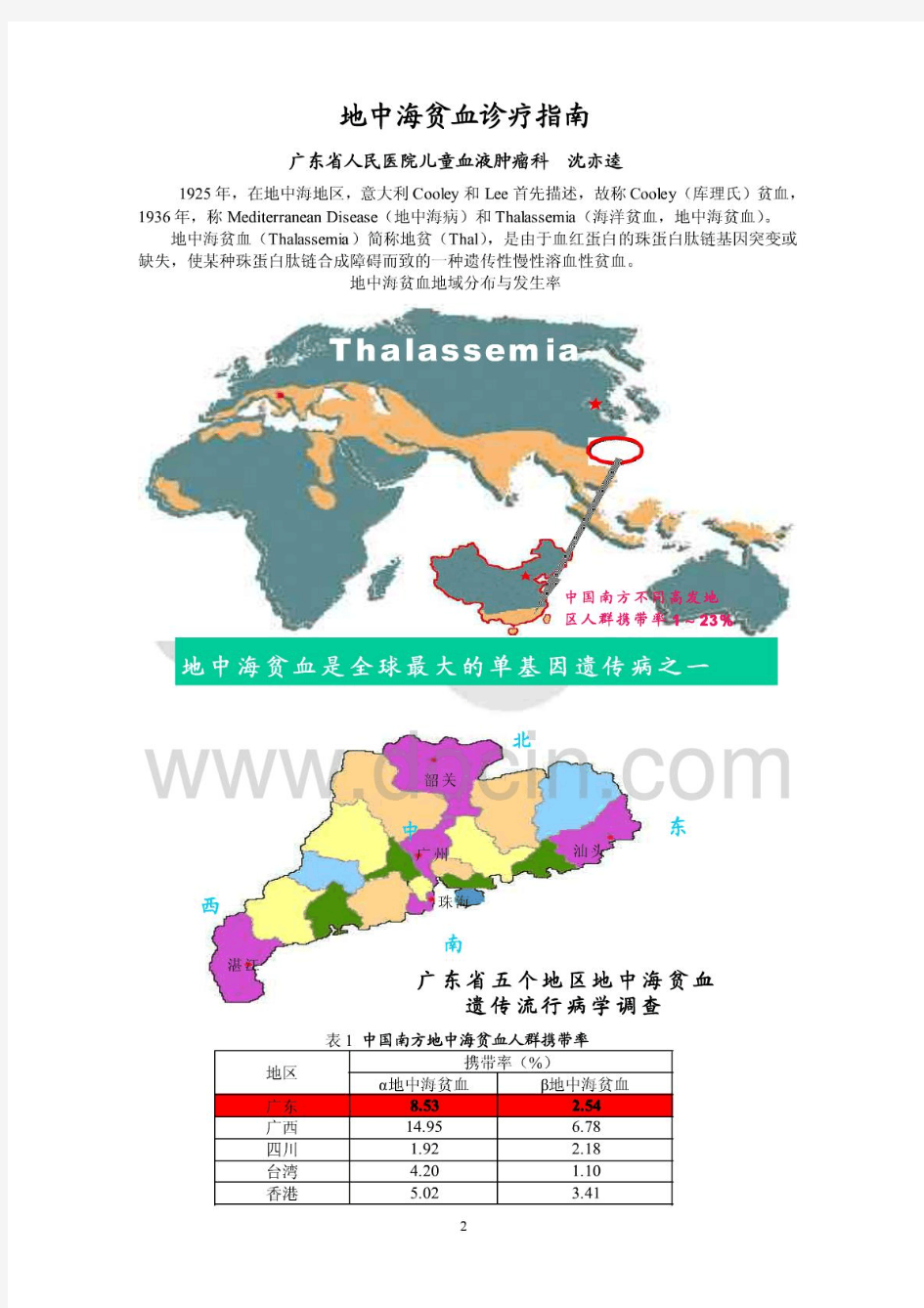
地中海贫血诊断与治疗指南

地中海贫血相关基因检测试剂注册技术审查指导原则
附件3 地中海贫血相关基因检测试剂 注册技术审查指导原则 本指导原则旨在指导注册申请人对地中海贫血相关基因检测试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。 本指导原则是对地中海贫血相关基因检测试剂的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。 本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,也不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但需要提供详细的研究和验证资料,相关人员应在遵循法规的前提下使用本指导原则。 本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善以及科学技术的不断发展,本指导原则相关内容也将适时进行调整。 一、适用范围 地中海贫血(简称地贫)是一种常染色体隐性遗传病,是由于珠蛋白基因发生突变(包括点突变和缺失等),致使珠蛋白肽 —1 —
链合成减少或完全不能合成而导致的一组单基因遗传性溶血性疾病,轻者可无临床表现,重者以进行性溶血性贫血为主要特征。根据珠蛋白肽链合成受到抑制的类型,地贫分为α-,β-和γ-地贫等,临床上最常见的是α-和β-地贫。地贫主要分布在地中海沿岸、东南亚和少数非洲地区,具有明显的种族特性和地域分布差异。地贫是我国南方地区最常见、危害最大的遗传病之一,尤以广西、云南、广东、海南、四川和贵州等省份为甚,云南、海南和广东的地贫基因人群携带率可达10%以上,广西更是达到了20%以上。 α-地贫主要是由于α-珠蛋白基因发生突变而引起。α-珠蛋白基因簇位于16p13.3,包括胎儿期和成人期表达的2个基因(α2和α1),基因的排列顺序为5’-α2-α1-3’。根据单倍型的情况,可将α地贫分为3类:(1)缺失型α+地贫(-α/),缺失1个α基因;(2)缺失型α0地贫(--/),2个α基因同时缺失;(3)非缺失型α+地贫(αTα/或ααT/ ),1个α1或α2基因发生点突变或少数几个碱基的缺失。中国现已发现至少19种α-珠蛋白基因大片段缺失和38种α-珠蛋白基因点突变。其中,6 种α-珠蛋白基因的突变:--SEA/、-α3.7/、-α4.2/、αWSα/(HBA2:c.369C>G)、αQSα/(HBA2:c.377 T>C);αCSα/(HBA2:c.427T>C)占患病人群总数的98%。 α-地贫基因型和临床分型的关系已经基本阐明。α-地贫的表型严重程度随着功能性α-珠蛋白基因的减少而加重:(1)1个α-基因缺失或点突变(-α/αα或ααT/αα或αTα/αα),称为静止型α-地贫,通常不贫血,血液学表现为小细胞低色素;(2)2个α- —2 —
中国急性肺栓塞诊断与治疗指南(2020年)
中国急性肺栓塞诊断与治疗指南(2020年) 急性肺栓塞(pulmonary embolism,PE)是我国常见的心血管系统疾病[1],在美国等西方国家也是常见的三大致死性心血管疾病之一[2]。PE是内源性或外源性栓子阻塞肺动脉引起肺循环障碍的临床和病理生理综合征,包括肺血栓栓塞症、脂肪栓塞综合征、羊水栓塞、空气栓塞、肿瘤栓塞等。其中肺血栓栓塞症(pulmonary thromboembolism,PTE)是最常见的PE 类型,指来自静脉系统或右心的血栓阻塞肺动脉或其分支所致疾病,以肺循环和呼吸功能障碍为主要临床表现和病理生理特征,占PE的绝大多数,通常所称的PE即指PTE。深静脉血栓形成(deep venous thrombosis,DVT)是引起PTE的主要血栓来源,DVT多发于下肢或者骨盆深静脉,脱落后随血流循环进入肺动脉及其分支,PTE常为DVT的合并症。由于PTE与DVT在发病机制上存在相互关联,是同一种疾病病程中两个不同阶段的临床表现,因此统称为静脉血栓栓塞症(venous thromboembolism,VTE)。本指南相关推荐主要针对血栓性PE,非血栓性PE发病率低,临床证据很有限,暂不做相关推荐。 近年来,对PE的认识不断提高,但临床实践中仍存在误诊、漏诊或诊断不及时,以及溶栓和抗凝治疗等不规范问题。2010年中华医学会心血管病学分会肺血管病学组、中国医师协会心血管内科医师分会专家组编写了“急性肺血栓栓塞症诊断治疗中国专家共识”[3],对规范我国急性PE的诊断流程和治疗策略,提高我国急性PE的诊治水平起到了极大了推动作用。在此后的5年中,肺血管疾病领域发展迅速,尤其在PE患者诊断、评估和治疗等方面,大量临床试验结果的发表,提供了新的循证医学证据,过去的指南已不能满足临床医师的需要。为此,中华医学会心血管病学分会肺血管病学组专家组在2010年专家共识的基础上,对新近出现的临床证据系统研判,并参考国际最新急性PE诊断和治疗指南[4],经认真研究讨论,达成共识,制订了本指南。与我国2010年专家共识相比,本指南在易患因素、危险分层、诊断治疗策略、新型口服抗凝剂、慢性血栓栓塞性肺高压等方面进行了更新,并对妊娠期和肿瘤患者PE的治疗给出正式推荐,旨在为PE的诊治提供依据和原则,帮助临床医师作出医疗决策,但在临床实践中面对每一个具体患者时,应该根据个体化原则制定诊疗措施。 