4. 1处的宽峰,经重水交换后消失。上述事实确定,未知物分子 中存在着羟基。 未知物核磁共振谱中δ0.9处的单峰,积分值相当3个质子,可 看成是连在同一碳原子上的3个甲基。δ3.2处的单峰,积分值 相当2个质子,对应1个亚甲基,看来该次甲基在分子中位于特 丁基和羟基之间。 质谱中从分子离子峰失去质量31(- CH2 OH)部分而形成基 峰m/e57的事实为上述看法提供了证据,因此,未知物的结构 CH3 是
H3C
C
CH3
CH2OH
根据这一结构式,未知物质谱中的主要碎片离子得到了如下 解释。
CH 3
2. 某未知物,它的质谱、红外光谱以及核磁共振谱如图,它的 紫外吸收光谱在210nm以上没有吸收,确定此未知物。
CH2
+ OH m/e31 -2H
+ . CH2OH
H3C
CH3
H3C
C
CH 3
C+
CH3
m/e88 -CH3 m/e29 m/e73
m/e57 -CH3 -H CH 3 C + CH 2
m/e41
[解] 在未知物的质谱图中最高质荷比131处有1个丰度很小的峰,应 为分子离子峰,即未知物的分子量为131。由于分子量为奇数,所以未 知物分子含奇数个氮原子。根据未知物的光谱数据中无伯或仲胺、腈、 酞胺、硝基化合物或杂芳环化合物的特征,可假定氮原子以叔胺形式存 在。 红外光谱中在1748 cm-1处有一强羰基吸收带,在1235 cm-1附近有1典型 的宽强C-O-C伸缩振动吸收带,可见未知物分子中含有酯基。1040 cm-1处的吸收带则进一步指出未知物可能是伯醇乙酸酯。 核磁共振谱中δ1.95处的单峰(3H),相当1个甲基。从它的化学位移来 看,很可能与羰基相邻。对于这一点,质谱中,m/e43的碎片离子 (CH3C=O)提供了有力的证据。在核磁共振谱中有2个等面积(2H)的三重 峰,并且它们的裂距相等,相当于AA’XX'系统。有理由认为它们是2个 相连的亚甲-CH2-CH2,其中去屏蔽较大的亚甲基与酯基上的氧原子 相连。 至此,可知未知物具有下述的部分结构:
O CH 2 CH 2 O C CH 3
从分子量减去这一部分,剩下的质量数是 44,仅足以组 成1个最简单的叔胺基。
CH 3 CH3 N
正好核磁共振谱中δ2. 20处的单峰(6H ),相当于2个连到氮原子上 的甲基。因此,未知物的结构为:
CH3 CH3 O N CH2 CH2 O C CH3
此外,质谱中的基峰m /e 58是胺的特征碎片离子峰,它是由氮原子 的β位上的碳碳键断裂而生成的。结合其它光谱信息,可定出这个 碎片为
CH3 CH3 N CH 2
1
热分析常用方法及谱图
常用的热分析方法 l热重法(Thermogravimetry TG) l 差示扫描量热仪(Differential Scanning Calorimetry DSC)l 差热分析(Differential Thermal Analysis DTA) l 热机械分析(Thermomechanical Analysis TMA) l 动态热机械法(Dynamic Mechanical Analysis DMA) 谱图分析的一般方法 《热分析导论》刘振海主编 《分析化学手册》热分析分册 TGA DSC 分析图谱的一般方法——TGA 1. 典型图谱 分析图谱的一般方法——TGA的实测图谱
I、PVC 35.26% II、Nylon 6 25.47% III、碳黑14.69% IV、玻纤24.58% 已知样品的图谱分析 与已知样品各方面特性结合起来分析 如:无机物(黏土、矿物、配合物)、生物大分子、高分子材料、金属材料等热分析谱图都有各自的特征峰。 与测试的仪器、条件和样品结合起来分析 仪器条件样品 应用与举例 TGA DSC/DTA TMA 影响测试图谱结果的因素——测试条件 TGA 升温速率 样品气氛
扫描速率 样品气氛 升温速率对TGA 曲线的影响 气氛对TGA 曲线的影响 PE TGA-7 测试条件: 扫描速率:10C/min 气氛:a. 真空 b. 空气 流量:20ml/min 样品:CaCO3(AR) 过200目筛,3-5mg 扫描速率对DSC/DTA曲线的影响气氛对DSC/DTA曲线的影响 气氛的性质
