药物合成工艺路线的设计和选择

第二章 药物合成工艺路线的设计和选择 药物生产工艺路线是药物生产的基础和依据。一个化学合成药物往往具有多种不同的合成途径,通常将具有工业生产价值的合成途径称为该药物的工艺路线。人们习惯上将化学合成药物的合成按起始原料的不同分为全合成和半合成两类:以结构简单的化工产品为起始原料,经一系列化学反应和物理处理过程制备的方法称为全合成(total synthesis );由具有一定基本结构的天然产物经化学结构改造和物理处理过程制备的方法称为半合成(semi synthesis )。一个药物具体采用何种方法合成主要取决于经济的合理性。
药物生产工艺路线的技术先进性和经济合理性是衡量生产技术水平高低的尺度。在创新药物研究中,人们通过筛选发现先导化合物,进而合成一系列目标化合物,优选出最佳的有效化合物作为新药(new chemical, NCE )。在此过程中经济问题居于次要地位,需要主要考虑的是如何最为快捷地合成所需化合物以进行进一步研究;但是一旦研究中新药(investigational drug, IND )在临床实验中显示出优异性质,便要加紧进行生产工艺研究,寻求合成药物的最佳途径,并根据社会的潜在需求量确定生产规模──这时必须把药物工艺路线的工业化、最优化和降低生产成本放在首位,同时考虑清洁化生产等诸多问题。
进行药物生产工艺路线的设计和选择必须首先对该药物或结构类似的化合物进行国内外文献资料的调查研究和论证,然后优化一条或多条技术先进、操作条件切实可行、设备条件容易解决和原辅料有可靠来源的技术路线,最后写出文献综述报告和生产研究方案,作为大规模工业化生产的基础。
第一节 药物生产工艺路线设计的基本方法──逆合成分析
合成是指从某些原料出发,经过若干步反应,最后制备出所需的产物,最后产物就是合成目标物(药物),或叫目标分子(target molecule ,TM)。实际上,进行合成路线设计时是反其道而行之。
考虑对一个特定药物进行合成,第一步是对这个药物分子的结构特征和理化性质进行收集和考察,由此可以简化合成中的问题或避免不必要的弯路。例如非甾体雌激素药物已烯雌酚(diethylstilbestrol, 2-1)的分子带有明显的对称性,因此可以考虑只合成一部分结构单元,采用分子对接的方法合成目标药物分子,从而减化合成步骤(详见分子对称法);而在考虑前列腺素E 2的合成时,由于已知分子中β-羰基酮体系是不稳定的,因此可以安排在合成的最后几步形成这一结构单元,使其避免经历较多的化学反应。
2-2 , 前列腺素E 2
OH
HO
2-1, 已烯雌酚
进行药物分子合成的第二步是以以上分析为基础,从药物本身出发,一步步倒推出合成此药物的各种合成路线和起始原料,也就是我们通常所说的逆合成法(retrosynthesis)。
逆合成法是药物生产工艺路线设计的最基本的方法,也叫做反合成法(antithetic synthesis),其他一些更为复杂的设计方法都是建立在此方法基础上的,所以首先要掌握逆合成法。逆合成法的整个设计思路也被称为逆(反)合成分析,即从目标分子的结构出发,逐步
考虑,层层分解,先考虑由哪些中间体合成目标物,再考虑由哪些原料合成中间体……最后的原料就是起始物(starting matcrial,SM)。
逆合成分析过程要求:①每步都有合理又合适的反应机理和合成方法;②整个合成要做到最大可能的简单化;③有被认可的(即市场能供应的)原料。
逆合成分析是药物合成的基础,分析思路与真正的合成正好相反:合成是使用各种各样的反应来形成分子骨架,改变分子骨架上的官能团,从而最终获得目标分子;在逆合成中则是利用一系列“转化(transformation)”来推导出一系列中间体和适宜的起始原料。转化用
所得的结构单元称为“合成元”(synthon),由合成元继续推导(用虚线“┄”表示)得到相应的试剂或中间体,有时合成元本身即是中间体。
一、转化的类型
一般来说,有机药物分子由碳骨架和官能团两部分组成。目标分子碳骨架的转化包括分拆(disconnection)、联接(connection)和重排(rearrangement)三种类型。
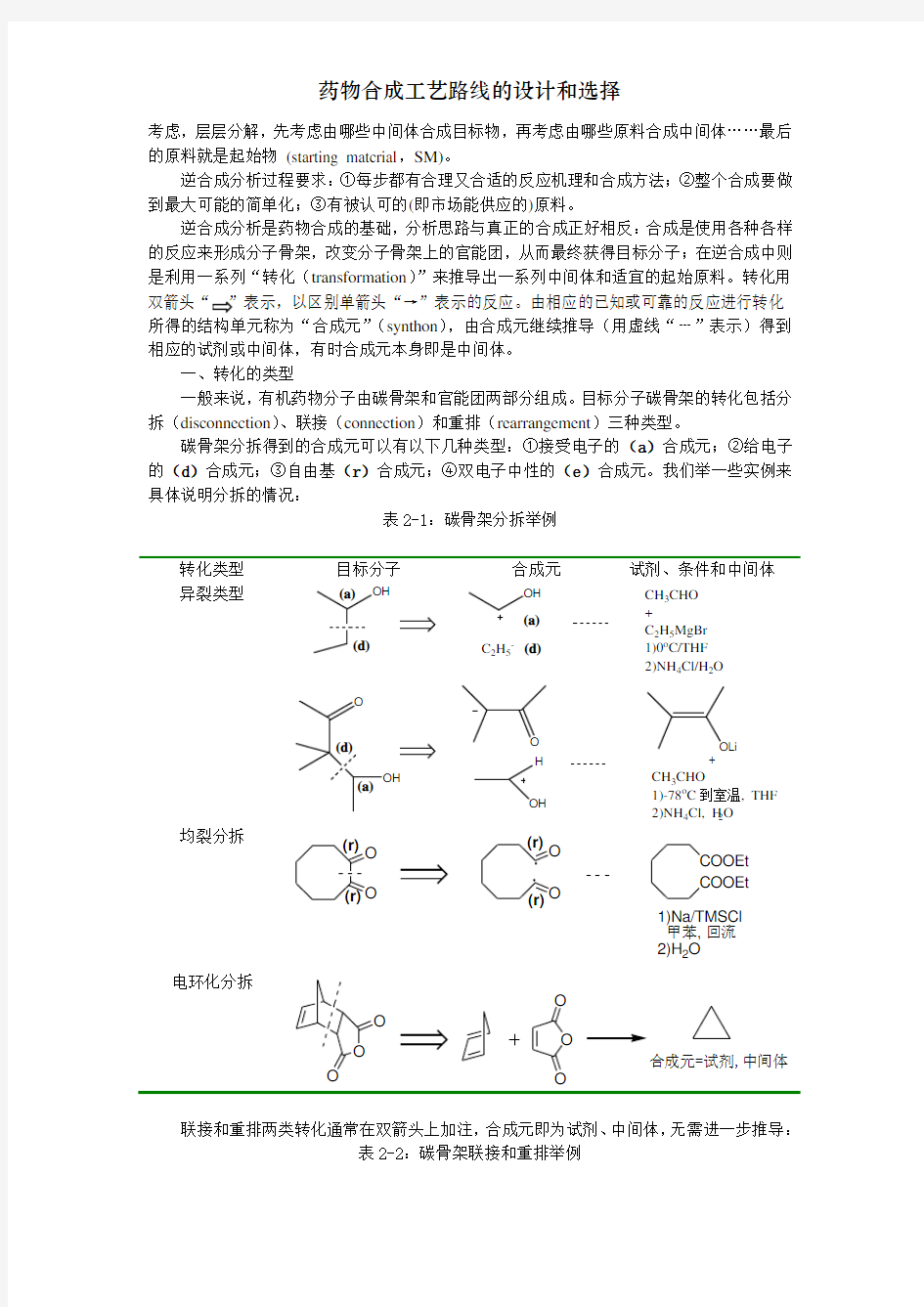
碳骨架分拆得到的合成元可以有以下几种类型:①接受电子的(a)合成元;②给电子的(d)合成元;③自由基(r)合成元;④双电子中性的(e)合成元。我们举一些实例来具体说明分拆的情况:
表2-1:碳骨架分拆举例
转化类型目标分子合成元试剂、条件和中间体异裂类型OH OH
(a)
C2H5-(d)
CH3CHO
+
C2H5MgBr
1)0o C/THF
2)NH4Cl/H2O
(a)
(d)
OH
O
(d)
(a)
O
H
OH
OLi
+
CH3CHO
1)-78o C 到室温, THF
2)NH4Cl, H2O
均裂分拆
O
O
(r)
(r)
COOEt
COOEt
1)Na/TMSCl
甲苯, 回流
2)H2O
电环化分拆
O
O
O
O
O
O
合成元=试剂,中间体
联接和重排两类转化通常在双箭头上加注,合成元即为试剂、中间体,无需进一步推导:
表2-2:碳骨架联接和重排举例
转化类型 目标分子
合成元(试剂、中间体)
反应条件
联接
CHO
CHO O 3/Me 2S
CH 2Cl 2, -78o C 联接
重排
NH
O N
HO
H 2SO 4
重排
官能团的转化也存在与合成反应中相同的三种情况:变换(interconversion, FGI )、引入(addition, FGA )和消除(removal, FGR )。
表2-3:官能团的转化举例
转化类型 目标分子 合成元(试剂、中间体) 反应条件 官能团变换
(FGI ) HgCl 2, MeCN FGI
O
OH CrO 3, H 2SO 4acet.
S
S
HgCl 2, H 2SO 4, aq.
官能团引入(FGA )
FGA
H 2, Pd-C, EtOH
O
O
O
COOH
官能团消除(FGR )
FGR
1) LDA, THF, -25o C 2) O 2, -25o C 3)rt, H 2O
O
O
OH
重复和交替使用上述转化过程,就可以推导出合成目标药物分子所需的起始原料。具体做法就是一步一步地进行逆合成分析,最终推导出合成此目标化合物的可能路线和易得的起始原料。每一步逆合成可以得出若干合成元,由合成元再推导得到试剂或反应底物,如果此试剂或反应底物仍然难得,则再进行进一步的逆合成。例如局麻药物普鲁卡因(procaine, 2-3)的转化分别经历了两次官能团的转化和一次分子骨架的转化,最终得到起始原料对硝基甲苯(2-7):
图2-1:普鲁卡因的逆合成分析
H 2N
O
O
N Et Et
O 2N
O
O N
Et
Et
O 2N O (d)O
N Et Et
(a)
HO
N Et Et
O 2N
O
OH
O 2N
O
HN
Et
Et
FGI
FGR
2-3
2-4
2-5
2-6
2-7
在合成普鲁卡因(2-3)的过程中,以重铬酸钠氧化对硝基甲苯(2-7),生成对硝基苯甲酸(2-5),再与二乙胺基乙醇(2-6)进行酯化反应,经二甲苯共沸脱水得硝基卡因(2-4),2-4于稀盐酸中用铁粉还原即得产物。
图2-2:普鲁卡因的合成
H 2N
O
O
N Et Et
2-3
O 2N
O
O N
Et Et
HO
N Et Et O 2N
O
OH
O 2N
2-4
2-5
2-6
2-7
Na Cr O H 2SO 4
二甲苯
HCl
推导合成元的目的是为合成设计服务,由于推导出的有些合成元所依据的转化、分拆还不存在相应的反应,因此一般没必要推导出所有可能的合成元。为从逆合成分析过程中得到实用的合成元和易得的中间体或原料,有机合成化学家已从合成工作实践中总结了许多规律,可以作为我们药物合成设计的有益借鉴。
设计药物分子的合成路线是比较困难的问题,即使结构不太复杂的药物分子,在它们的合成过程中也总包含有骨架与官能团的变化,这样就产生了一个问题:在解决骨架与官能团都有变化的合成问题时应该首先考虑什么?