为了便于读者了解诊疗措施的价值或意义,多因素权衡利弊,本指南对推荐类别的表述仍沿用国际上通常采用的方式: I类:指那些已证实和(或)一致公认有益、有用和有效的操作或治疗,推荐使用。 Ⅱ类:指那些有用/有效的证据尚有矛盾或存在不同观点的操作或治疗。 Ⅱa类:有关证据/观点倾向于有用/有效,应用这些操作或治疗是合理的。 Ⅱb类:有关证据/观点尚不能充分证明有用/有效,可以考虑应用。 Ⅲ类:指那些已证实和(或)一致公认无用和(或)无效,并对一些病例可能有害的操作或治疗,不推荐使用。 对证据来源的水平表达如下: 证据水平A:资料来源于多项随机临床试验或荟萃分析。 证据水平B:资料来源于单项随机临床试验或多项非随机对照研究。 证据水平C:仅为专家共识意见和(或)小规模研究、回顾性研究、注册研究。 一、流行病学 急性PE是VTE最严重的临床表现,多数情况下PE继发于DVT,现有的流行病学多将VTE作为一个整体进行危险因素、自然病程等研究,其年发病率100-200/10万人[5,6]。 PE可以没有症状,有时偶然发现才得以确诊,甚至某些PE患者的首发表现就是猝死,因而很难获得准确的PE流行病学资料。根据流行病学模型估计[6],2004年总人口为4.544亿的欧盟6国,与PE有关的死亡超过317,000例。其中,突发致命性PE占34%,其中死前未能确诊的占59%,仅有7%的早期死亡病例在死亡前得以确诊。PE的发生风险与年龄增加相
地中海贫血临床路径.doc
地中海贫血临床路径 (2016 年版) 一、地中海贫血临床路径标准住院流程 ( 一 )地中海贫血诊断 A.目的 确立地中海贫血(Thalassanemia ,简称地贫)一般诊疗的标准操作规程,确保患者诊疗的正确性和规范性。 B.范围 适用于地贫患者的诊断及其治疗 C.诊断依据 根据《血液病诊断及疗效标准》(第三版,科学出版社)及《血液病学》(第三版,人民卫生出版社)。 D.进入路径标准 (1)(2)第一诊断为地中海贫血 当患者同时具有其他疾病诊断,但在住院期间不需要 特殊处理,也不影响第一诊断的临床路径流程实施时,可以进入路径。 E.分型 ( 1)α珠蛋白生成障碍性贫血(α地中海贫血)是α珠蛋白链合成不足的结果,α珠蛋白基因缺失数目多少与α珠蛋白
链缺乏程度及临床表现严重性平行。当正常人与α地中海贫血 基因携带者结合,或是夫妇双方都是α地中海贫血基因携带者, 就会产生四种表现型: a)α+基因与正常α基因携带者结合,α / β链合成比值基 本正常,产生静止型α地中海贫血 ( α2杂合子 ) 。 b)α0基因与正常α基因携带者结合,α/ β链合成比值减 少到 0.7 ,产生α地中海贫血特征 ( α1杂合子 ) 。静止型携带者 及α地中海贫血特征者无任何症状及特征。 c)HbH病 ( α1与α2双重杂合子 ) : HbH患者出生时与正常婴儿一样,未满 1 岁前多无贫血症状,随着年龄增长逐渐出现典 型的 HbH病特征,表现为轻至中度的慢性贫血,约2/3 以上患 者有肝脾肿大,无地中海贫血外貌,生长发育正常。 d)Hb Bart ’ s 胎儿水肿综合征:α0基因的纯合子,往往在 妊娠 30~40 周成为死胎,流产或早产后胎儿绝大部分在数小时内 死亡。 ( 2)β珠蛋白生成障碍性贫血(β地中海贫血)是由于β 珠蛋白基因突变导致β珠蛋白链合成不足而引起的溶血。 a)轻型β地中海贫血:为杂合子β地中海贫血,多数患者无贫血,贫血可因感染、妊娠等情况加重,脾脏可轻度肿大。 b)中间型β地中海贫血:不依赖输血,临床表现介于重型 与轻型β地中海贫血之间的β地中海贫血患者。 c)重型β地中海贫血:为纯合子β地中海贫血,β珠蛋白链
地中海贫血表型筛查和基因诊断的现状及展望
地中海贫血表型筛查和基因诊断的现状及展望 作者:周玉球, ZHOU Yu-qiu 作者单位:519001,珠海市妇幼保健院检验科珠海市医学遗传研究所 刊名: 中华检验医学杂志 英文刊名:Chinese Journal of Laboratory Medicine 年,卷(期):2012,35(5) 被引用次数:3次 参考文献(22条) 1.Patrinos GP;Kollia P;Papadakis MN Molecular diagnosis of inherited disorders:lessons from hemoglobinathies 2005 2.Rund D;Rachmilewitz E Beta-thalassemia 2005 3.徐湘民地中海贫血预防控制操作指南[外文期刊] 2011 4.Weatherall DJ;Clegg JB The thalassemia syndromes 2001 5.NHS Sickle Cell and Thalassaemia Screening Programme.Handbook for laboratories 6.Ryan K;Bain BJ;Worthington D Significant haemoglobinopathies:guidelines for screening and diagnosis[外文期刊] 2010(1) 7.Galanello R;Eleftherious A;Tarager-Synodinos J Prevention of thalassemias and other haemoglobin disorders 2003 8.Old JM Screening and genetic diagnosis of haemoglobin disorders[外文期刊] 2003 9.