两个氧化分解峰 曲线b: 一个氧化分解峰, 和一个热裂解峰 影响测试图谱结果的因素——样品方面 TGA/DSC/DTA 样品的用量 样品的粒度与形状 样品的性质 样品用量对TGA/DSC/DTA曲线的影响 样品的粒度与形状对曲线的影响——TGA/DSC/DTA 样品的性质对曲线的影响——TGA/DSC/DTA TGA/ DSC/DTA 热分析曲线的形状随样品的比热、导热性和反应性的不同而不同。即使是同种物质,由于加工条件的不同,其热谱图也可能不同。如PET树脂,经过拉伸过的PET树脂升温结晶峰就会消失。 PET 树脂的DSC 曲线 TGA应用 成分分析 无机物、有机物、药物和高聚物的鉴别与多组分混合物的定量分析。游离水、结合水、结晶水的测定,残余溶剂或单体的测定、添加剂的测定等。 热稳定性的测定 物质的热稳定性、抗氧化性的测定,热分解反应的动力学研究等 居里点的测定 磁性材料居里点的测定 可用TGA测量的变化过程
气相色谱分析方法的建立
气相色谱分析方法的建立
内标法与外标法 一、内标法 什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样
四大谱图综合解析
3 待鉴定的化合物(I)和(II)它们的分子式均为C8H12O4。它们的质谱、红外光谱和核磁共振谱见图。也测定了它们的紫外吸收光谱数据:(I)λmax223nm,δ4100;(II)λmax219nm,δ2300,试确定这两个化合物。 未之物(I)的质谱 未之物(II)质谱
化合物(I)的红外光谱 化合物(II)的红外光谱 化合物(I)的核磁共振谱
化合物(II)的核磁共振谱 [解] 由于未知物(I)和(II)的分子式均为C8H12O4,所以它们的不饱和度也都是3,因此它们均不含有苯环。(I)和(II)的红外光谱呈现烯烃特征吸收,未知物(I):3080cm-1,(υ=C-H),1650cm-1(υ=C-C) 未知物(II)::3060cm-1 (υ=C-H),1645cm-1(υ=C-C) 与此同时两者的红外光谱在1730cm-1以及1150~1300 cm-1之间均具有很强的吸收带,说明(I)和(II)的分子中均具有酯基; (I)的核磁共振谱在δ6.8处有1单峰,(II)在δ6.2处也有1单峰,它们的积分值均相当2个质子。显然,它们都是受到去屏蔽作用影响的等同的烯烃质子。另外,(I)和(II )在δ4. 2处的四重峰以及在δ1.25处的三重峰,此两峰的总积分值均相当10个质子,可解释为是2个连到酯基上的乙基。因此(I)和(II)分子中均存在2个酯基。这一点,与它们分子式中都含有4个氧原子的事实一致。 几何异构体顺丁烯二酸二乙酯(马来酸二乙酯)和反丁烯二酸二乙酯(富马酸二乙酯)与上述分析结果一致。现在需要确定化合物([)和(II)分别相当于其中的哪一个。 COOEt COOEt COOEt EtOOC 顺丁烯二酸二乙酯反丁烯二酸二乙酯 利用紫外吸收光谱所提供的信息,上述问题可以得到完满解决。由于富马酸二乙酯分子的共平面性很好,在立体化学上它属于反式结构。而在顺丁烯二酸二乙酯中,由于2个乙酯基在空间的相互作用,因而降低了分子的共平面性,使共轭作用受到影响,从而使紫外吸收波长变短。
色谱分析谱图
A5000气相色谱工作站分析报告 样品信息: 样品名称: 乙酸乙酯、甲苯盲样样品编号: 样品来源: 省职防院邮寄采样人: 稀释倍数: 0.0 样品量: 0.0 含量单位: 取样时间: 仪器条件: 仪器名称: 气相色谱仪柱子型号: FFAP 检测器: FID 积分参数: 最小值: 10.00 漂移: 0.02 mV/min 噪声: 0.05 mV 最小峰宽: 2.00 S 相对窗宽: 5% 计算方式: 峰面积 色谱条件: 柱箱温度: 50 (℃)程序升温载气流速: 30 (ml/min) 检测器温度: 130 (℃)空气流速: 300 (ml/min) 气化室温度: 200 (℃)氢气流速: 30 (ml/min) 谱图: 分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.91 9726895 366254 9726895 BB 2 乙酸乙酯0.00 0 0 0.000000 BB