化合物的性质主要是由分子的官能团决定的,但是在解决骨架与官能团都有变化的合成问题时,要优先考虑骨架的形成,这是因为官能团是附着于骨架上的,骨架建立不起来,官能团就没有根基。
考虑骨架的形成时,首先研究目标分子的骨架是由哪些较小单元的骨架,通过哪些成键反应结合而成的,较小单元的骨架又是由哪些更小的碎片的骨架通过何种成键反应结合成而的……依此顺序推断下去,直到得出最小碎片的骨架,也就是应该使用的原料的骨架。
但是考虑骨架形成的过程中又不能脱离官能团。碳骨架的形成和官能团的运用是两个不同的方面,二者相对独立但又相互联系:碳骨架只有通过官能团的运用才能装配──反应是在官能团上发生的,或是在由于官能团的影响所产生的活动部位(例如羰基或双键)上发生的,因此,在建立碳碳键之前应首先建立碳杂键。
二、逆合成转化的一般顺序
逆合成分析过程如同数学运算:数学运算是从已知条件开始,最终获得正确答案,虽然解题的过程只要逻辑正确可以因人而异,却有繁简之分;而任何一条合成路线的设计,只要能合成出所需要的化合物,应该说都是合理的,但是合成的技巧、路线设计水平的高低却体现在路线的简洁、产率的高低、原料的来源方便与否、操作的难易等诸多方面。为了设计一条高水平的合成路线,应该科学、合理地做好逆合成转化工作。一般来说,逆合成转化工作应遵循以下顺序:
1. 由目标分子结构和反应性决定逆合成顺序。在进行药物分子合成过程中,首先需要对目标分子有充分认识,并对有机反应有深入了解,通过对目标分子的结构考察,分析其结构特征及化学反应性质,从而设计出有针对性的合成路线。在目标分子的分拆过程中,应首先分拆对称部分(见分子对称法);然后分拆分子中不稳定部分或影响分子反应性及选择性的部分。
目标分子中C-N 、C-S 、C-O 等碳杂键通常是该分子的拆键部位,即分子的连接部位。例如抗真菌药益康唑(econazole, 2-8)分子中有C-O 和C-N 两个拆键部位,可从这两处追溯其合成的前一步中间体:
O
N
Cl
N
Cl
Cl
2-8, 益康唑
a b
如图2-3所示,从虚线a 处断开,益康唑的前体为对氯甲基氯苯(2-11)和1-(2,4-二氯苯基)-2-(1-咪唑基)乙醇(2-9);从虚线b 处断开,前体为咪唑(2-12)和2-(4-氯苯甲氧基)-2-(2,4-氯二苯)氯乙烷(2-10)。进一步分拆,断开C-N 键或C-O 键,2-9的前体为1-(2,4-二氯苯基)-2-氯代乙醇(2-13)和咪唑(2-12);2-10的前体为对氯甲基氯苯(2-11)和2-13。综上所述,无论按照a 途径或b 途径分拆,得到的合成益康唑的基本原料都为咪唑(2-12)、对氯甲基氯苯(2-11)和1-(2,4-二氯苯基)-2-氯代乙醇(2-13),问题是先合成C-O 键还是先合成C-N 键有利呢?
按照b 途径分拆,2-13与对氯甲基氯苯(2-11)在碱性条件下制备中间体2-10时,不可避免地将发生2-13自身分子的烷基化反应,从而使反应复杂化,降低的2-10收率。因此,先形成C-N 键,再形成C-O 键的a 途径对合成益康唑分子更为有利。
图2-3:益康唑的逆合成分析
O
N
Cl
N Cl
Cl
2-8
a
b
N
Cl
N
Cl
Cl Cl
a 途径
O HN
Cl
N
Cl
Cl b 途径
HO Cl
2-9
2-10
HN
N
Cl
Cl HO Cl
Cl HO
2-13
2-11
Cl
Cl
Cl
Cl
2-11
2-122-12
2-13
1-(2,4-二氯苯基)-2-氯代乙醇(2-13)是一个仲醇,可由相应的酮还原制得,而其前体α-氯代-2,4-二氯苯乙酮(2-14)可由2,4-二氯苯(2-15)与氯乙酰氯(2-16)经Friedel-Crafts 反应制备。
图2-4:1-(2,4-二氯苯基)-2-氯代乙醇(2-13)的逆合成分析
2-15
2-16
Cl
Cl HO 2-13
Cl
Cl
Cl O
2-14
Cl
Cl
Cl O
Cl
Cl
FGI
图2-5:益康唑的合成
2-112-15
2-16Cl Cl HO
2-13
Cl
Cl
Cl O
2-14
Cl
Cl O
Cl
Cl AlCl 3
4
O
N
Cl
N
Cl
Cl
2-8
a b
N
N
Cl Cl HO
2-9
HN N
Cl
Cl
CH 3OH
CH 3ONa
2.从合成角度考虑逆合成转化顺序
一般来说,碳杂键易于合成,在分拆过程中处于优先考虑的地位。但是有时候首先分拆碳碳键可简化合成过程,提高目标药物分子的合成收率。例如中枢神经镇痛药哌替啶(pethidine, 2-17)是含脂环叔胺的药物,在其分子分拆中,首先经过两次官能团的转化,然后从分子环键结合处分拆C-C 键,再经过一次官能团转化和分拆C-N 键,即可得到起始原料环氧乙烷和甲胺。
图2-6:哌替啶的逆合成分析
N CO 2Et
2-17, 哌替啶
FGI
酯化
N
CO 2H
FGI 水解
N
CN
C-C CN
Cl
N Cl
FGI
HO
N OH
O
CH 3NH 2
2-182-19
2-21
2-22
2-20
分拆
C-N 分拆
在本品的合成过程中,首先是使用环氧乙烷和甲胺发生氮烷基化反应,生成二(β-羟乙基)甲胺(2-22),然后使用氯化亚砜氯化2-22,生成二(β-氯乙基)甲胺(2-21),2-21与苯乙腈(2-20)在碱性缩合剂钠胺存在下环合生成4-苯基-4-氰基哌啶(2-19),2-19在酸性条件下水解,再酯化生成哌替啶(2-17)。
图2-7:哌替啶的合成
CN
2-20
N
CO 2Et
2-17
N
CO 2H
N CN
Cl N
Cl
HO
N
OH
O
CH 3NH 2
2-182-19
2-21
2-22
2
NaNH 2
H 2O H 2SO 4
C 2H 5OH H 2SO
4
不论碳碳键或碳杂键,从合成角度考虑逆合成转化顺序,特别是对一些比较复杂的药物分子,应着重强调从分子的中部分拆以获得汇聚法的合成(见第三节);从分子中环键结合处或从分子的交叉点进行分拆。
例如1998年上市的非甾体抗炎药环氧化酶-2选择性抑制剂西来曲葆(celebrex, 2-23)的逆合成路线,首先选择在位于分子中部的吡唑处分拆分子,形成二酮2-24与4-磺酸氨基苯肼盐酸盐(2-25),二酮2-24可由4-甲基苯乙酮(2-26)和三氟乙酸乙酯(2-27)通过Claisen 缩合反应制得。
图2-8:西来曲葆的逆合成分析
3
2-23, 西来曲葆
3
O
O
H N
NH 2.HCl
S O
O
NH 22-24
2-25
O
Et
3O 2-26
2-27
将4-甲基苯乙酮(2-26)和三氟乙酸乙酯(2-27)在甲醇钠存在下、甲醇中回流,制备二酮2-24,2-24与4-磺酸氨基苯肼盐酸盐(2-25)在乙醇中回流缩合即得目标分子西来曲葆(2-23)。
图2-9:西来曲葆的合成
2-23
2-242-252-26
2-27
N
N
S O
O
NH 2
CF 3
3
O
O
O
Et
3O NaOCH 3
CH 3OH 回流, 24h
EtOH 回流
从合成角度考虑逆合成转化顺序,还应注意首先安排相应反应产率高的转化,或相应反应成功把握大的转化,这是因为越到合成工作的最后环节,原料越为珍贵,失败的代价越为高昂,因此要尽一切可能增加成功率。要做到这一点需要对有机反应有切实深入的理解。
3.从合成反应优化合成转化顺序
一个目标药物分子往往有多种分拆方法,分拆方法不同导致所应用的合成反应不同、合成路线的长短不同、反应条件不同,原辅料和产率也有所差别。因此可以尝试从合成反应优化合成转化顺序。
从合成反应优化合成转化顺序首先可以寻求多键分拆的策略:通过一步合成反应同时建立多个化学键是简化合成步骤的有效方法。上文谈到的哌替啶和西来曲葆的合成实际上都应用了这个策略。在设计降血脂他汀类药物美伐他汀(mevastatin,
2-28)的逆合成路线时也应用了基于协同反应的多键分拆策略。
2-32
2-312-29
2-30
2-28,
美伐他汀
Diels-Alder 转化
Johnson 甾体合成法利用了含氧基团电性效应引发的仿生-烯烃多重环合反应,可在 此反应中可同时建立三个碳碳键和三个脂环,是合成甾体药物的理想方法。
图2-9:Johnson 甾体合成法
OH OCOF 3
H H H
O
H
H H
O
TFA/hexane DCE/0o C, 1h
78%
1)O 3/MeOH/CH 2Cl 2 H 2O/rt 48%
Nu
H
另外,可以尝试使用重排法和连接法。苯并二氮类镇静催眠药氯氮(Chlordiazepoxide ,2-36)的合成就是利用了2-氯甲基-4-苯基-6-氯-喹唑啉-3-氧化物(2-33)的扩环重排获得的。
图2-10:氯氮的合成
N N
O
CH NH N N
NH O
2-36, 氯氮
2-33
N H
N O
HN
N
N
Cl
OH
NH Cl
2-34
2-35
连接法也是十分有用的逆合成分析方法,如经典的甾体全合成中D 环(2-37)的形成──将带有甲基酮侧链的D 环推导得醛酮的中间体(2-38),而此中间体可用连接法推导得到其前体甲基环已烯(2-39),这里依据的是臭氧氧化反应。
2-38
2-39
2-37
O
D
O
O
CHO
O
O 3
con.