周玉球;商璇;尹保民1998-2010年珠海市地中海贫血大规模人群的遗传筛查和产前诊断结果分析 2012 10.Demir A;Yarali N;Fisgin T Most reliable indices in differentiation between thalassemia trait and iron deficiency anemia[外文期刊] 2002 11.Stephens AD;Angastiniotis M;Baysal E ICSH recommendations for the measurement of Haemoglobin A2 2012 12.Stephens AD;Angastiniotis M;Baysal E ICSH recommendations for the measurement of Haemoglobin F 2012 13.Joutovsky A;Hadzi-Nesic J;Nardi MA HPLC retention time as a diagnosis tool for hemoglobin variants and hemoglobinopathies:a study of 60000 samples in a clinical diagnosis laboratory 2004 14.Van Delft P;Lenters E;Bakker-Verweij M Evaluating five dedicated automatic devices for haemoglobinopathy diagnostics in multi-ethnic populations 2009 15.Higgins T;Mack M;Khajuria A Comparion of two methods for the quantification and identification of hemoglobin variants 2009 16.Waneesorn J;Panyasai S;Kongthai K Comparison between capillary electrophoresis and high performance liquid chromatography for detection and quantification of Hh constant spring[Hb CS; α142,Term → Gln (TAA 》 CAA IN α2)] 2011 17.Munkongdee T;Pichanun D;Butthep P Quantitative analysis of Hb Bart's in cord blood by capillary electrophoresis system 2011 18.Chan LC;Ma SK;Chan AYY Should we screen for globin gene mutations in blood samples with mean corpuscular volume (MCV) greater than 80 fl in areas with a high prevalence of thalassemia 2001
地中海贫血的诊断方法.doc
地中海贫血的诊断方法 B-地中海贫血的筛查和诊断主要依赖实验室检查,方法主要有: 1 血常规检测 地中海贫血的重要特征之一是小细胞低色素性贫血,如MCV≤80 fl,MCH≤25.0 Pg,则可疑为地中海贫血患者或基因携带者,可同时测定血清铁和铁蛋白,以排除缺铁性贫血。 2 红细胞渗透脆性试验(一管法) 其原理是地中海贫血红细胞膜表面粗糙、凹陷、折叠和浆膜扩展,膜与内容物之比增大,对渗透溶解的抗性增加,在0.32%(或0.36%)NaC1中溶解度降低(脆性降低)。一管法可用于地中海贫血群体筛查。 3 血红蛋白(Hb)电泳Hb电泳 是检测地中海贫血、异常血红蛋白最常用的方法,可观察到HbE、HbH等异常血红蛋白区带,同时可定量检测HbF、HbA2的含量并区分常见类型的地中海贫血。有研究显示MCV、Hb电泳和红细胞脆性实验三者联合检测的灵敏度可达100%,阴性预告值达100%,联合特异度可达100%,阳性预告值达100%DS。 4 高效液相色谱技术(HPLC) 原理:采用微柱法离子交换层析和梯度洗脱技术,全自动分析仪可分离血红蛋白的变异体与亚型,容易发现重型和轻型B地中海贫血。在操作上,HPLC采用的是全血标本,不需要制备Hb液,只要将全血标本直接放在仪器上,通过电脑操作便能实现HbA、HbA2、HbF等定
量检测。优点:所需样本量少,自动化程度高,操作简单,快速,能消除人为误差,结果准确。HPLC也可用于胎儿脐带血的产前诊断,可诊断出重型B地中海贫血,但不能区分正常胎儿和杂合子胎儿。近年来,地中海贫血高发地区也采用此法进行携带者检测。 5 基因诊断 近年来,随着分子生物学研究领域的不断发展,从最初的B珠蛋白基因簇限制性酶切多态性检测至目前的聚合酶链反应(PCR)技术结合其他分子生物学方法,B地中海贫血的诊断已逐步改进和完善。基因诊断方法有下列几种: 5.1 限制性片段长度多态性连锁分析(RFLP连锁分析) 原理:DNA限制性内切酶可识别并切割DNA上特定的核苷酸序列,得到一定长度的DNA片段,而碱基的突变可导致酶切位点的丢失或形成,从而改变酶切片段的大小。突变基因在经过相应的限制性内切酶水解后,其电泳条带的数量和大小就会发生改变,根据这些改变可判断出突变是否存在。缺点:由于单独使用该方法,不能直接测出受试者突变基因的类型,必须结合寡核苷酸探针等技术,故其应用范围有一定限制,且操作繁琐。如果母亲或父亲在所有的多态性位点上都为纯合子,无法用此方法进行产前诊断,或者患儿和父母所有位点上都是杂合状态,只能进行50%的排除性诊断。 5.2 探针斑点杂交技术(allele—specific oligonucleotide ASO)应用引物扩增珠蛋白基因,同时合成与正常序列和突变序列完全互补的寡核苷酸探针。将PCR扩增产物点在尼龙膜上,分别与