3 甲苯0.00 0 0 0.000000 BB 谱图: 分析结果: 定量方法:归一法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.87 9287219 363551 9287219 BB 2 乙酸乙酯 5.40 67436 4449 25.265 BB 3 甲苯8.2 4 63476 13403 8.777 B B 谱图:
分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.88 9515607 362744 9515607 BB 2 乙酸乙酯 5.42 68086 4510 25.508 B B 3 甲苯8.25 58293 13600 8.061 BB 谱图: 分析结果: 定量方法:外标法 序号组分名保留时间峰面积峰高含量峰型 1 二硫化碳 3.88 9231735 354067 9231735 BB 2 乙酸乙酯 5.41 67415 4556 25.256 B B 3 甲苯8.25 59548 13601 8.235 BB 谱图:
布鲁克质谱说明书
300-MS and 320-MS Quadrupole Mass Spectrometer Hardware Operation Manual
Legal and Regulatory Notices Copyright ? 2010 Bruker All other trademarks are the sole property of their respective owners. All Rights Reserved Reproduction, adaptation, or translation without prior written permission is prohibited, except as allowed under the copyright laws. Warranty The information contained in this document is subject to change without notice. Bruker makes no warranty of any kind with regard to this material, including, but not limited to, the implied warranties of merchantability and fitness for a particular purpose. Bruker is not liable for errors contained herein or for incidental or consequential damages in connection with the furnishing, performance or use of this material. Bruker assumes no responsibility for the use or reliability of its software on equipment that is not furnished by Bruker. Use of Trademarks The names of actual companies and products mentioned herein may be the trademarks of their respective owners.
实验 醇系物的气相色谱分析