路线设计是药物合成工作的起始工作,也是最重要的一环。在对分子结构特征和理化性质收集、考察及进行逆合成分析,设计出初步的合成路线之后,路线设计的第三步工作就是在此逆合成分析设计的基础上进行正向检查,确定合成路线的切实可行性,这时主要是对合
成所涉及的化学反应进行进一步的考查,保证合成路线的顺利完成。
由于各种原因,按照有机合成基本原理设计的合成路线在实际执行过程中常常会遇到一些始料未及的困难,因此在合成设计时一定要留有机动灵活的余地,最理想的情况是为每一个中间体的合成都准备两到三套方案。
第二节 药物生产工艺路线设计常用方法
逆合成分析方法是合成药物的最基本方法,但是通过对药物分子结构和性质的考察,我们能够发现许多更为便捷有效的合成途径,而没有必要对每一个药物都进行繁琐的逆合成分析。下面介绍几种常用的合成药物方法。
一、分子对称法
对某些药物或中间体进行结构剖析时,常可发现其中存在分子对称性(molecular symmetry ),这对于合成路线的设计工作是非常有益处的。具有分子对称性的化合物往往可由两个相同的分子经化学合成反应制得,或在同一步反应中将分子的相同部分同时构建起来,这就是分子对称法。
例如已烯雌酚(2-1)可以通过两分子的对硝基苯丙烷(2-40)在氢氧化钾存在的情况下与水合肼作用,缩合与还原同时发生,生成3,4-双对氨基苯基已烷(2-41),再经重氮化、水解反应,转变官能团制得。
NH 2
H 22-41
O 2N
H NNH /KOH/H 2O
OH
2-1
1. NaNO 2/H 2SO 4/H 2O
2. dilute H 2SO 4
2-40
目标药物分子的对称性,包括轴和面的对称性都可用于简化合成设计,对于简单分子而言这一点是明显的。但有些药物分子本身并没有对称因素,只有经过一定的逆合成转化,才能得到一个对称的分子或一条对称的合成路线,从而简化合成设计,这叫做分子的潜在对称性(potential molecular symmetry )。例如抗麻风药物氯法齐明(clofazimine ,2-42)可看作吩嗪亚胺类化合物,即是2-对氯苯氨基-5-对氯苯基-3,5-二氢-3-亚胺基吩嗪(2-43)的衍生物,2-43从虚线处可看成两个对称分子。
N
N Cl
N Cl
2-42,氯法齐明
2-43
因此,可使用两分子的N -对氯苯基邻苯二胺(2-44)在三氯化铁存在下进行缩合反应,然后与异丙胺进行加压反应,即可制得氯法齐明(2-42)。
N
N Cl
N
Cl
2-42
NH 2Cl
N H Cl
2-44
NH 2FeCl /EtOH
N
N Cl
NH Cl
2-43
H 2NCH(CH 3)2
98%
2-44
除草剂地衣酸(usnic acid ,2-45)本身也无对称性,但可分拆为相同的两部分,即可由相同两片段汇聚合成,这时的合成是左右对称的(这种情况被称为自反性)。
OH
HO
OH
EtOOC
COOEt
O
O
OEt Cl
O 2-45, 地衣酸
MeCN
2-46
2-472-48
2-49
2-50
二、类型反应法
对于一些药物或关键中间体,可根据它们的化学结构类型和官能团性质采用类型反应法进行药物工艺路线的设计。类型反应法就是指利用常见的典型合成反应和合成方法进行路线设计的方法。类型反应法在逆合成分析方法出现之前曾在有机合成领域发挥重要作用,既包括各类化学结构的有机合成通法,又包括官能团的形成、转换或保护等合成反应。对于有明显化学结构特征和官能团的化合物,可考虑采用这种方法进行合成路线设计。
抗真菌药物克霉唑(clotrimazole, 2-51)的关键中间体邻氯苯基二苯基氯甲烷(2-52)具有多种合成方法:首先可参考叔醇的合成方法,采用邻氯苯甲酸乙酯(2-53)与溴苯进行Grignard 反应,先合成叔醇(2-54),再氯化得到:
Cl Ph
Ph Cl
HN
N
2-51,克霉唑
2-52
O
OEt Cl PhBr/Mg Et 2O
OH Ph
Ph Cl
SOCl 2
2-53
2-54
此种合成方法得到的邻氯苯基二苯基氯甲烷(2-52)质量较好,但由于应用了Grignard 试剂,因此需要严格的无水条件,对原辅料质量要求比较严格;同时由于使用的溶剂乙醚易燃、易爆,故要求使用的反应设备要有严格的安全措施,使大规模生产受到一定限制。
鉴于此,参考四氯化碳与苯通过Friedel-Crafts 反应可生成三苯基氯甲烷的类型反应,人们又设计了以邻氯苯基三氯甲烷(2-56)为关键中间体的合成路线:
Cl Ph
Ph Cl 2-52
Cl CCl 4
C 6H 6Ph
Ph Cl
Ph
3CCl 3Cl
Cl /PCl 3C 6H 6
2-55
2-56
这种方法合成路线简短,原辅料来源方便,曾为工业化生产所采用。但在邻氯甲苯(2-55)经氯化反应制得邻氯苯基三氯甲烷(2-56)的过程中,一步反应需要引入三个氯原子,反应温度较高,反应时间较长,未反应的氯气易逸出,不易吸收完全,存在环境污染和设备腐蚀等严重问题。
第三种合成邻氯苯基二苯基氯甲烷(2-52)的方法是以邻氯苯甲酸(2-57)为原料,经两步氯化、两步Friedel-Crafts 反应完成。这种方法虽然工艺路线较长,但原辅料来源方便,反应条件温和,各步反应产率较高,成本也较低,而且没有上述氯化反应的缺点,因此为工业化生产所广泛采用。
2-52
2-57
Cl COOH Cl Cl 3
C 6H 6
Cl Ph
Cl Cl SOCl O
Cl O Ph
5
3C 6H 6
2-58
2-59
2-60
应用类型反应法进行药物或中间体的工艺路线的设计过程中,如果官能团的形成与转化等单元反应的排列方式可能出现两种或两种以上的不同方式时,不仅要从理论上考虑排列顺序的合理性,而且要从实际情况出发,着眼于原辅料、设备条件等因素,通过实验反复比
较确定。化学反应类型相同,但进行顺序不同,则所应用的原辅料不同;原辅料不同,即反应物料的化学组成与理化性质不同,将导致反应的难易程度和反应条件不同,因此往往带来完全不同的结果:药物质量、收率、“三废”治理、反应设备和生产周期等方面都会有较大差异。
例如β-受体阻断剂塞利洛尔(celiprolol, 2-61)的合成,对氨基苯乙醚(2-62)与N ,N -二乙胺基甲酰氯(2-63)作用,生成N -酰化物2-64,降低了的氨基的邻、对位定位作用,同时增大了氨基邻位空间位阻,使后面进行的Friedel-Crafts 酰化反应发生在酚羟基的邻位,得到2-65;若先进行Friedel-Crafts 反应,N -酰化反应居后,则乙酰化反应可同时发生在酚羟基和氨基的邻位,产物复杂。
2-62
NH 2
OEt Cl
N Et
Et O KHCO 3
HN
OEt N Et O
3AlCl 3
HN
OH N Et
Et O O
Et
O 2-632-642-65
三、模拟类推法
对于化学结构复杂、合成路线设计困难的药物,可模拟类似化合物的合成方法进行合成设计——实际上从初步的合成设想开始,通过文献调研,改进他人尚不完善的概念和方法来进行药物工艺路线设计,是药物合成设计中最为广泛使用和实用的方法。
中药黄连中的抗菌有效成分小檗碱(黄连素,berberine ,2-66)与镇痛药帕马丁(palmatine, 2-77)都是具有母核二苯并[a, g]喹嗪,含有稠合的异喹啉环结构的化合物。
O O
O O
O
O
O
N N a
b
c d e
f g
1
23456
8
2-66, 小檗碱
2-67, 帕马丁
二苯并[a, g]喹嗪
4H -喹啉
小檗碱(2-66)可以3,4-二甲氧基苯乙酸2-68为起始原料,采用合成异喹啉环的方法,经Bischler-Napieralski 反应及Pictet-Spengler 反应先后两次环合全合成:
O
O
O O
2-66
HOOC
O O
ClOOC
O O
O
O
NH 2
NH
O
O
O O
O
N
O
O
O O
NH
O O
O O
NH
O O
O
O
Br
N
O
O
O
O
Br SOCl 2POCl 2AcOH
N
O O O O
Zn NaOH 2-68
2-69
2-71
2-72
2-73
2-74
2-75
2-76
在Pictet-Spengler 环合反应前进行溴化,是为了提高环合位置的选择性,最后一步氧化反应可采用电解氧化或HgI 做氧化剂。从合成化学的观点考察,这条路线合理可行,但由于路线较长,收率不高,且使用试剂昂贵,因而不适于工业化生产。
1969年Muller 等人发表了帕马丁(2-67)的合成方法:使用3,4-二甲氧基苯乙胺(2-77)与2,3-二甲氧基苯甲醛(2-78)进行脱水缩合生成Schiff 碱2-79,并立即将其双键还原转变成苯乙基苯甲基亚胺的骨架2-80;然后与乙二醛反应,一次引进两个碳原子而合成二苯并[a, g]喹嗪环,按此合成途径得到二氢帕马丁高氯酸盐2-81与帕马丁高氯酸盐2-67混合物。
O O
2-77
H 2N
CHO
O
O N
O O
O O
HN
O O
O O
O
O
O O
-O O
O O
O
O
HCOOH HClO 4
2-79
2-80
2-81
2-67
2-78
参照上述帕马丁的合成方法,从胡椒乙胺2-70与2,3-二甲氧基苯甲醛(2-78)出发,可快速合成小檗碱(2-66):
2-82
2-83
2-78
O
O
O O
O O
NH 2
2-70
CHO
O
O
N
O O
HN
O O
O
HCOOH
HCl
2-66
O O
O O
按此工艺路线制得的小檗碱(2-66)不含二氢化衍生物,产物的理化性质与抑菌能力同天然提取的黄连素完全一致,符合药典要求,并较前述路线更为简捷,所用原料2,3-二甲氧基苯甲醛(2-78)是工业生产香兰素的副产物,因此较适于大规模生产。
应用模拟类推法的要点在于适当的类比和对有关化学反应的了解,使用时注意比较已有合成方法、类似化学结构及化学活性的差异。
喹诺酮类抗菌药物(2-54-2-90)具有相似的基本骨架,合成多以多取代苯胺为起始原料构建吡酮酸环。构建方法是在诺氟沙星(norfloxacin, 2-84)和环氧沙星(ciprofloxacin, 2-85)等早期品种的合成基础上发展而来的,主要有取代苯胺与乙氧基亚甲基丙二酸二乙酯(EMME, 2-91)缩合成环和经取代芳胺环上的亲核取代反应成环两种。
X
N R 1F
R 2
O
OH
O N
N
R 3
R 4 R 1 R 2 R 3 R 4 X 诺氟沙星, 2-84 Et H H H CH 环氧沙星, 2-85 cyclo -C 3H 5 H H H CH 洛美沙星, 2-86 Et H H Me CF 依诺沙星, 2-87 Et H H H N 氟罗沙星, 2-88 CH 2CH 2F H Me H CF
加替沙星, 2-89 cyclo -C 3H 5 H H H C-OCH 3格雷沙星, 2-90 cyclo -C 3H 5 Me H Me CH
1.取代苯胺与EMME 缩合成环
NH 2
R
EtO 2C
CO 2Et OEt 2-91
N
H
R
CO 2Et
EtO 2C N H
R
CO 2Et
O
诺氟沙星的合成以3-氯-4-氟苯胺(2-92)为原料,先与EMME 脱乙醇缩合,然后在250-260o C 加热环合形成吡酮酸结构的2-93,溴乙烷为烃化试剂完成N 原子上的乙基化,然后水解,引入哌嗪基,得到诺氟沙星2-84。
NH 2
N H
CO 2Et
EtO 2C
N H CO 2Et
OH
F Cl
F F Cl
(C 6H 5O)2O
C H Br K 2CO 3DMF
N
COOH
O
F Cl
HN
NH
py
N
COOH O
F
N
HN
2-842-92
2-93
2-94