《缺铁性贫血营养防治专家共识》(2019)要点
《缺铁性諾血营养防治专家共识》(2019)要点 m匕目 —、冃景 贫血是人体血液红细胞不能满足生理功能需求而产生的一类疾病。贫血会影响疾病治疗的预后,增加妇女、儿童的死亡率及患病率,影响儿童的认知发育,造成劳动能力下降并影响高风险地区的经济増长,导致贫困。 贫血可以分为多种类型,包括营养性贫血、再生障碍性贫血、地中海贫血等,其中营养性贫血较为晋遍。营养性贫血是指由于营养不良,导致参与血红蛋白和血红细胞形成的营养素包括铁、叶酸、维生素Bl2、维生素B6、维生素A、维生素C、蛋白质及铜等营养素不足而产生的贫血,其中又以铁缺乏引起的缺铁性贫血(IDA )最为常见。铁缺乏造成体内贮存铁耗竭,血红蛋白合成减少,进而影响红细胞生成所弓I起的贫血。据世界卫生组织(WHO)报告z在儿童及孕妇等主要贫血人群中,IDA发病率高于50%。 第一部分研究证据 —、缺铁性贫血筛查 二、膳食指导 (-)—般人群
(二)婴幼儿 (三)孕妇和乳母 (四)老年人 三、食用铁强化食品 四、营养素补充剂 五、营养补充食品 六、营养宣传教育 七、其他与缺铁性贫血 八、缺铁性贫血的临床诊断与治疗 第二部分推荐意见 —、缺铁性贫血的筛查方法 1 ?贫血筛查方法见《WS/T 441 -2013人群贫血筛查方法》。米用氧化咼
铁法、hemocue s或血常规测定检验血红蛋白。对于正常出生婴幼儿,推荐6月、9月和12月时筛查,以后每年一次;早产儿建议在3个月时就开始筛查,此后转入正常筛查。孕妇应于孕早期、孕中期、晚期进行筛查;其他人群应每年至少进行一次筛查。不同年龄、性别人群贫血判定标准见表2、3。(A) 2.铁缺乏筛查方法见《WS/T 465-2015人群铁缺乏筛查方法》表4。( B ) ( B ) 3.通过以上方法判定的既贫血又缺铁的为IDA o —、个体和群体的防治指导建议 (—)个体防治建议 1.—般人群 (1 )通过食物多样性和平衡膳食,达到《中国居民膳食营养素参考摄入量》中建议的各种营养素的摄入量。(A ) (2 )如确定为IDA ,应增加摄入富含铁、维生素C等微量营养素的食物。减少摄入植酸、多酚含量较高的食物。同时应增加富含叶酸、维生素A、
地中海贫血临床路径
地中海贫血临床路径 一、地中海贫血临床路径标准住院流程 (一)适用对象 第一诊断为地中海贫血(ICD10:D56.0/D56.100/D56.102/D56.900) (二)诊断依据。 根据《诊断和疗效标准》(张之南、沈悌主编,科学出版社,2008年,第三版)、《诸福棠实用儿科学(第七版)》(人民卫生出版社)、《临床诊疗指南–血液病学分册》(中华医学会编着,人民卫生出版社)。 1.有贫血、黄疸、肝脾肿大、发育不良、“地贫外貌”等临床表现。 2.血象轻型患者Hb常在80g/L以上,重型患者Hb<60g/l,呈小细胞低色素性贫血,红细胞形态不一,大小不均,可见靶形红细胞,网织红细胞增多。 3.骨髓象呈溶血性贫血骨髓象,红细胞系极度增生。 4.血红蛋白分析轻型HbA2 >3.5%,HbF正常或轻度增加(不超过5%),重型HbF增多,多在30%以上,少数HbF为10%~30%,HbA2多正常。 5.家系调查重型患者父母均为β地中海贫血杂合子。轻型患者父母中一方为β地中海贫血杂合子。中间型患者父母均为β地中海贫血杂合子;或父母中一方为β地中海贫血杂合子,但其中一方HbF持续存在;或父母中一方为β地中海贫血杂合子,而另一方为aβ地中海贫血。 6.轻型患者应除外缺铁性贫血。重型及中间型患者应除外HbF增加的其他类型地中海贫血。 (三)治疗方案的选择。 根据《诸福棠实用儿科学(第七版)》(人民卫生出版社)、《临床诊疗指南–血液病学分册》(中华医学会编着,人民卫生出版社)。 (四)标准住院日为14天内。 (五)进入路径标准。 1.第一诊断必须符合ICD10:D56.0/D56.100/D56.102/D56.900地中海贫血疾病编码。 2.血液检查指标符合需要住院指征:血红蛋白<70g/L,或伴有明显缺氧症状,或血红蛋白下降过快。 3.当患者同时具有其他疾病诊断,但在住院期间不需要特殊处理,也不影响第一诊断的临床路径流程实施时,可以进入路径。 (六)明确诊断及入院常规检查需2-3天(工作日)。 1.必需的检查项目:(1)血常规(包括网织红细胞计数);(2)骨髓象; 2.根据患者情况可选择的检查项目:(1)血红蛋白电泳;(2)基因分析; (七)治疗开始于诊断第1天。 (八)治疗方案与药物选择。 轻型无临床症状者无需治疗,中间型及重型患者应用下列措施: 1.输血重型患者目前主张应用高量输血法,长期规则输血,维持患者Hb在100~120g/L之间,一般每4周输血1次,每次每千克体重输12ml洗涤红细胞或浓缩红细胞(或20ml全血),可使Hb升高40g/L左右。