实验10 醇系物的气相色谱法定性、定量分析 【实验目的】 (1)理解用已知纯物质对照定性的方法。 (2)理解用气相色谱归一化法进行定量分析的方法和特点。 (3)了解CP-3800气相色谱仪的使用及软件的操作。 (4)掌握微量进样器进样技术。 (5)了解程序升温气相色谱法的原理及基本特点。 【实验原理】 气相色谱法是以气体作为流动相(简称载气)的色谱法。 气相色谱法具有如下的特点: 1.高效能、高选择性可分离性质相似的多组分混合物,如同系物、同分异构体等;分离制备高纯物质,纯度可达99.99%。 2.灵敏度高可检出10-13-10-11g的物质; 3.分析速度快通常一个样品的分析可在几分钟到几十分钟内完成; 4.应用范围广气体样品、低沸点、易挥发或可转化为易挥发的液体或固体样品,不仅可分析有机物,也可以分析部分无机物。 气相色谱仪一般由气路系统、进样系统、分离系统、检测记录系统和温度控制系统(图中未显示)五部分组成(见图1-8-1)。 1.气路系统包括气源(高压气瓶)、气体净化、 气体流量控制等 图1-8-1 气相色谱过程示意图 1.高压钢瓶 2.减压阀 3.净化管 4.流量控制 5.进样口 6.色谱柱 7.检测器
部分组成,其作用是为仪器提供纯洁、稳定的载气。常用的载气有氮气和氢气,也可用氦气、氩气或空气。 2.进样系统包括进样装置和气化室。其作用是将样品在进入色谱柱前迅速气化,并定量转入到色谱柱中。要想获得良好的分离结果,进样速度应极快,且样品应在气化室瞬间气化。液体样品一般都采用微量进样器,可根据进样量的不同选用不同体积的进样器。对气化室的要求是热容量要大,温度要足够高且无催化效应。 3.分离系统该部分由色谱柱组成,是色谱仪的心脏,其作用是分离样品。色谱柱分为填充柱和毛细管柱两种: (1)填充柱:由不锈钢或玻璃作为柱管,内填固定相制成,一般内径为2~4mm,长1~3m。形状有U型和螺旋型两种。 (2)毛细管柱又叫空心柱。毛细管材料可以是不锈钢、玻璃或石英。内径有0.53mm、0.32mm、0.25mm等几种规格,长度一般为10~30m。它的固定相可以直接涂布或通过化学交联键合在预先经过处理的管壁上。按照所用的色谱柱不同又可分为:填充柱色谱和毛细管柱色谱。 4.温度控制系统在气相色谱法中,温度直接影响到色谱柱的分离选择、检测器的灵敏度和稳定性。因此在仪器中主要是对色谱柱箱、气化室、检测器三处的温度进行控制。 5.检测和放大记录系统当样品经色谱柱分离后,各组分按保留时间不同随载气进入检测器,检测器将有关各组分含量的信息转化为易于测量的信号(一般为电信号),经过必要的放大传递给记录仪,最后得到该样品的色谱流出曲线。
气相色谱分析中色谱峰的特征
气相色谱分析中色谱峰的特征 质检部油品分析小班的色谱岗的样品分析,都是使用气相色谱仪和液相色谱仪等等。在色谱分析中,色谱图中的色谱峰不仅是我们得出最终数据的主要依据,而且一定程度上还可以精准的反映出仪器的性能和平稳性。因此,作为分析人员必须深入和充分的理解色谱峰的特征,以进一步提高分析数据的准确性。 为理解色谱峰的特征,首先必须能清楚气相色谱分析、色谱图和色谱峰之间的关系。 气相色谱(GC) 是一种把混合物分离成单个组分的实验技术它被用来对样品组分进行鉴定和定量测定。和物理分离(比如蒸馏和类似的技术)不同,气相色谱(GC) 是基于时间差别的分离技术。气相色谱仪将分析的样品分离、鉴定后,由记录仪绘出样品中各个组分的流出曲线图,即色谱图。色谱图是以组分的流出时间(t)为横坐标,以检测器对各组分的电讯号响应值(mv)为纵坐标。色谱图上可得到一组色谱峰,每个峰代表样品中的一个组分(见下图)。 图1 色谱图 对于一个色谱峰,我们可以获得以下四个基本的测量数据特征: (1)进样后到色谱峰被检测到的时间——定性分析 色谱图中色谱峰的出峰时间反映了被分析的组分因与色谱柱中固定相发生相互作用,而在色谱柱中滞留的时间,从本质上更准确的表达了被分析组分的保留特性。色谱峰的峰位与气相色谱分离过程的热力学性质密切相关,是进行气相色谱定性分析的主要依据。 (2)色谱峰的大小——定量分析
色谱峰的大小指峰高或峰面积的大小,其和每个组分在样品中的含量相关。即若色谱峰的峰面积大,则该峰代表的组分在样品中的含量高;反之,则该峰代表的组分在样品中的含量低。色谱峰的峰高或峰面积是气相色谱进行定量分析的重要依据。 (3)色谱峰的宽窄——色谱柱柱效的高低 色谱峰的宽窄可用来说明色谱分离过程的动力学性质——色谱柱柱效率的高低,色谱峰形愈窄说明柱效愈高,色谱峰形愈宽表明柱效愈低,但是色谱峰的宽窄只能定性的表达柱效。 (4)色谱峰间的距离——色谱柱的选择性 在色谱图上,两个色谱峰之间的距离大,表明色谱柱对各组分的选择性好;两个色谱峰之间的距离小,表明色谱柱对各组分的选择性差。 深入和充分的理解色谱峰特征,是为了进一步获得色谱分析的重要信息,保证分析数据准确无误与及时。