与氟诺沙星的合成类似,氟罗沙星(fleroxacin , 2-88)可以2,3,4-三氟硝基苯(2-95)为起始原料,经还原、与EMME 缩合、高温环合、氟乙基化、引入N-甲基哌嗪、酸水解等6步反应得到。
NO 2
2-91N H
CO 2Et
EtO 2C
N H CO 2Et
O
F F
F F
F F
NH 2
F F
F 300o C
TsOCH
2CH 2F 2N
2Et
O
F F
F F 22-88
2-95
2-96
2-97
2-98
2-99
2-100
2.经取代芳胺环上的亲核取代反应成环,离去基团为卤素或硝基
R 2
R 1
CO 2Et
N H O
R
3
R 1
CO 2Et
O
N R 3
(R 2=F, Cl, NO 2)
环丙沙星(2-85)与诺氟沙星(2-84)的结构差异在于1位取代基分别为环丙基和乙基,但两者的合成路线却有很大不同。环丙沙星的合成从2,4-二氯-5-氟苯甲酸(2-101)开始,成酰氯后与β-环丙胺丙烯酸甲酯(2-103)缩合,环合、水解,再引入哌嗪基。
2-101
HN NH
F Cl
Cl N
O
F 2-85
COOH SOCl 2
F Cl
Cl COCl
COOMe
N H F Cl
Cl
COOMe N H
O
Cl
N
COOH
O
F Cl
N
COOH
O
F N
HN
CF COOH py DMF
2-1022-103
2-104
2-105
2-106
按照上述环丙沙星的合成方法,加替沙星(2-89)可以2,4,5-三氟-3-甲氧基苯甲酸(2-107)为起始原料,成酰氯后与3-二甲氨基丙烯酸乙酯(2-109)反应后,再用环丙胺(2-110)置换得到关键中间体2-112。这时若将2-112水解直接与2-甲基哌嗪缩合,由于8位甲氧基的强推电子作用,使得7位氟作为亲核取代反应的离去基团活性大大降低,故缩合收率仅为19.4%;因此将2-112先与硼化物反应生成络合物2-113,由于4位羰基上氧原子的p 电子向硼原子的空轨道发生转移,使得4位羰基的吸电子效应进一步增强,从而提高了7位氟对亲核试剂的反应活性,与2-甲基哌嗪缩合(2-114),然后水解得到加替沙星,按此路线改进后缩合与水解两步产率可提高到75.5%。
2-107
HN
N
F
F
O
2-89
2
F F
O
COOEt
NMe 2F F
F N H
O
N O
F F
F
or (COCl)2F
O COOH COCl NH 2
O
O O B
OAc AcO
N
O F O
O
O B
OAc AcO H 3BO 32Ac 2O
DMF
2-112
2-1082-1092-1102-1112-113
2-114
上述合成喹诺酮类抗菌药物的两条工艺路线都比较成熟,但工艺步骤相对较多,特别是都将7位引入哌嗪基的反应放在路线的最后,且收率较低,技术难度较大。近年来人们对喹诺酮类抗菌药物的合成又有新的改进,尤其是对成环工艺改进很大:以2-氯-4-氨基-5-氟苯甲酸乙酯(2-115)为起始原料,后4步总收率可达94.5%。
2-115
F H 2N
Cl
F N
Cl
HN
F N
Cl
HN
COOEt COOEt
O
COOEt
F
N
Cl
HN
O
COOEt
OEt
F
N Cl
HN
O
COOEt
NH R
F N
N HN
O
COOEt
R F N
N HN
O
COOH R
HN(CH 2CH 2OH)
286.2%
2281.5%
3
RNH 2
环合
水解
2-84 R = Et
2-85 R = cyclo -C 3H 5
2-1162-117
2-118
2-119
2-120
第三节 药物工艺路线的评价与选择
一个药物的工业生产路线往往不只一条,特别是对一些临床使用多年的非创新类药物,由于需求量大、生产企业众多,曾经使用或正在使用的生产路线更是不胜繁举。因此如何在众多已存在和新设计的工艺路线中选择一条适应当前实际情况、技术先进的生产路线,是降低成本、提高企业竞争力的关键。
一、药物工艺路线的评价标准
一个药物通常具有多条合成路线,它们各有特点。对于哪条路线可以发展成为适于工业化生产的工艺路线,必须通过深入细致的综合比较和充分论证才能确定。要想对工艺路线做出正确选择,首先需要对现有的工艺路线做出正确评价。综合考虑生产过程中涉及到的各种情况,一条理想的药物合成工业生产路线应该具有以下基本特征:
1.化学合成途径简洁,即原辅料转化为药物的路线要简短; 2.所需原辅料品种少且易得,并有足够数量的供应;
3.中间体容易提纯,质量符合要求,最好是多步反应连续操作; 4.反应在易于控制的条件下进行,如常温、常压、安全无毒等; 5.设备条件要求较低,不苛刻; 6.“三废”较少且易治理;
7.操作简便,经分离、纯化易达到质量标准; 8.收率最佳、成本最低、经济效益最好。
以上几条标准并不是每一条实际使用的生产路线都能具备的,但却是我们在设计工艺路线时应重点考虑并不断追求的。当不能全部满足所有要求时,最重要的是满足第八条,即收率最佳、成本最低、经济效益最好──实际上其它各条要求也都是为了实现这一条标准而展开的。当然,当未能找到现有路线或现有路线不能满足需要时,可参照前文所述原则和方
法进行重新设计。
抗菌增效剂甲氧苄啶(trimethoprim ,TMP ,2-121)能够抑制细菌二氢叶酸还原酶,阻止细菌核酸合成,主要用作磺胺类抗菌药物的增效剂。它与磺胺甲噁唑(sulfamethoxazole, SMZ )组成的复方片剂(1:5)称为复方新诺明,与磺胺嘧啶(sulfadiazine, SD )合用(1:10)组成的双嘧啶片都是国内目前应用很广的抗菌制剂,但在欧美一些经济发达国家,甲氧苄啶已逐渐退出临床使用,主要作为牲畜用药。
甲氧苄啶最早由Hitchings 等人于1959年合成成功,1962年Stenbuck 等人应用于工业生产,目前全世界有几十个国家生产。在我国,甲氧苄啶生产企业最多时达到40多家, 1997年年产量曾达1737吨。
图2-11:甲氧苄啶合成的剖析
2
O O
O 2O
2-121, 甲氧苄啶
2-122
2-123
从甲氧苄啶化学结构考察,可分为I 和II 两部分。I 部分是合成甲氧苄啶的重要中间体,它的化学结构应具有形成C-C 的活性功能基,目前应用最多的是3,4,5-三甲氧基苯甲醛(2-122)(也有使用3,4,5-三甲氧基氯苄的合成路线);II 部分为2,4-二氨基嘧啶衍生物,若先合成2,4-二氨基嘧啶(2-123)再与I 部分缩合,则由于嘧啶环上的氨基须先加以保护和在5位引入基团的困难,使工艺路线冗长。所以在生产过程中采用逐步形成嘧啶环的途径:即将嘧啶开剖析成a 和b 两部分,将I 部分与a 部分首先缩合,最后与b 部分环合:
图2-12:甲氧苄啶的合成
2-122
N
N O O
O NH 2
NH 2
O O
O O
O
CN
CH 3ONa
O O
O O
CN
O O
O O
CN
O
CH OH CH 3ONa
22
NH HNO 33CH 3ONa
2-125
2-124
2-121
关键中间体3,4,5-三甲氧基苯甲醛(2-122)的合成按原料来源与化学反应中功能基的转化又可分为以下几条途径:
1.以没食子酸为原料 上世纪80年代,我国的甲氧苄啶生产主要以没食子酸(2-126)为原料。
抑制肿瘤细胞增殖的药物筛选方法
抑制肿瘤细胞增殖的药物筛选方法 09级生科3班余振洋200900140156 一、【实验原理】 1.关于恶性肿瘤和抗肿瘤药物: 恶性肿瘤是一种常见病,严重威胁着人类的生存质量,被称为人类健康的第一杀手。多年来人类一直在不断的进行抗肿瘤药物的研究,抗肿瘤药物的筛选是整个研究过程中很重要的个环节,而进行药物的筛选首先离不开合理的筛选方法和系统。寻找选择性强、对实体瘤有效的新型抗肿瘤药物,是摆在抗肿瘤药物研究人员面前的重要任务。世界各国对抗肿瘤药物的筛选都非常重视,投入了大量的人力、物力、财力,每年都有大量的化合物(合成药、天然产物和微生物发酵产物)待筛,抗肿瘤药物筛选方法的发展经历了一个探索的过程。 8O年代中期以前,普遍采用的筛选方法是以体内小鼠白血病/淋巴瘤模型P388和L1210为基础的 J,所有化合物在进一步的临床研究之前必须通过这种小鼠肿瘤模型的筛选。即小鼠白血病P388和L1210作为第一轮初筛,能通过第一轮初筛的化合物才能被允许进入第二轮筛选。这种方法有一个很明显的缺陷就是一些在临床上有活性的药物将被筛选掉,无法保证所有具有抗肿瘤作用的药物都能通过筛选。鉴于以前的筛选方法存在较大的缺陷,1985年之后以NCI为首的一些研究单位普遍开始采用针对疾病的筛选方法来代替针对化合物的筛选方法,即放弃体内小鼠筛选,代之为体外代表各种常见实体瘤的人类肿瘤细胞株筛选。这种筛选系统是一种高通量的抗肿瘤筛选体系,其主要优势有两点:其一是多种细胞株初筛有可能筛选出对特殊的人类肿瘤或对特殊组织亚型有活性的物质;其二是这种体外筛选尤其适合于复杂天然产物提取物中有效成份的证实,过去动物筛选需较大量的天然产物,而现在天然产物的需要量就大大减少,可以指导有效成份的进一步分离纯化,使得从天然产物中发现新的抗肿瘤药物更加便利。 2.关于筛选方法: 下面为现阶段较为普遍采用的一些抗肿瘤药物的筛选方法的实验原理。 1)以端粒酶活性为作用靶点筛选抗肿瘤药物 端粒是染色体特殊结构,起着保护染色体的完整和稳定性的作用,端粒酶是一种核糖核蛋白返转录酶,由RNA和蛋白质组成,可以以自身的RNA为模板合成端粒末端。已发现在正常的体细胞和良性肿瘤组织中端粒酶活性是阴性,而在人体恶性肿瘤组织和人的肿瘤细胞株中都表达了很高的活性。因此,认为端粒酶与恶性肿瘤的发生发展有密切的关系,有可能成为肿瘤治疗的靶点。 2)应用快速荧光素测定法筛选抗肿瘤药物 快速荧光素测定法是一种近几年发展起来的应用非常广泛的体外药物敏感性测定方法,其原理为采用一些特殊的荧光染料,对细胞的特定成份进行染色或标记。或通过细胞酶的作用使无荧光性的材料分解或转换为荧光材料,通过测定荧光强度从而测定出活细胞的量。现在普遍采用一种特殊的荧光染 FDAL1u(Fluoreseein diacetate),在正常情况下它不具有荧光,但当它加人到具有完整细胞膜的肿瘤细胞的营养液中时,由于细胞分泌的水解酶的作用,FDA
设计药物合成路线的方法
设计药物合成路线的方法 一.主要思路 二.主要步骤 1药物结构的剖析:在设计药物的合成路线时,首先应从剖析药物的化学结构入手,然后根据其结构特点,采取相应的设计方法。 2药物剖析的方法:对药物的化学结构进行整体及部位剖析时,应首先分清主环与侧链,基本骨架与功能基团,进而弄清这些功能基以何种方式和位置同主环或基本骨架连接。 研究分子中各部分的结合情况,找出易拆键部位。键易拆的部位也就是设计合成路线时的连接点以及与杂原子或极性功能基的连接部位。如:C -O 、C -S 、C -N 键等。 3考虑基本骨架的组合方式,形成方法;如:基本骨架是芳香环,可采用苯或者苯的同系物或衍生物为原料合成; 基本骨架为杂环化合物的,有一部分可以以天然来源的杂环化合物为原料,例如吡啶,但大部分需要采用缩合或者环合的方式合成。 以此化合物的合成为 例: 4.类型反应法 类型反应法—指利用常见的典型有机化学反应与合成方法进行的合成设计。 主要包括各类有机化合物的通用合成方法,功能基的形成、转换、保护的合成反应单元。 对于有明显类型结构特点以及功能基特点的化合物,可采用此种方法进行设计。 利用典型有机化学反应:如烷基化反应、酰基化反应、酯化反应、缩合反应等等。 例1 抗霉菌药物克霉唑(邻氯代三苯甲基咪唑) 药物合成工艺路线 和引入次序功能基和侧链形成方法功能基一侧链架组合方式主环形成方法或基本骨主环与基本骨架工艺路线设计??? ? ???????→→?