中间型患者无须长期规则输血,仅在应激、感染、妊娠期间需输血。 2.铁鳌合剂治疗继发性血色病是地中海贫血的主要并发症及死亡原因。一般主张3岁后或接受10~20单位红细胞输血,或血清铁蛋白浓度在l000ug./L以上时开始去铁治疗;常用去铁胺(desferal,DF())20~50mg/(kg.d)静脉或皮下缓慢持续10~12小时滴注,每周5~6天;也可用口服去铁剂奥贝安可(ferriprox),同时服用维生素C 50~l00mg/d可增加体内铁的排泄,但心衰患者慎用。 3.异基因造血干细胞移植是目前临床治愈的唯一方法,对有HLA相合同胞供体的重型患者来说应作为首选治疗。 4.抗氧化剂治疗如维生素E、阿魏酸钠等能稳定红细胞膜,减轻溶血。 5.γ珠蛋白基因激活剂治疗羟基脲、白消安、5一氧胞苷、丁酸钠等药物能激活γ珠蛋白基因的表达,增加HbF的合成,改善贫血。对中间型患者疗效较好,对重型患者疗效较差。(九)出院标准。 不输红细胞情况下,血红蛋白≥70g/L且无缺氧症状,并且持续3天以上。 (十)变异及原因分析。
2019年整理esc急性肺栓塞诊断和管理指南中文完整版资料
2014ESC急性肺栓塞诊断和管理指南中文完整版 2014ESC急性肺栓塞诊断和管理指南中文完整版是由欧洲心脏病学学会(ESQ)袖珍指南之一,网上主要是关于它的解读,很少有完整版本,这个中文完整版是胡大一教授主审的,希望对大家有帮助! 2?概述 2.1流行病学静脉血栓栓塞症(VTE)包括深静脉血栓形成(DVT)和肺栓塞(PE),其年发病率为100-200/10万,为第三大常见心血管疾病。其中急性肺栓塞是VTE最严重的临床表现,是其发病、死亡及住院的主要因素。2004年,通过对六个欧洲国家的总人数约四亿五千四百余万人口流行病学调查中发现有超过317,000人死于VTE。其中,34%的患者死于突发致命性的PE,59%的患者死于生前未诊断出的PE,在早期死亡的患者中仅有7%在死前明确诊断出PE。年龄超过40岁的患者发生PE的风险较高,并且其危险度每十年将会提高近一倍,预计在未来越来越多的患者被诊断出(或者死于)PE。 2.2危险因素VTE是患者自身因素(长期因素)及环境因素(临时因素)相互作用的结果。在一些临时或者可逆的危险因素(如手术、创伤、制动、妊娠、口服避孕药或激素替代治疗等)的作用下,6周到3个月内发生的VTE被认为是诱发型,其它则被称为非诱发型。PE也可能发生在没有任何已知危险因素的情况下。与临时因素不同,长期因素可能影响PE患者的长期抗凝治疗方案的选择。
2.3病理生理 急性肺栓塞血液动力学障碍的关键因素
2.4临床分期和初始危险分层 急性PE发生的严重程度的临床分级是根据对PE患者院内发生的早期死亡风险或者30天死亡率。这个分层是根据患者临床表现来划分,对临床诊断及治疗方案发挥重要作用。存在休克或者持续的动脉低压的情况为高危PE。
患者血液管理术前贫血诊疗专家共识【最新版】
患者血液管理术前贫血诊疗专家共识 贫血在术前患者中较常见,如未有效治疗将会影响患者的手术及其预后,因此术前贫血的及时诊断和治疗非常重要。目前临床上对术前贫血未予足够重视,通常仅以输血纠正贫血或不干预直接手术,这不仅增加围手术期输血风险和手术风险,也会增加术后并发症的发生率和病死率。随着对围手术期患者血液管理的不断深入认识,国内外陆续发布了相关共识和指南。2016年,关节置换术安全性与效果评价项目组发布了《中国髋膝关节置换术加速康复--围手术期贫血诊治专家共识》,为骨科手术的围手术期贫血管理提供了指导。然而术前贫血不仅限于骨科手术患者,在其他手术科室亦常见,因此我们检索了国内外文献,结合已公开发表的相关共识和指南,广泛征询相关专业专家意见,制定了本共识,旨在为术前贫血的规范化诊疗提供依据(本共识不包括儿科手术患者)。 第一部分概述 一、流行病学 不同疾病的术前贫血情况存在一定的差异。一项系统性
回顾研究显示,术前贫血发生率为5%~76%[1]。心脏手术术前贫血发生率为24%~37%[2,3],非心脏手术术前贫血的发生率为30%[4],其中膝髋关节手术为25%~45%[1],肿瘤患者为30%~90%[5],结直肠癌为30%~67%[6],妇科手术为24%~45%[7,8]。术前贫血的发生率随年龄增长而增加,80岁以上男性择期心脏手术患者中40%有术前贫血[9],女性由于月经及生育更容易发生贫血。国内20 308例关节置换术的数据表明,全髋关节置换术、全膝关节置换术和股骨头置换术的术前贫血发生率分别为30%、26%和44%[10]。 二、病因 1.造血原料缺乏: 以缺铁最为常见,约占术前贫血的1/3。缺铁是结直肠癌患者、妇科肿瘤患者以及围产期孕妇最常见的术前贫血原因,其次为叶酸、维生素B12缺乏。 2.慢性病性贫血: 见于感染、急慢性炎症、恶性肿瘤、自身免疫性疾病等,可引起红细胞寿命缩短、骨髓对贫血的反应障碍、铁的利用
地中海贫血基因检测案例
地中海贫血基因检测案例 ——从罕见地贫基因到双线检测 钦州市妇幼保健院 基因科学与遗传医学诊断中心 汇报人:龚菲菲组员:龙驹、龚菲菲、施狄秋、张城鸿
引言 地中海贫血是一种单基因遗传疾病,广西人群中地贫基因携带率约为25%。 目前常规地贫基因分析试剂盒所检测的范围是4种α缺失基因、3种非缺失型α地贫和17种非缺失型β地贫。 研究表明,人群中有一定的地贫基因携带者,其携带的基因型不在常规地贫基因检测试剂盒检测范围内。 本案例将阐述一例由罕见地贫基因的检出而改进地贫基因检测分析流程,进而降低地贫基因检测漏诊风险的事例。
2015年6月,一对夫妇来我院进行地中海贫血基因检测。在检查过程中,我们发现了一些问题。 先证者(来自A家系)是一个28岁的男性个体,其妻子在孕期4个月时检出为--SEA携带者。其丈夫血液学数据如表所示。 结果显示MCH稍低,于是采用MLPA进行检测以排除罕见型。 AⅡ-1 性别-年龄M-28 MCV(fL) 82.3 MCH(pg) 26.5 Hb(g/dL) 16.1 HbF (%) 0.8 HbA2 (%) 2.4 Hb Bart’s+Hb H (%) 0 Ferritin (μg/L)306.8 α 常规基因型αα/ααβ基因型βA/βA 一例罕见地贫家系的检出 该先证者的临床表型
MLPA结果图 MLPA结果显示其缺失的断裂点位于337和142探针,以及283和310探针之间。同时采集了他们家系进行分析。此时,也发现一例患儿疑似携带该变异,合并研究。结果显示,该家系疑似携带2.4KB缺失型基因
电泳和测序验证-α2.4等位基因的确诊。 (A)家系A和一例HbH 病患者的琼脂糖电泳图 (wt表示野生型)。3个 个体检出300bp的PCR产 物。 (B)测序结果以及-α2.4等 位基因的示意图。
地中海贫血的诊断方法
地中海贫血的诊断方法 Document number:PBGCG-0857-BTDO-0089-PTT1998
地中海贫血的诊断方法 B-地中海贫血的筛查和诊断主要依赖实验室检查,方法主要有: 1 血常规检测 地中海贫血的重要特征之一是小细胞低色素性贫血,如MCV≤80 fl,MCH≤25.0 Pg,则可疑为地中海贫血患者或基因携带者,可同时测定血清铁和铁蛋白,以排除缺铁性贫血。 2 红细胞渗透脆性试验(一管法) 其原理是地中海贫血红细胞膜表面粗糙、凹陷、折叠和浆膜扩展,膜与内容物之比增大,对渗透溶解的抗性增加,在0.32%(或0.36%)NaC1中溶解度降低(脆性降低)。一管法可用于地中海贫血群体筛查。 3 血红蛋白(Hb)电泳Hb电泳 是检测地中海贫血、异常血红蛋白最常用的方法,可观察到HbE、HbH等异常血红蛋白区带,同时可定量检测HbF、HbA2的含量并区分常见类型的地中海贫血。有研究显示MCV、Hb电泳和红细胞脆性实验三者联合检测的灵敏度可达100%,阴性预告值达100%,联合特异度可达100%,阳性预告值达100%DS。 4 高效液相色谱技术(HPLC) 原理:采用微柱法离子交换层析和梯度洗脱技术,全自动分析仪可分离血红蛋白的变异体与亚型,容易发现重型和轻型B地中海贫血。在操作上,HPLC采用的是全血标本,不需要制备Hb液,只要
将全血标本直接放在仪器上,通过电脑操作便能实现HbA、HbA2、HbF等定量检测。优点:所需样本量少,自动化程度高,操作简单,快速,能消除人为误差,结果准确。HPLC也可用于胎儿脐带血的产前诊断,可诊断出重型B地中海贫血,但不能区分正常胎儿和杂合子胎儿。近年来,地中海贫血高发地区也采用此法进行携带者检测。 5 基因诊断 近年来,随着分子生物学研究领域的不断发展,从最初的B珠蛋白基因簇限制性酶切多态性检测至目前的聚合酶链反应(PCR)技术结合其他分子生物学方法,B地中海贫血的诊断已逐步改进和完善。基因诊断方法有下列几种: 5.1 限制性片段长度多态性连锁分析(RFLP连锁分析) 原理:DNA限制性内切酶可识别并切割DNA上特定的核苷酸序列,得到一定长度的DNA片段,而碱基的突变可导致酶切位点的丢失或形成,从而改变酶切片段的大小。突变基因在经过相应的限制性内切酶水解后,其电泳条带的数量和大小就会发生改变,根据这些改变可判断出突变是否存在。缺点:由于单独使用该方法,不能直接测出受试者突变基因的类型,必须结合寡核苷酸探针等技术,故其应用范围有一定限制,且操作繁琐。如果母亲或父亲在所有的多态性位点上都为纯合子,无法用此方法进行产前诊断,或者患儿和父母所有位点上都是杂合状态,只能进行50%的排除性诊断。
地中海贫血基因检测试剂结果判读
地中海贫血基因检测试剂结果判读 亚能生物技术(深圳)有限公司 1.α-地中海贫血基因检测试剂结果判读 病理生理改变非常轻微,临床一般无症状。红细胞形态正常,出生时脐带血中Hb Bart's含量为~ 但3个月后即消失(少部分可见MCV<79fl,MCH<27pg,红细胞脆性试验阳性)。 遗传学:父母中至少一方为α地中海贫血。 左缺失涉及整个α2-珠蛋白基因缺失,要比右缺失贫血程度要严重。 轻型地贫无需特殊治疗。 尚能代偿性地合成相当数量的α链,临床上亦无症状或有轻度贫血症状,肝脾无肿大。红细胞形 态有轻度改变,血液学检查可呈现平均红细胞体积和平均红细胞血红蛋白的降低 (如大小不等、中央 浅染、异形等;红纽胞渗透脆性降低;变性珠蛋白小体阳性; HbA2和HbF含量正常或稍低。患儿脐 血Hb Bart's含量为~,于生后6个月时完全消失。) 实验室检查:出生时Hb Bart’s可占5%~15%,几个月后消失,红细胞有轻度形态改变,可见 靶形红细胞,血红蛋白稍降低或正常,MCV<79fl,MCH<27pg,红细胞脆性试验阳性。 遗传学:父母一方或双方为α地中海贫血。 一般不需要治疗。 慢性溶血性贫血,此型临床表现差异较大,出现贫血的时间和贫血轻重不一(少数患者血红蛋白可 低于60g/L或高于100g/L)。大多在婴儿期以后逐渐出现贫血,疲乏无力,肝脾大,轻度黄疽;年 龄较大患者可出现类似重型β地贫的特殊面容。合并呼吸道感染或服用氧化性药物,抗疟药物等可诱
发急性溶血而加重贫血,甚至发生溶血危象。 实验室检查:红细胞渗透脆性减低;变性珠蛋白小体阳性;HbA2及HbF 含量正常。红细胞形态基 本同重型β地中海贫血所见,红细胞内可见包涵体。骨髓中红细胞系统增生极度活跃。血红蛋白电泳 出现HbH 区带,HbH 成分占5%~30%(个别患者HbH 成分可小于5%或高达40%),也可出现少量Hb Bart ’s (出生时Hb Bart ’s 可达15%以上)。随年龄增长,HbH 逐渐取代Hb Bart's ,其含量约为 ~。包涵体生成试验阳性。非缺失型血红蛋白H 病可出现微量Hb Constant Spring 。 遗传学:父母双方均为α地中海贫血。 胎儿常于30~40周时流产,死胎或娩出后半小时内死亡,胎儿呈重度贫血,黄疽,水肿,肝脾肿大,腹水,胸水。体腔积液,胎盘巨大且质脆。孕妇可有妊娠高血压综合征。 实验室检查:脐血血红蛋白明显降低,红细胞中心浅染、形态不一、大小不均,有核红细胞显著增多,靶形红细胞增多。血红蛋白电泳:Hb Bart ’s 成分>70%,少量Hb Portland ,可出现微量 HbH 。无HbA 、HbA2和HbF 。 2.β-地中海贫血基因检测试剂结果判读
地中海贫血症的治疗方法
地中海贫血症的治疗方法 简单明了,不说大话,不做药贩子,不做骗子,借助这个贴吧平台,希望能够有更多人得到治疗,也希望吧主能够打开一扇大门,让更多的人有机会看到,而受到帮助。 婆婆,祖传中医方法,治疗地中海贫血症 真实例子:医院已经不能再输血的患者,最后找到了婆婆,在婆婆的慢慢调理下,现在已经结婚生孩子了。 地中海贫血症一次性无法治疗,只能慢慢的调理,需要一段过程。。急于求成治好的人,可能无法满足,中药调理需要疗程的,如果有需要帮助的人,可以联系我,希望能够治疗更多的患者。联系QQ 675561594 1症状体征 1.α球蛋白生成障碍性贫血出生时即可贫血,临床表现为轻~中度的慢性贫血,伴有黄疸、肝脾肿大。继发感染、服用氧化剂药物可加重HbH的不稳定性而促发溶血。合并妊娠可加重贫血。患者的发育一般不受影响,骨骼改变也不明显。 2.纯合子(β0)球蛋白生成障碍性贫血患儿出生时正常,出生数月后,HbF逐渐被HbA(α2β2)替代,患儿出现贫血,呈进行性加重,可有发热、纳差、腹泻、黄疸、肝、脾逐渐肿大。3~4
岁时,表现生长发育迟缓,精神萎靡,面无表情,体弱无力。骨髓造血代偿性增生使骨髓腔变宽骨皮质变薄,导致患儿额部、顶部隆起,头颅增大,面颊隆起,鼻梁塌陷,上颌及牙齿前突,形成特殊面容。患者发生下肢慢性溃疡。发病愈早,症状愈重。 3.杂合子(β)珠蛋白生成障碍性贫血此类患者又称静止型 或微型β珠蛋白生成障碍性贫血,因为大多数患者无贫血或其他症状,多在普查、家系调查或合并其他疾病进行检查时发现。查血或可发现少数靶形红细胞、红细胞渗透脆性轻度降低、HbA2轻度增多。少数患者可有贫血,尤其当合并妊娠或继发感染时,表现为轻度至中度贫血,可出现黄疸、轻度脾大。 2用药治疗 1.α珠蛋白生成障碍性贫血多数患者在病情稳定时不需要治疗,当贫血加重时,应注意排除诱发因素如感染、服用氧化剂药物(磺胺类、亚硝酸盐类、氯喹等),必要时给予输浓缩红细胞。对严重贫血及巨脾、肝功能亢进者,可行脾切除术。因脾是本病红细胞破坏场所,故手术效果较好。 2.纯合子(β0)珠蛋白生成障碍性贫血主要治疗措施是输红细胞、防止感染、防止继发性血色病及脾切除手术。这些治疗措施可以取得改善临床症状、提高生存质量、延长生命的效果,而不能根治本病。对有HLA(人类白细胞抗原)相合骨髓供者、难以
妊娠期贫血指南
妊娠期铁缺乏和缺铁性贫血诊治指南 中华医学会围产医学分会 推荐1-1:妊娠合并贫血是指妊娠期Hb浓度<110 g/L。 推荐1-2:铁缺乏指血清铁蛋白浓度<20 μg/L。 推荐1-3:妊娠期IDA是指妊娠期因铁缺乏所致的贫血,Hb浓度<110 g/L。 推荐2-1:小细胞低色素的贫血患者首选铁剂治疗试验,治疗2周后Hb升高,则提示为IDA。铁剂治疗无效者应进行鉴别诊断(推荐级别Ⅰ-B)。 推荐2-2:铁剂治疗无效者,应进一步检查是否存在吸收障碍、依从性差、失血及叶酸缺乏症等情况,并转诊至上一级医疗机构(推荐级别Ⅰ-A)。 推荐2-3:广东、广西、海南、湖南、湖北、四川及重庆等地中海贫血高发地区,应在首次产前检查时常规筛查地中海贫血。 推荐2-4:有条件的医疗机构对所有孕妇检测血清铁蛋白。 推荐2-5:患血红蛋白病的孕妇,应检测血清铁蛋白(推荐级别Ⅰ-B)。 推荐2-6:检测C-反应蛋白有助于鉴别诊断因感染造成的血清铁蛋白增高(推荐级别Ⅱ-B)。 推荐3-1:所有孕妇应给予饮食指导,以最大限度地提高铁摄入和吸收(Ⅰ-A)。 推荐3-2:一旦储存铁耗尽,仅仅通过食物难以补充足够的铁,通常需要补充铁剂(推荐级别Ⅰ-A)。 推荐3-3:诊断明确的IDA孕妇应补充元素铁100~200 mg/d,治疗2周后复查Hb 评估疗效(推荐级别Ⅰ-B)。 推荐3-4:治疗至Hb恢复正常后,应继续口服铁剂3~6个月或至产后3个月(推荐级别Ⅰ-A)。 推荐3-5:非贫血孕妇如果血清铁蛋白<30 μg/L,应摄入元素铁60 mg/d,治疗8周后评估疗效(推荐级别Ⅱ-B)。 推荐3-6:患血红蛋白病的孕妇如果血清铁蛋白<30 μg/L,可予口服铁剂。
肺栓塞诊断治疗指南
肺栓塞诊断治疗指南 肺动脉栓塞(pulmonary embolism ,肺栓塞) 肺栓塞是常见的心血管急症 ,它可阻塞肺动脉床而导致危及生命的右室衰竭。其初始治疗旨在恢复血流挽救生命 ,长期抗凝在预防复发中极为重要 ,但常因缺乏特异的临床表现而被误诊。 1 流行病学 PE 及深静脉血栓形成 (DVT ) 是静脉栓塞 (VTE ) 的两种临床表现 ,并具有相同的易患因素 ,大多数情况下二者伴随发生。其在美国的死亡率仅次于恶性肿瘤和心肌梗死而排在第3位。肺栓塞在我国一直被认为是少见病,但从20世纪90年代中对部分医院进行临床流行病学调查,发现病例数呈稳步上升趋势,2001年中华医学会呼吸病分会公布的《肺血栓栓塞症的诊断与治疗指南(草案)》以来,部分医院病例增长10倍。最新的前瞻性研究显示急性 PE 的致死率约为 7 % ~11 % 。 1.1 易患因素 诱发因素包括:年龄、VTE 史、恶性肿瘤、下肢麻痹的神经系统疾病、长期卧床、激素替代治疗及服用避孕药等。80岁以上人群的发病率是50岁以下人群的8 倍。 1.2 自然病程 大多数情况下PE 是DVT 的并发症 , 约1 /3 的V T E 在数天后可自愈 ,约40 %左右病情不会进展 , 但25 %可发展成为中心DVT 和PE 。 1.3 病理生理学 急性 PE 主要是血流动力学改变 ,尤其当 30 % ~50 %的肺血管床被栓塞后症状较为明显 。 P E 常伴的 呼吸功能不全也是血流动力学紊乱的结果 : 低心输出量影响了肺静脉的血 氧交换 , 进而 导 致了 低 氧 血 症 的 发 生 。 较 小的和 远 1.4 风险评估 PE 应进行个体化的死亡风险评估 ,这远比栓塞的解剖形态和面积重要 。低血压或者休 克、右室功能障 碍 ( R VD )及心肌损伤标志物升高可用于对PE 进行危险分层 :高危PE 危及生命(短期病死率>15 % ) ,需溶栓治疗或外科手术摘除栓子;非高危PE 可根据其死亡风险评估高低而选择住院治疗和院外治疗 。 2 诊断 2,1 临床表现 肺栓塞是内源性或外源性栓子堵塞肺动脉引起肺循环障碍的临床和病理生理综合征, 肺栓塞引起的肺出血或坏死称为肺梗死(pulmonary infarction)。其临床表现可从无症状到咯血乃至猝死,症状与栓子大小、栓塞发生速度及基础心、肺功能相关。典型症状为呼吸困难、胸痛和咯血,被称为肺梗死三联征。呼吸困难发生率高达84%一90%,多表现为劳力性呼吸困难。临床医生应注意呼吸困难的诱因、性质、程度和持续时间。以胸憋闷为主诉的呼吸困难须与劳力性心绞痛鉴别。胸痛发生率40%~70%。多为胸膜痛,为肺梗死累及到胸膜所致,4%~12%病人表现为“心绞痛样痛”,可能由于冠状动脉痉挛或右心室肥厚缺血所致。咯血发生率11%~30%,血量不多,鲜红色,数日后变为暗红色,提示有肺梗死。其他症状有咳嗽,发生率53%,多表现为干咳,可伴哮鸣音;凉恐,发生率55%,由胸痛或低氧血症所致。当大块肺栓塞或重症肺动脉高压时,可引起一时性脑缺血,表现为晕厥,其发生率11%~20%,可为肺梗死的首发症状。应特别强调的是,临床表现为典型肺梗死三联征的患者不足30%。 肺梗死体格检查可发现体温正常或升高,呼吸和脉搏加快。血压下降通常提示大块肺栓塞,发绀提示病情严重。胸部检查可无任何异常体征,如一侧肺栓塞范围
地中海贫血简介
地中海贫血简介 地中海贫血(简称地贫)是我国南方各省最常见、危害最大的遗传病,人群发生率高达 10%以上,以广东、广西为主。地贫主要分α和β地贫两种,以α地贫较常见。本病的发生是由于血红蛋白分子中的珠蛋白肽链结构异常或合成速率异常,造成肽链不平衡而产生以溶血性贫血为主的症状群。 地中海贫血的表现 地贫的诊断主要依据临床表现、实验室检查和基因诊断。地贫的临床表现呈多样性,轻者本人多无自觉症状而不易察觉,常因体检时发现轻度贫血或家系分析时才诊断。重型α地贫胎儿,又称 Bart's水肿胎,因4个α基因全部缺失,α珠蛋白链不能合成,常于怀孕中期开始发病,表现为胎儿全身水肿,心脏畸形,体腔积液,胎盘巨大而水肿,多于怀孕晚期死于宫内。即使能怀孕至足月,也多于出生后数分钟内死亡,而且孕妇常合并妊高征,胎盘早剥,子痫抽搐,产时或产后大出血等产科危重并发症。另一种次严重的α地贫,又称血红蛋白H病,与重型β地贫表现相似或贫血症状稍轻,这两类地贫在胎儿期无特殊临床表现,可以怀孕至足月分娩,出生时与正常胎儿几无分别,多于生后6个月左右开始发病。表现为进行性溶血性贫血,血色素可低至2-4g/dl,肝脾肿大,脸色萎黄,苍白无力。此病目前国内外尚无有效的治疗方法,仅能依靠输血维持生命。由于极易发生各种并发症,多于青少年期死亡,给家庭和社会带来沉重的负担。 地中海贫血的遗传方式 夫妇双方若同为轻型地贫(即地贫基因携带者),怀孕以后,对子代的遗传几率是: 1/4为正常胎儿,1/2为轻型地贫(同父母一样),而1/4为重型地贫患者。地中海贫血的遗传与性别无关,男胎女胎发病机率均等。 地中海贫血的处理方法 轻型地贫患者无需特殊处理,主要是针对重型地贫,需进行产前诊断。未在婚前进行有关地贫检查的夫妇,要求怀孕后早筛查,早诊断,确诊后流产是目前防止重型地贫胎儿出生、保障母体安全的唯一办法。对遗传咨询后确认为高风险地贫胎儿时,应尽早通过羊水穿刺或绒毛取样后,进行基因检测,可诊断地贫胎儿的基因型。 常见咨询问题 1.夫妇双方只有一方为轻型地贫患者,需要做产前诊断吗? 不需要。他们所怀的胎儿只有 1/2几率为轻型地贫,其余1/2是正常胎儿,不会孕育重型地贫胎儿。 2.孕检时,女方未检出地贫,需要男方来做地贫检查吗? 一般不需要。这种情况即使男方为重型地贫,所怀胎儿至多为轻型地贫患者,没有大碍。