实验-醇系物的气相色谱分析
实验10醇系物的气相色谱法定性、定量分析 【实验目的】 (1)理解用已知纯物质对照定性的方法。 (2)理解用气相色谱归一化法进行定量分析的方法和特点。 (3)了解CP-3800气相色谱仪的使用及软件的操作。 (4)掌握微量进样器进样技术。 (5)了解程序升温气相色谱法的原理及基本特点。 【实验原理】 气相色谱法是以气体作为流动相(简称载气)的色谱法。 气相色谱法具有如下的特点: 1.高效能、高选择性可分离性质相似的多组分混合物,如同系物、同分异构体等;分离制备高纯物质,纯度可达99.99%。 2.灵敏度高可检出10-13-10-11g的物质; 3.分析速度快通常一个样品的分析可在几分钟到几十分钟内完成; 4.应用范围广气体样品、低沸点、易挥发或可转化为易挥发的液体或固体样品,不仅可分析有机物,也可以分析部分无机物。 气相色谱仪一般由气路系统、进 样系统、分离系统、检测记录系 统和温度控制系统(图中未显示) 五部分组成(见图1-8-1)。 1.气路系统包括气源(高压气 图1-8-1 气相色谱过程示意图瓶)、气体净化、气体流量控制等 1.高压钢瓶 2.减压阀 3.净化管
部分组成,其作用是为仪器提供纯洁、稳定的载气。常用的载气有氮气和氢气,也可用氦气、氩气或空气。 2.进样系统包括进样装置和气化室。其作用是将样品在进入色谱柱前迅速气化,并定量转入到色谱柱中。要想获得良好的分离结果,进样速度应极快,且样品应在气化室瞬间气化。液体样品一般都采用微量进样器,可根据进样量的不同选用不同体积的进样器。对气化室的要求是热容量要大,温度要足够高且无催化效应。 3.分离系统该部分由色谱柱组成,是色谱仪的心脏,其作用是分离样品。色谱柱分为填充柱和毛细管柱两种: (1)填充柱:由不锈钢或玻璃作为柱管,内填固定相制成,一般内径为2~4mm,长1~3m。形状有U型和螺旋型两种。 (2)毛细管柱又叫空心柱。毛细管材料可以是不锈钢、玻璃或石英。内径有0.53mm、0.32mm、0.25mm等几种规格,长度一般为10~30m。它的固定相可以直接涂布或通过化学交联键合在预先经过处理的管壁上。按照所用的色谱柱不同又可分为:填充柱色谱和毛细管柱色谱。 4.温度控制系统在气相色谱法中,温度直接影响到色谱柱的分离选择、检测器的灵敏度和稳定性。因此在仪器中主要是对色谱柱箱、气化室、检测器三处的温度进行控制。 5.检测和放大记录系统当样品经色谱柱分离后,各组分按保留时间不同随载气进入检测器,检测器将有关各组分含量的信息转化为易于测量的信号(一般为电信号),经过必要的放大传递给记录仪,最后得到该样品的色谱流出曲线。
综合谱图解析
1、某未知物分子式为CHO,它的质谱、红外光谱以及核磁共振谱如图,它的125紫外吸收光谱在200 nm以上没有吸收,试确定该化合物结构。 1 : 2 : 9 [解] 从分子式CHO,求得不饱和度为零,故未知物应为饱和脂肪族化合物。125未知物的红外光谱是在CCl溶液中测定的,样品的CCl稀溶液的红外光谱44-1处有1尖峰,这是游离O H基的特征吸收峰。样品的在3640cmCCl浓溶液在4word
编辑版. -1宽峰,但当溶液稀释后复又消失,说明存在着分子间氢键。未知13360cm处有处的宽峰,经重水交换后消失。上述事实确定,未知物分4. 1物核磁共振谱中δ子中存在着羟基。个质子,可看成是连在同3未知物核磁共振谱中δ0.9处的单峰,积分值相当个亚甲基,12个质子,对应个甲基。一碳原子上的3δ3.2处的单峰,积分值相当看来该次甲基在分子中位于特丁基和羟基之间。的事OHCH)部分而形成基峰m/e57质谱中从分子离子峰失去质量31(-2实为上述看法提供了证据,因此,未知物的结构是CH3OHCHC CH23CH3根据这一结构式,未知物质谱中的主要碎片离子得到了如下解释。CHCH3+3.++CCHOH CH OHCHC CH m/e31CHCH33m/e88m/e57-2H-CH-H-CH33m/e29CHCCHm/e7323+m/e41 3232 2、某未知物,它的质谱、红外光谱以及核磁共振谱如图,它的紫外吸收光谱在210nm以上没有吸收,确定此未知物。 word 编辑 版.