路线一: 路线二: Cl C 6 H C 6 H 5 5 N H N Cl CH 3 Cl CCl 3 Cl C 6 H 5 C 6 H 5 Cl Cl COOC 2 H 5 Cl C 6 H 5 C 6 H 5 OH Cl C 6 H 5 C 6 H 5 Cl Cl COOH Cl COCl Cl COC 6 H 5 Cl Cl C 6 H 5 Cl Cl C 6 H 5 C 6 H 5 Cl
菌种筛选方法 (2)
菌种筛选方法 在实际工作中,为了提高筛选效率,往往将筛选工作分为初筛和复筛两步进行。初筛的目的是删去明确不符合要求的大部分菌株,把生产性状类似的菌株尽量保留下来,使优良菌种不致于漏网。因此,初筛工作以量为主,测定的精确性还在其次。初筛的手段应尽可能快速、简单。复筛的目的是确认符合生产要求的菌株,所以,复筛步骤以质为主,应精确测定每个菌株的生产指标,测得的数据要能够反映将来的生产水平。 1 从菌体形态变异分析有时,有些菌体的形态变异与产量的变异存在着一定的相关性,这就能很容易地将变异菌株筛选出来。尽管相当多的突变菌株并不存在这种相关性,但是在筛选工作中应尽可能捕捉、利用这些直接的形态特征性变化。当然,这种鉴别方法只能用于初筛。有人曾统计过3,484个产维生素B2的阿舒假囊酵母(Eremoth ecium ashbyii)的变异菌落,发现高产菌株的菌落形态有以下特点:菌落直径呈中等大小(8-10毫米),凡过大或过小者均为低产菌株;色泽深黄色,凡浅黄或白色者皆属低产菌株。又如,在灰黄霉素产生菌荨麻青霉(Penicillium urticae)的育种中,曾发现菌落的棕红色变深者往往产量有所提高,而在赤霉素生产菌藤仓赤霉(Gibberell a fujikuroi)中,却发现菌落的紫色加深者产量反而下降。 2 平皿快速检测法平皿快速检测法是利用菌体在特定固体培养基平板上的生理生化反应,将肉眼观察不到的产量性状转化成可见的
"形态"变化。具体的有纸片培养显色法、变色圈法、透明圈法、生长圈法和抑制圈法等,见图。这些方法较粗放,一般只能定性或半定量用,常只用于初筛,但它们可以大大提高筛选的效率。它的缺点是由于培养平皿上种种条件与摇瓶培养,尤其是发酵罐深层液体培养时的条件有很大的差别,有时会造成两者的结果不一致。图平皿快速检测法示意图平皿快速检测法操作时应将培养的菌体充分分散,形成单菌落,以避免多菌落混杂一起,引起"形态"大小测定的偏差。 1) 纸片培养显色法将饱浸含某种指示剂的固体培养基的滤纸片搁于培养皿中,用牛津杯架空,下放小团浸有3%甘油的脱脂棉以保湿,将待筛选的菌悬液稀释后接种到滤纸上,保温培养形成分散的单菌落,菌落周围将会产生对应的颜色变化。从指示剂变色圈与菌落直径之比可以了解菌株的相对产量性状。指示剂可以是酸碱指示剂也可以是能与特定产物反应产生颜色的化合物。 2) 变色圈法将指示剂直接掺入固体培养基中,进行待筛选菌悬液的单菌落培养,或喷洒在已培养成分散单菌落的固体培养基表面,在菌落周围形成变色圈。如在含淀粉的平皿中涂布一定浓度的产淀粉酶菌株的菌悬液,使其呈单菌落,然后喷上稀碘液,发生显色反应。变色圈越大,说明菌落产酶的能力越强。而从变色圈的颜色又可粗略判断水解产物的情况。 3) 透明圈法在固体培养基中渗入溶解性差、可被特定菌利用的营养成分,造成浑浊、不透明的培养基背景。将待筛选在菌落周围就
-合理药物设计
合理药物设计 合理药物设计(rational drug design)是依据与药物作用的靶点即广义上的受体,如酶、受体、离子通道、抗原、病毒、核酸、多糖等,寻找和设计合理的药物分子。主要通过对药物和受体的结构在分子水平甚至电子水平上全面准确地了解,进行基于结构的药物设计和通过对靶点的结构功能与药物作用方式及产生生理活性的机理的认识进行基于机理的药物设计。合理药物设计是化学、生物学、数学、物理学以及计算机科学交叉的产物,是在社会对医药需求的强大推动下逐步发展起来的,主要应用各种理论计算方法和分子图形模拟技术来进行合理药物设计。合理药物设计方法包括3类:①基于配体的药物设计②基于受体结构的药物设计③基于药物作用机理的药物设计。 1.基于配体的药物设计方法 合理药物分子设计必须在已知受体结构模型的条件下才能进行但到目前为止许多已知药物作用的受体结构是未知的在未知受体结构时应用合理药物设计的原理和概念开始药物设计也有了不少的尝试,这方面的研究大致可分为两类;探索系列小分子药物三维结构与活性的关系---主要有3D-QSAR;根据已知药物结构反推受体结构模型,再行合理药物设计,如药效团模型(Pharmacophore Modeling)方法。 1.1定量构效关系(3D-QSAR) 从对药物与受体相互作用的研究可以知道药物的作用是依赖自身空间形状的,其与受体的作用一般为非共价性质虽然在未知受体结
构时无法进行常规意义上的合理药物设计,但可以在对已知药物研究的基础上进行受体形状推测(receptor-mapping),将与药物本身形状有关的参数引入到定量构效关系中,称之为3D-QSAR。该方法是基于被研究的分子结合在同一个靶标生物大分子的相同部位的基本假定,将药物的结构信息、理化参数与生物活性进行拟合计算,建立合理的定量关系的数学模型,再以此关系设计新的化合物。不同方法采用不同的结构性质来确定构效关系。 利用小分子三维结构作为参数的三维定量构效关系方法在预测小分子与生物大分子的相互作用时非常有用,各种在化合物三维结构基础上进行三维定量构效关系研究的方法(3D-QSAR),在药物研究中己经越来越广泛地应用。主要方法为距离几何(Distance Geometry, DG)、分子形状分析(Molecular Shape Analysis, MSA)、比较分子场分析(Comparative Molecular Field Analysis, CoMFA)以及虚拟受体(Pseudo Receptor)方法。 在3D-QSAR中,CoMFA是目前应用最为广泛的方法,它采用化合物周围的静电场、范德华力场等的空间分布作为化合物结构描述变量,通过最小二乘法建立化合物的生物活性与化台物周围各种力场空间分布之间关系的模型。CoMFA是在不了解受体结构的情况下,通过将分子势场图示到网格点上来表示分子的周围环境,比较它们与药物分子的生物活性定量关系,用以推测受体的某些性质,并可依次建立起作用模型来设计新的化合物,定量地预测其活性强度。 1.2药效基团模型方法
(完整版)制药工艺学元英进课后答案
第一章论绪 第二章1-1:分析制药工艺在整个制药链中的地位与作用。 答:制药工艺学的工程性和实用性较强,加之药品种类繁多,生产工艺流程多样,过程复杂。即使进行通用药物的生产,也必须避开已有专利保护,要有自主知识产权的工艺。制药工艺作为把药物产品化的一种技术过程是现代医药行业的关键技术领域,在新药的产业化方面具有不可代替的作用;制药工艺学是研究药的生产过程的共性规律及其应用的一门学科,包括制配原理,工艺路线和质量控制,制药工艺是药物产业化的桥梁与瓶颈,对工艺的研究是加速产业化的一个重要方面。 1-2.提取制药、化学制药、生物技术制药的工艺特点是什么,应用的厂品范围是什么? 答:提取制药工艺的特点:以化工分离提取单元操作组合为主,直接从天然原料中用分离纯化等技术制配药物;应用的产品范围包括:氨基酸、维生素、酶、血液制品、激素糖类、脂类、生物碱。 化学制药工艺的特点:生产分子量较小的化学合成药物为主,连续多步化学合成反应,随即分离纯化过程;应用产品范围包括;全合成药物氯霉素,半合成药物多烯紫杉醇,头孢菌素C等。 生物技术制药工艺特点:生产生物技术制药、包括分子量较大的蛋白质、核酸等药物。化学难以合成的或高成本的小分子量药物。生物合成反应(反应器,一步)生成产物,随后生物分离纯化过程;应用的产品范围包括:重组蛋白质、单元隆抗体、多肽蛋白质、基因药物、核苷酸、多肽、抗生素等。 1-3化学制药产品一定申报化学制药吗?生物技术制药产品一定申报生物制药吗?为什么?举例说明。 答:化学制药产品和生物制药产品均不一定申报化学药物和生物制药制品:
有些药物的生产工艺是由化学只要和生物技术制药相互链接有机组成的。如两步法生产维生素C,首先是化学合成工艺,之后是发酵工艺,最后是化学合成工艺;有些药物经过化学合成工艺,最后是生物发酵工艺,如氢化可的松。 1-4从重磅炸弹药物出发,分析未来制药工艺的趋势。 答:重磅炸弹药物是指年销售收入达到一定标注,对医药产业具有特殊贡献的一类药物。未来制药工艺的趋势:(1)主要药物的类型将会增加(2)研发投入加大(3)企业并购与重组讲促进未来只要工艺的统一化(4)重磅炸弹药药物数量增加,促进全球经济的发展。 1-5世界销售收入排前十位的制药是什么?它们属于哪类药物?采取的制药工艺是什么? 答:(1)抗溃疡药物(219亿美元),属于内分泌系统药物,采取化学制药工艺,(2)降低胆固醇和甘油三酯药物(217亿美元),属于生物合成药物,采取生物技术制药工艺.(3)抗抑郁药物(170亿美元)属于中枢神经系统药物,采用化学制药工艺(4)非甾体固醇抗风湿药物(113亿美元)属于生物制品,采用生物制药工艺(5)钙拮抗药物(99亿美元)属于化学合成药物,采用化学合成工艺(6)抗精神病药物(95亿美元)中枢神经系统药物,化学制药工艺(7)细胞生成素(80亿美元)血液和造血系统药物,化学制药工艺(8)口服抗糖尿病药物(80亿美元)生物制药,生物制药工艺(9)ACE抑制药(78亿美元)化学合成药物,化学制药工艺(10)头孢菌素及其组合(76亿美元)生物制品,提取制药工艺 1-6列举出现频率较高的制药工艺技术 答:生物制药技术发展迅速,出现频率较高,该工艺包括微生物发酵制药,酶工程技术制药,细胞培养技术制药 1-7化学药物,生物药物,中药今年来增长情况怎样? 答:随着现代科技技术改造和发展,世界正处于开发新药过程中,而化学药物,生物药物,中药今年来增长依然迅速,起着主导作用,尤其是生物药物为人
药物合成工艺论文
阿普斯特是治疗银屑病的一种药物,其作用原理是磷酸二酯酶-4作为一种抑制剂,抑制参与引起银屑病病发的多个炎症的标志性活性位点,使其活性降低甚至于不参与病发,从而起到抑制治疗银屑病的效果。在临床试验中,用阿普斯特片治疗患者中观察到的副作用主要表现为腹泻,恶心和头痛,且产生副作用的人群数较少,多为孕妇和免疫缺陷的人群,因此该药物被批准使用,阿普斯特使FDA批准的首个也是唯一一个用于斑块型银屑病治疗的PDE-4抑制剂。 阿普斯特化学名为N-【2-[C(S)-1-(3-乙氧基-4-甲氧苯基)-2-(甲基磺酰基)乙基]-2,3-二氢-1,3-二氧-1H-异吲哚-4-】-乙酰胺,其为白色片状,分子式为C22H24N2O7S,分子量为460.5,是以3-乙氧基-4-甲氧基苯腈为原料制备而成的,下面为其合成路线: 由于3-乙氧基-4-甲氧基苯腈在市面上可以购买到且经过一系列的化学反应,可以得到较多的需求产物,反应过程操作方便,反应比较温和,所需的基础物易得,所以适合于工业生产。 一实验内容 1 主要试剂于仪器 3-乙氧基-4-甲氧基苯腈、二甲亚砜、正丁基里、硼氢化钠、N-乙酰-L-亮氨酸(上述均购自上海达瑞化学有限责任公司),3-N-乙酰氨基邻苯二甲酸酐(自制)。四氢呋喃、甲醇、乙酸,柱色谱所用固定相为100~200目的硅胶。 NMR测定:德国BrukerAVIII400M核磁共振仪、集热式恒温加热磁力搅拌器、低温恒温搅拌反应浴、旋转蒸发仪、循环水式多用真空泵、真空干燥箱、三用紫外线分析仪、热风枪(均购自郑州科泰实验设备有限公司)。 2合成路线 (1)以3-乙氧基-4-甲氧基苯甲醛为原料 3-乙氧基-4-甲氧基苯甲醛在六甲基二硅胺基锂四氢呋喃溶液里,原料中 的醛基上的氢原子被氮所取代,因为醛基上的氧上有一对孤对电子,双 键容易被强还原性原子还原成单键,达到引入原子的作用,因为正丁基 锂上的锂离子有强的还原性,所以双键被还原为单键锂与氧连接在一起。 在二甲基砜的作用下,氧原子脱去,二甲基砜上的硫原子与碳相连,再 在三氟化硼的乙醚溶液中还原,氮被还原为亚胺。该过程所需的原料较 为昂贵且其合成出的亚胺产率仅有41%,合成环境需要在-78摄氏度下进 行,不适合工业化生产,所以该合成路线被舍弃。 (2) 3-乙氧基-4-甲氧基苯腈为原料
药物合成与制药工艺
药物合成与制药工艺课程设计 指导老师: ☆药物化学:李家明 ☆制药工艺学:李传润 设计名称:罗美昔布的合成工艺流程设计设计时间: 2012.3.1-2012.5.12 班级: 09 制药工程 小组成员:边术梓(09313001) 蔡华代(09313002) 陈捷(09313003)
课程设计说明书目录 一.前言 (3) (一)基本介绍………………………………………………-- (二)相关信息………………………………………………-- (三)药理学作用……………………………………………-- 二.部分GMP要求…………………………………………………-- 三.设计资料……………………………………………………-- 四.工艺路线选择………………………………………………-- 五.制法及流程说明……………………………………………-- 六.物料衡算……………………………………………………-- (一)物料衡算基准………………………………………-- (二)物料衡算过程………………………………………-- (三)物料平衡表………………………………………………-- (四)原料消耗定额……………………………………………-- (五)物料衡算图………………………………………………-- 七.能量衡算……………………………………………………-- (一)设备的热量衡算………………………………………-- (二)加热剂、冷却剂、压缩空气的计算……………………-- (三)设备选型………………………………………………-- (四)设备流程图…………………………………………--八.总结…………………………………………………………--九.参考文献…………………………………………………………--
第三章化学合成药物的工艺分析研究99
第三章化学合成药物的工艺研究 第一节概述 在药物合成工艺路线的设计和选择之后,接下来要进行工艺条件研究。 <1)一个药物的合成工艺路线通常可由若干个合成工序组成,每个合成工序包含若干个化学单元反应,每个单元反应又包括反应和后处理两部分,后处理是产物的分离、精制的物理处理过程,只有经过适当而有效的后处理才能得到符合质量标准的药物。<2)对这些化学单元反应进行实验室水平的工艺<小试工艺)研究,目的在于优化和选择最佳的工艺条件;同时,为生产车间划分生产岗位做准备。 <3)药物的制备过程是各种化学单元反应与化工单元操作的有机组合和综合应用。 另:在合成工艺上多倾向于在同一反应器中,连续地加入原辅材料,以进行一个以上的化学单元反应,成为一个合成工序;即多个化学单元反应合并成一个合成工序的生产工艺,习称为“一勺烩”工艺。 本章讨论的具体内容:研究反应物分子到产物分子的反应过程,深入探讨药物化学合成工 艺研究中的具体问题及其相关理论。 <1)在了解或阐明反应过程的内因<如反应物和反应试剂的性质)的基础上,探索并掌握影响反应的外因<即反应条件);只有对反应过程的内因和外因以及它们之间的相互关系深入了解后,才能正确地将两者统一起来,进一步获得最佳工艺条件。 药物化学合成工艺研究的过程也就是探索化学反应条件对反应物所起作用的规律性的过程。 <2)化学反应的内因,主要是指反应物和反应试剂分子中原子的结合状态、键的性质、立体结构、官能团的活性,各种原子和官能团之间的相互影响及物化性质等,是设计和选择药物合成工艺路线的理论依据。 <3)化学反应的外因,即反应条件,也就是各种化学反应的一些共同点:配料比、反应物的浓度与纯度、加料次序、反应时间、反应温度与压力、溶剂、催化剂、pH值、设备条件,以及反应终点控制、产物分离与精制、产物质量监控等等。在各种化学反应中,反应条件变化很多,千差万别,但又相辅相成或相互制约。有机反应大多比较缓慢,且副反应很多,因此,反应速率和生成物的分离、纯化等常常成为化学合成药物工艺研究中的难题。 反应条件和影响因素<7个方面):
药物合成技术
课程超市入选课程申请表
《制药工艺学》课程教学大纲 适用专业 _________ 制药工艺学 ______________ 学 时 ___________ 72 __________________ 学 分 ___________ 4 ___________________ 第一部分 课程说明 课程代码:050053 课程名称:制药工艺学 开课学期:4 课程类型:职业方向课 课程的性质和任务: 本课程是生物化工工艺专业的必修课。通过这门课程的理论和实践教学的学 习,使学生能够掌握药物合成及相关职业所必需的基本理论知识、基本方法和基本 操作技能,为毕业后很快胜任药厂不同制剂岗位群工作奠定坚实的基础。 本门课程与其他课程关系: 本课程是生物化工工艺类专业学生学习药物合成的专业课程。前导课程为《生 物化学》、《微生物》、《药物化学》和其专业相关的生产工艺类知识 第二部分教学内容纲要 教学内容及要求 第1部分: 课程内容:绪论 教学要求:1、药物合成技术的研究现象和内容 2、 制药工业的特点 3、 制药工业的现状及发展趋势 第2部分: 课程内容: 酰化反应技术 教学要求: 1、 酰化反应 2、 应用实例 阿司匹林的合成 3、 实验实训 阿司匹林的合成和精制 第3部分: 课程内容:还原反应技术 推荐教材及参考书: 《现代生物制药工艺学》 《现代生物制药工艺学》 (高职高专教材) 辛秀兰化学工业出版社 (高职高专教材) 李家洲中国轻工业出版社
教学要求: 1、还原反应 2、应用实例——对乙酰氨基酚的合成 3、实验实训——对乙酰氨基酚的实验室合成 第4 部分: 课程内容:卤化反应技术 教学要求: 1、卤化反应 2、应用实例——诺氟沙星的合成 3、实验实训——诺氟沙星的实验室合成 第5 部分: 课程内容:烃化反应技术 教学要求:1、烃化反应 2、应用实例——磺胺甲噁唑的合成 第6 部分: 课程内容:缩合反应技术 教学要求:1 、缩合反应 2、光学异构药物的拆分 3、应用实例——氯霉素的合成技术 4、氯霉素生产中的综合利用与“三废”处理 5、实验实训——氯霉素的实验室合成 第7 部分: 课程内容:氧化反应技术 教学要求:1、氧化反应及常用氧化剂 2、消除反应 3、应用实例——氢化可的松的合成技术 第8 部分: 课程内容:发酵制药技术 教学要求:1、微生物发酵制药技术 2、维生素C 概述 3、应用实例——莱氏法生产维生素 C 工艺原理和过程 4、应用实例——两步发酵法生产维生素 C 工艺过程 5、莱氏法和两步发酵法的比较及维生素 C 收率的计算 6、维生素C 生产中“三废”治理和综合利用 第9 部分: 课程内容:溶剂和催化剂应用技术 教学要求:1、溶剂对化学反应的影响 2、催化剂对化学反应的影响 3、抗生素的概述 4、半合成青霉素的合成技术
药物筛选方法
药物筛选的方法 分子水平筛选 Microbead FCM联合筛选 原理:FCM(流式细胞术)(flow cytometry)是一种可以对细胞或亚细胞结构进行快速测量的新型分析技术和分选技术,它可以根据穿过毛细管的细胞荧光强度或类型分离细胞。由于不同的分子与标记有不同荧光素的受体或抗体结合,利用和细胞大小相似的Microbead作为固相载体取代细胞通过FCM,不同荧光标记的Microbead就被分离出来,于是靶分子或目标分子就很容易的被分离、纯化。 应用:Lanza等利用这项技术测定了患有脊髓发育不良综合症和急性骨髓源白血病病人体内的各种细胞因子受体CR表达量的变化。 蛋白质-蛋白质;蛋白质-RNA相互作用。 免疫共沉淀(co-immunoprecipitation) 原理:它是利用抗原和抗体的特异性结合以及细菌的Protein A或G特异性地结合到抗体(免疫球蛋白)的Fc片段的现象开发出来的方法。其基本原理是,在细胞裂解液中加入抗兴趣蛋白的抗体,孵育后再加入与抗体特异结合的结合于Agarose珠上的Protein A或G,若细胞中有与兴趣蛋白结合的目的蛋白,就可以形成这样一种复合物:“目的蛋白—兴趣蛋白—抗兴趣蛋白抗体—Protein A或G”,经变性聚丙烯酰胺凝胶电泳,复合物又被分开。然后经免疫印迹或质谱检测目的蛋白。这种方法得到的目的蛋白是在细胞内与兴趣蛋白天然结合的,符合体内实际情况,得到的结果可信度高。 应用:免疫共沉淀一般用于低丰度蛋白的富集和浓缩,为SDS-PAGE(聚丙烯酰胺凝胶电泳)和MS质谱分析鉴定准备样品,常用于测定两种目标蛋白质是否在体内结合;也可用于确定一种特定蛋白质的新的作用搭档。主要用于检测大分子和大分子相互作用。 放射免疫性检测(RIA)(与EIA,FIA相比灵敏度更高,但由于放射性元素,RIA的使用受到了限制)原理: (1)竞争结合分析:Ag(非标记抗原)+ Ab(特异性抗体) - AgAb *Ag(标记抗原)+ Ab(特异性抗体)- *AgAb (2)IRMA(免疫放射分析) 应用: (1)肿瘤相关抗原的测定:AFP,CEA,CA199,CA125,CA153,CA724,CYFRA211,PSA等
第三章 化学合成药物的工艺研究
第二章药物合成工艺路线的设计和选择 第一节概述 工艺路线:一个化学合成药物往往可通过多种不同的合成途径制备,通常将具有工业生产价值的合成途径称为该药物的工艺路线。 工艺研究的首要任务:在化学合成药物的工艺研究中,首先是工艺路线的设计和选择,以确 定一条经济而有效的生产工艺路线。 工艺路线设计与选择的研究对象: (1)即将上市的新药 在新药研究的初期阶段,对研究中新药(investigational drug,IND)的成本等经济问题考虑较少,化学合成工作一般以实验室规模进行。当IND在临床试验中显示出优异性质之后,便要加紧进行生产工艺研究,并根据社会的潜在需求量确定生产规模。这时必须把药物工艺路线的工业化、最优化和降低生产成本放在首位。 (2)专利即将到期的药物 药物专利到期后,其它企业便可以仿制,药物的价格将大幅度下降,成本低、价格廉的生产企业将在市场上具有更强的竞争力,设计、选择合理的工艺路线显得尤为重要。 (3)产量大、应用广泛的药物 某些活性确切老药,社会需求量大、应用面广,如能设计、选择更加合理的工艺路线,简化操作程序、提高产品质量、降低生产成本、减少环境污染,可为企业带来极大的经济效益和良好的社会效益。 第二节药物合成工艺路线的设计 药物合成工艺路线设计属于有机合成化学中的一个分支,从使用的原料来分,有机合成可分为全合成和半合成两类: (1)半合成(semi synthesis):由具有一定基本结构的天然产物经化学结构改造和物理处 理过程制得复杂化合物的过程。 (2)全合成(total synthesis):以化学结构简单的化工产品为起始原料,经过一系列化学
第九章 药物合成设计原理和方法 练习题
第九章药物合成设计原理和方法练习题 一、名词解释 1、靶分子: 2、合成子: 二、完成下列反应 1、生物碱鹰爪豆碱的合成 2、喜树碱中间体的喹啉环的合成 3、β- 咔啉的合成 4、合成 ArCHCOOH CH3 5、全身麻醉药氟烷的合成。 F3C CHBrCl
三、按要求完成下列化合物全合成。 1、采用逆合成分析法完成布洛芬(Ibuprofen)的逆推过程并写出合成的反应。 i-Bu COOH 2、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 CHO OH 3、采用逆合成分析法完成茉莉酮的逆推过程并写出合成的反应。 O 4、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 MeO2C CHO COOMe
5、采用逆合成分析法完成下面化合物的逆推过程并写出合成的反应。 CO 2Me OH 6、完成非那西丁(Phenacetin)的合成反应。 NHCOCH 3 OH 7、完成吲哚美辛(Indometacin)的合成反应。 CH 2COOH CH 3 CO Cl O H 3C 8、 完成芬太尼(Fentany Citrate)的合成。 CH 3CH 2CON N CH 2CH 2
9、完成盐酸多巴(Dopamine Hydrochloride)的合成。 OH OH NH2 HCl 10、完成盐酸可乐(Clonidine Hydrochloride)的合成。 Cl Cl N H H N HCl 11、完成硫酸沙丁胺醇(Sulbutamol sulfate)的合成。 OH CH2OH CHCH2NH(CH3)3 OH 1/2 H2SO4
药物筛选
药物筛选 药物筛选是现代药物开发流程中检验和获取具有特定生理活性化合物的一个步骤,系指通过规范化的实验手段从大量化合物或者新化合物中选择对某一特定作用靶点具有较高活性的化合物的过程。药物筛选的过程从本质上讲就是对化合物进行药理活性实验的过程,随着药物开发技术的发展,对新化合物的生理活性实验从早期的验证性实验,逐渐转变为筛选性实验,即所谓的药物筛选。作为筛选,需要对不同化合物的生理活性做横向比较,因此药物筛选的实验方案需具有标准化和定量化的特点。随着组合化学和计算化学的发展,人们开始有能力在短时间内大规模合成和分离多种化合物,因而在现代新药开发流程中药物筛选逐渐成为发现先导化合物的主要途径之一。 筛选模型: 筛选模型就是在药物筛选实验中所应用的药理实验模型,由于药物筛选要求实验方案有标准化和定量化的特征,因而在传统药理实验中常见的动物实验在药物筛选中较少应用,根据实验模型的不同,药物筛选可以分为生化水平的筛选和细胞水平的筛选。 生化水平的药物筛选用拟开发药物作用的靶点设计实验,一般而言这种作用靶点是具有特定生理功能的蛋白质,如酶和受体等,此外一些编码功能明确的DNA也越来越多地成为药物作用的靶点。候选化合物与靶点混合后,可以通过酶连免疫、荧光显色、核磁共振等方法定量测定化合物与靶点的相互作用,从而成为筛选化合物的依据。 细胞水平的药物筛选是更接近生理条件的一种药物筛选模型,其模型是拟设计药物作用的靶细胞,应用细胞培养技术获取所需细胞,将这些细胞与候选化合物相互作用,通过与生化水平筛选类似的检测技术测定化合物的作用能力,从而对化合物进行筛选。 生化水平的药物筛选操作相对简单,成本较低,但是由于药物在体内的作用并不仅仅取决于其与靶酶的作用程度,吸收、分布、代谢、排泄均会对药物的作用产生极大的影响,仅仅一道薄薄的细胞膜就能够阻挡住许多候选化合物成为药物的道路,因而生化水平的药物筛选不确定因素更多,误筛率更高。细胞水平的药物筛选模型更接近生理条件,筛选的准确率更高,但是需要建立细胞模型,操作更复杂,成本更高,数据之间的平行形较差,另外由于技术的限制,有些靶标还不能进行细胞水平的药物筛选。 高通量筛选 高通量筛选最初是伴随组合化学而产生的一种药物筛选方式。1990年代末,组合化学的出现改变了人类获取新化合物的方式,人们可以通过较少的步骤在短时间内同时合成大量化合物,在这样的背景下高通量筛选的技术应运而生。高通量筛选技术可以在短时间内对大量候选化合物完成筛选,经过近十年的发展,已经成为比较成熟的技术,不仅仅应用于对组合化学库的化合物筛选,还更多地应用于对现有化合物库的筛选。目前世界各大药物生产商都建立有自己的化合物库和高通量筛选机构,对有潜力形成药物的化合物进行篦梳式的筛选。 一个高通量药物筛选体系包括微量和半微量的药理实验模型、样品库管理系统、自动化的实验操作系统、高灵敏度检测系统以及数据采集和处理系统,这些系统的运行保证了筛选体系能够并行操作搜索大量候选化合物。高通量筛选技术结合了分子生物学、医学、药学、计算科学以及自动化技术等学科的知识和先进技术,成为当今药物开发的主要方式。完整的高通量筛选体系由于高度的整合和自动化,因而又被称作“药物筛选机器人系统”
药物筛选药物筛选方法学概论药物筛选概况一
第二篇药物筛选 第五章药物筛选方法学概论 第一节药物筛选概况 一、药物筛选定义 药物筛选:是对可能作为药用的物质进行初步药理活性的检测和试验,以求发现其药用价值和临床用途,为新药研究和开发提供最初始的依据和资料。成功的筛选能够缩短创新药物研究与开发的周期、降低成本、减少风险和提高效率。 虽然偶然发现的药物在药物研究中具有一定的作用,但过程是不可控的,因而不可能成为发现药物的主要途径。新药的发现,必须依赖主动寻找的过程,或称为广义的药物筛选过程。 二、药物筛选形式 (一)定向筛选 即采用特定的方法,专门筛选防治某种疾病的药物。这种方法是现代医学研究过程中长期使用的方法,并在药学研究中取得了巨大的成就,如治疗心血管疾病的药物、抗肿瘤药物等。定向筛选对于发现某一类型的药物行之有效,但对于被筛选的物质来讲,却不能全面反映出内在的作用,因此理想的方法是在定向筛选的同时能够实现一药多筛,从多方面发现这些物质的作用。 (二)对特定样品的筛选 其特点在于利用已有信息,在特定的样品范围内进行筛选。例如抗生素类药物的筛选,筛选多种细菌产物的抗菌活性,从而发现了大量新的抗生素。对中药的研究也是采取这种方法,根据中药已有的相关信息,筛选特定中药的有效成分。这种方式具有较高的成功率,但被筛选的范围受到限制,忽略了广泛的资源,样品间对比的范围较小,易造成对低效样品的高投入研究,特别是信息资料不可靠时可能产生误导。 (三)比较筛选 根据对现有药物的认识,以确定的模型进行筛选,由此发现同类型而作用更好的新药物,其中包括“me-too”药。可利用的药物信息包括药物作用机制、药物代谢过程以及病理机制等。例如根据甾体激素类药物的结构,找到了大量抗炎药物;根据阿片类镇痛作用原理,发现了新的镇痛药物等。 (四)随机筛选 是对可能作为药用的样品进行药理活性的广泛筛选。这种筛选方法是新药发现的最基本方式,也是在医药发展过程中人们一直进行的方式。特点是能够发现全新的药物,但成功率不可预测。要保证药物随机筛选的成功率,就必须有足够的被筛选样品量和广泛的药物作用筛选方法。 (五)计算机筛选 计算机筛选实际上是根据药理学、药物化学、计算机科学等多学科的知识和理论,应用
药物筛选
一.高通量药物筛选在新药研发中的应用 高通量药物筛选的原理,就是药物多是通过特异地作用于体内的靶点蛋白质(受体、酶、离子通道等)而重新调整病人的生理状态,达到治疗目的。该技术是指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机对实验数据进行分析处理,同一时间对数以千万样品检测,并以相应的数据库支持整体系运转的技术体系。高通量筛选在创新先导物的发现过程中具有高效、快速、微量等特点。 高通量药物筛选的基本模式是以单一的筛选模型对大量样品的生物活性进行评价,从中发现针对某一靶点具有活性的样品。随着人类基因组计划的完成,潜在药物靶点不断被发现,新的药物靶点不断出现,不仅为创新药物的发现提供了机遇,也对高通量筛选效率提出新的要求。 1 高通量药物筛选开发新药的基本过程 药物筛选与新药发现的基本过程由于高通量筛选获得的结果是分子水平或细胞水平的活性结果,多数情况下只能反映出样品的作用机制的信息,并不能完全证明对某种疾病具有防治作用,这是其与传统筛选方法的重要区别。传统的药理学研究一般是首先研究其客体作用进而研究作用机制。而采用高通量筛选方法则是从分子水平开始,这一过程又称作反向药理学。采用高通量筛选方法发现和开发药物一般要经过药物的初筛和复筛、深入筛选、确证筛选过程。 1.1 初筛和复筛 是在分子或细胞水平筛选样品,证明某一样品对该靶点具有药理活性(或亲和力)。初筛以后,选择具有活性的化合物,采用系列浓度,进行同一模型的复筛,阐明其对该靶点的作用特点、作用强度和量效关系,由此发现活性化合物(样品)。 1.2 深入筛选 在初筛和复筛的基础上,将得到的样品,采用与初筛不同但相关的分子、细胞模型作进一步的筛选,包括证明样品的选择性、细胞毒性,以及其他性质。经过深入筛选,为比较全面地评价活性化合物的药用价值提供更充分的实验资料。根据这些资料,并结合活性化合物的化学结构、性质特点,进行综合分析,确定在结构和作用方面具有新颖性和开发价值的化合物,作为先导化合物。同时也可以结合组织器官或整体动物模型,证明其药理作用,为样品提供更加充分的实验依据。 1.3 确证筛选 对深入筛选获得的先导化合物或优化后被选定的活性最好的化合物进行更深入广泛的研究,包括药理作用、药物代谢过程、一般毒性等多方面的筛选,以确定其开发前景。将符合要求的样品确定为药物候选化合物,进入开发研究程序,即临床前研究,为临床研究准备必要的资料。 2 高通量药物筛选技术对药物开发策略的转变 新方法和新技术的应用,必然会对现有的操作方式产生冲击,在药物发现过程中更是如此,由于大量新技术的应用,传统的筛选模式肯定不能适应新技术发挥作用。因此,为了提高成功率和筛选速度,积极应用新技术是必须的,但是,在研究的策略方面不进行调整和改变,就会制约新技术的应用和研究进程的发展,甚至会导致错误的结果。 2.1 随机筛选策略的改变 高通量药物筛选技术的应用,改变了传统的药物发现的模式。由于筛选方法是建立在分子细胞水平,由此来分析药物的治疗作用,必然存在着大量需要解决的问题。分子水平的某点变化,在疾病的病理表现中可能是微不足道的,或是要受到多种干扰。因此,用这种方法发现药物,就必须改变传统的模式,建立全新的新药发现策略建立基于高通量药物筛选技术上的药物发现新模式,需要解决的主要问题是活性的评价、筛选模型与疾病的相关性、影响筛选
药物合成考试题
一、名词解释 1.亲电试剂亲点试剂一般都是带正电荷的试剂或具有空的p轨道或者d轨道,能够接受电子对的中性分子 2.亲核试剂一些带有未共享电子对的分子或负离子,与正电性碳反应时称为亲核试剂,所谓亲核试剂就是一种电子对供体,即路易威斯 3.硝化反应是向有机化合物分子中引入硝基(—NO2)的反应,硝基就是硝酸失去一个羟基形成的一价的基团 4.协同反应协同反应又称一部反应,是指起反应的分子—单分子或双分子—发生化学键的变化,反应过程中只有键变化的过渡态,一步发生键和断键,没有自由基或离子等活性中间体的产生。 5.自由基反应通过化合物分子中的共价键均型成自由基而进行的反应,在链反应中起了重要的引发、传递和终止过程的作用 6.重氮化反应芳香族伯胺和亚硝酸作用(在强酸介质下)生成重氮盐的反应称为重氮化反应 7.卤化反应卤化反应又称卤代反应,是指有机化合物中的氢或其他基团被卤素取代生成含卤有机化合物的反应 8.缩合反应两个或两个以上有机分子相互作用后以共价键结合成一个大分子,并常伴有失去小分子(如水、氯化氢、醇等)的反应 9.氧化反应有机化反应时把有机物引入氧或脱去氢的反应叫氧化反应 10.烃化反应用烃基取代分子中的氢原子(包括官能团或碳骨架上的氢原子)或通过加成而引入烃基的反应 11.酰化反应为有机化学中,氢或者其他基团被酰基取代的反应 12.重排反应取代基由一个原子转移到同一个分子中的另一个原子上的反应,分子的碳骨架发生重排生成结构异构体的化学反应 13.还原反应物质(分子、原子或离子)得到电子或电子对偏近的反应。有机物反应时把有机物引入氢或失去氧的反应 二、简答题 1.药物合成反应研究的内容有哪些? 讨论药物合成反应的机理,反应物结,反应条件和,方向,反应物之间的关系。反应的主要影响因素实际特点,应用范围与限制。讨论药物合成反应的一般规律和特殊性质一击各基本反映之间的关系。 2.学习药物合成的目的是什么? 答:使学生能系统的掌握药物制备中重要的有机药物合成单元反应和合成设计原理,使学生掌握药物合成的本质和一般规律以及现代药物合成领域中的新理论新试剂和新方法培养对典型药物合成过程中各种变化因素的分析能力及选择合理的工艺条件和控制方法的能力利用药物化学制药工艺学等后续课程的学习为将来从事药物合成生产操作实施常规生产与管理参与新药开发奠定基础 3.药物合成反应主要研究的内容是什么? 答:主要研究反应机理反应的主要影响因素应用范围以及在药物合成中的应用等,以探讨有机药物合成反应的一般规律 4.卤化反应,在药物合成反应中有哪些重要作用 以制备具有不同的生理活性的含卤素有机药物 在官能团转化中,卤化物,常常是一类重要的中间体 为了提高反应选择性卤素原子,可作为保护基,阻断基
药物设计合成
肿瘤多药耐药逆转剂的设计与合成 班级:药学四班姓名:王梦雪学号:2011092604 肿瘤是威胁人类健康的主要疾病之一,化疗药物为其治疗的主要手段,肿瘤细胞的多药耐药性的产生是目前肿瘤化学治疗失败的一个主要原因。目前尚无有效逆转多药耐药的药物,因此,研究设计新型逆转剂仍然是抗癌药物研究的重点课题。而海洋就像一座药用资源的宝库,开发海洋药物,已成为当前药物开发的热点。海洋生物为了生存繁衍和适应海洋的独特环境,在漫长的进化中各自形成了特殊的结构和奇妙的生理功能,为人类提供了众多结构新颖、功能独特和生理活性强的活性物质。 目的:恶性肿瘤为细胞性病变,多药耐药(MDR)的产生是肿瘤化疗中存在的主要问题。开发多药耐药逆转剂,逆转现有化疗药物的耐药性意义重大。 方法:以全甲基化的海洋生物碱Ningalin B为先导化合物,设计并合成了一类以2,5-吡咯二酮为骨架的结构新颖的类似物。以3,4-二甲氧基苯乙酮为起始原料,通过Mitsunobu反应合成了5种未见报道的新化合物。 结果:所有化合物均通过核磁共振氢谱、核磁共振碳谱和质谱进行了结构表征,抗多药耐药活性实验正在进行。 结论:所设计合成的化合物有望成为新型的低毒高效的肿瘤多药耐药逆转剂。 一.化合物的结构与生物活性讨论 海洋天然产物不仅是药物的重要来源,而且在药物开发中作为分子骨架而受到关注。从蓝绿藻Stigonema dendroideum分离得到的海洋天然产物Dendroamide A,以及从海洋海鞘Lissoclinum patella中分离得到的海洋天然产物Patellamides B、C和D是抗肿瘤MDR类化合物中比较典型的化合物。近来发现海洋生物碱Ningalins B的甲基化衍生物具有良好的抗肿瘤MDR的活性,受到药物化学家的关注。 N OH OH O HO HO OH OH O N OMe OMe permethyl Ningalin B O MeO MeO OMe OMe O A B C D E Ningalin B 图 1 Ningalin B及其甲基化衍生物 海洋生物碱Ningalins B是由Fenical在1997年从Ningaloo海角附近的西澳大利亚海岬附近的Didemnum类海鞘中分离得到的,Ningalins B及其全甲基化的结构式见图1。Ningalin B 的甲基化衍生物对L1210 和HCT116肿瘤细胞的细胞毒性比Ningalin B分别低5倍和2.5倍。而对治疗HCT116/VM46肿瘤细胞的抗肿瘤药长春碱和阿霉素有显著的逆转性。在1μM 的浓度条件下,全甲基化的Ningalin B达到100%的逆转。可见,Ningalin B多酚的结构全甲基化转变为芳甲基醚的类似物是一类新型的低毒高效的多药耐药(MDR)逆转剂。 P-gp的过量表达是产生肿瘤多药耐药的典型机理,Peace对全甲基化的Ningalin B的药效团进行了定义:两个平面芳香环与带有长的脂肪链的氮原子,并且两者处于尽量张开的状态,