3622 个丰度很小的峰,应为分子离处有在未知物的质谱图中最高质荷比1311] [解。由于分子量为奇数,所以未知物分子含奇数个子峰,即未知物的分子量为131氮 原子。根据未知物的光谱数据亚无伯或仲胺、腈、酞胺、硝基化合物或杂芳环化合物的特征,可假定氮原子以叔胺形式存在。-1-1典型的红外光谱中在1748 cm处有一强羰基吸收带,在1235 cm1附近有-1处的吸--宽强COC1040 cm伸缩振动 吸收带,可见未知物分子中含有酯基。word 编辑版. 收带则进一步指出未知物可能是伯醇乙酸酯。个甲基。从它的化学位移来看, 11.95处的单峰(3H),相当核磁共振谱中δ提供了C=O)很可能与羰基相邻。对于这一点,质谱中,m/e43的碎片离子(CH3并且它们的裂距相等,的三重峰,在核磁共振谱中有2个等面积(2H)有力的证据。,其中去-2个相连的亚甲-CHCH相当于AA'XX'系统。有理由认为它们是22屏蔽较大的亚甲基与酯基上的氧原子相连。至此,可知未知物具有下述的部分结构:OCHCHCHOC322个
综合热分析
寒假—综合热分析 物质加热后发生化学的或物理的变化时,会表现出吸热、放热等能量的转变,或重量、体积等的变化,不同的物质有不同的组成和结构,加热后有特定的热效应,当物质发生相变化时,就会在特定热效应中反应出来。因此,可以用对物质加热的方法进行相分析。 热重法 材料在加热过程中脱水、氧化、蒸发、升华或燃烧等都会发生重量的变化。 调节和控制加热速度,记录材料重量变化与时间或温度的关系、重量变化的大小,称为热重分析。 热差分析 用二种物质在一定的温度范围内加热,其中一种物质加热后不发生相变化,如果另一种物质加热过程也无相变化,则二种物质之问无热量差;如果其中有一种物质在加热过程中产生相变化,由于吸热或放热,会产生与另一种物质的热量差,即差热。量测产生差热时的温度和差热大小,可以定性或定量分析该物质。加热时无相变化的物质称为参比样 一、脱水 以各种不同状态存在于材料中的水.在加热后失水时要吸收热量,因此不同状态的水的脱除为吸热反应。材料结构不同,水的存在形态不同,则脱水吸热的温度也不同。脱水后,材料失重二脱水温度取决于水在物质中的结合力。 二、分解 加热后,物质由一种化合物变成二种以上的化合物称为分解,破坏了原来的结构,吸收热量成为破坏动能。分解温度和吸收的热量取决于晶格结合的牢固程度。 三、结晶 物质由无定形转变为晶态,即无序→有序,内能减少,放出热量。如果结晶破坏转变为非晶态,则为吸热反应。
硫酸盐对混凝土的侵蚀: 分为化学侵蚀与物理侵蚀。化学侵蚀主要是硫酸盐与水泥水化产物发生化学反应导致混凝土膨胀破坏。物理侵蚀是指硫酸盐结晶对混凝土产生的破坏,这种破坏来自于盐结晶后体积膨胀,其本身未与水泥的水化产物发生化学反应。硅酸盐水泥主要水化产物有水化硅酸钙、水化硅酸钙凝胶、氢氧化钙和水化铝酸钙。 硫酸盐侵蚀是一个复杂的物理化学过程,它是典型的膨胀性腐蚀。以硫酸钠为例,当硫酸根离子的浓度较低时,主要膨胀性产物为钙矾石当硫酸根的浓度很高时,还会生成另一种膨胀产物石膏。其反应如下: 3CaO·Al 2O 3 ·CaSO 4 ·18H 2 O+2CaSO 4 +14H 2 O → 3CaO·Al 2 O 3 ·3CaSO 4 ·32H 2 O(钙矾石) Na 2S0 4 ·10H 2 O+Ca(OH) 2 → CaSO 4 ·2H 2 O+2NaOH+8H 2 O(石膏) Biczok等认为,对硫酸钠侵蚀而言,当硫酸盐浓度比较小时(< 1000mg/L SO 4 2-)侵蚀产物以钙矾石为主,而在高浓度下(> 8000mg/L SO42-)以石膏为主,在1000 —8000mg/L SO 4 2-范围内,石膏和钙矾石都被观察到。 钙矾石(3CaO·Al 2O 3 ·3CaSO 4 ·32H 2 O)在87℃时失去6个结晶水,135℃时失去21~ 22 个结晶水,225℃时失去全部结晶水。石膏(CaSO 4·2H 2 O)在165.6℃转变为CaS04。 (1/2)H 2O,在233 . 7 ℃时转变为无水CaSO 4 ,也有文献报道是123℃和130℃。 因试验原材料、试验条件和仪器型号及参数设置等的不同,不同文献得出的结论也有所差异。 综合目前文献可知 钙钒石主要脱水温度区间是80—130℃ 石膏的主要脱水温度区间是130—150℃ 420-500℃区间的峰对应Ca(OH) 2 的分解 700-850℃区间的峰对应CaCO 3 分解 综合热分析曲线: