Recent Developments in the Medicinal Chemistry and Therapeutic Potential of Dihydroorotate Dehydroge

Mini-Reviews in Medicinal Chemistry, 2011, 11, 1039-1055
1039
1389-5575/11 $58.00+.00 ? 2011 Bentham Science Publishers
Recent Developments in the Medicinal Chemistry and Therapeutic Poten-tial of Dihydroorotate Dehydrogenase (DHODH) Inhibitors
V.K. Vyas* and M. Ghate
Department of Pharmaceutical Chemistry, Institute of Pharmacy, Nirma University, Ahmedabad, 382 481 Gujarat, India
Abstract: Dihydroorotate dehydrogenase (DHODH) is a flavin-dependent mitochondrial enzyme that catalyzes fourth re-action of pyrimidine de-novo synthesis. Pyrimidine bases are essential for cellular metabolism and cell growth, and are considered as important precursors used in DNA (thymine and cytosine), RNA (uracil and cytosine), glycoproteins and phospholipids biosynthesis. The significance of pyrimidines biosynthesis in DNA and RNA makes them ideal targets for pharmacological intervention. Inhibitors of DHODH have proven efficacy for the treatment of malaria, autoimmune dis-eases, cancer, rheumatoid arthritis and psoriasis. Plasmodium falciparum dihydroorotate dehydrogenase (Pf DHODH) rep-resents an important target for the treatment of malaria. Many of the clinically relevant anti-tumor and immunosuppres-sive drugs target human dihydroorotate dehydrogenase (h DHODH), and the two most promising drugs of such kinds are brequinar (antitumor and immunosuppressive) and leflunomide (immunosuppressive). X-ray crystal structures of DHODH in complex with inhibitors reveal common binding region shared by each inhibitor. A number of compounds are identified by high-throughput screening (HTS) of chemical libraries and structure-based computational approaches as se-lective DHODH inhibitors. Based upon the understanding of molecular interaction of DHODH inhibitors with binding site, some of the common structural features are identified like ability of compounds to interact with ubiquinone (CoQ) binding site and substituents linked to a variety of heterocyclic and heteroaromatic rings responsible for H-bonding with binding site. These findings provide new approaches to design DHODH inhibitors and highlights DHODH as a target for chemotherapeutics. This review is mainly focused on the recent developments in the medicinal chemistry and therapeutic potential of DHODH inhibitors as a target for drug discovery.
Keywords: Dihydroorotate dehydrogenase (DHODH), pyrimidines biosynthesis, DHODH inhibitors, cancer, arthritis, malaria.
1. INTRODUCTION
The flavoenzyme dihydroorotate dehydrogenase (DHODH) [EC 1.3.99.11] [1] is a fourth enzyme of
pyrimidine de-novo synthesis that catalyses oxidation of in-termediate dihydroorotate (DHO) to orotate (ORO).
Pyrimidines are required for the biosynthesis of DNA, RNA,
glycoproteins, and phospholipids, and are linked by phos-phodiester bridges to purine nucleotides in double-stranded DNA, both in nucleus and mitochondria [2]. There are two
routes for the synthesis of pyrimidines in most of the organ-isms; salvage pathways and de novo synthesis from small
metabolites. Requirement of pyrimidine depends on cell type
and developmental stage, involvement of de novo pathway is
small in resting or fully differentiated cells where cell ac-quire pyrimidine mainly by the salvage pathways [3]. In con-trast, activated T cells and other rapidly proliferating cells, in
order to meet their increased demand for nucleic acid precur-sors and other cellular components, depends heavily on de
novo nucleotide synthesis [4]. DHODH catalyzes conversion
of DHO to ORO (Fig. 1), which represents the rate limiting step in de novo pyrimidine biosynthesis [5].
*Address correspondence to this author at the Department of Pharmaceuti-cal Chemistry, Institute of Pharmacy, Nirma University, S.G. Highway, Chharodi, Ahmedabad, 382 481, Gujarat, India; Tel: +91 9624931060; Fax:
+91 2717 241916;
E-mail: vicky_1744@https://www.360docs.net/doc/547953823.html,, vivekvyas@nirmauni.ac.in The significance of pyrimidine for cell proliferation, me-tabolism and multiplication determines DHODH as targets
for the development of new drug candidate [6]. Inhibition of DHODH leads to reduced levels of essential pyrimidine nu-cleotides. Inhibitors of human DHODH (h DHODH) have
proven efficacy for treatment of cancer [7,8] and immu-nological disorders, such as rheumatoid arthritis and multiple
sclerosis [9-11]. DHODHs are attractive chemotherapeutic
targets in various pathogens, such as Plasmodium falcipa-rum, Helicobacter pylori , Enterococcus faecalis [12-15] and
as antifungal agents [16]. DHODH has a number of proper-ties that make it a particularly strong candidate as a new drug
target. Brequinar [17] and leflunomide [18, 19] (Fig. 2) are
two examples of such compounds. Brequinar is an antitumor
and immunosuppressive agent, while leflunomide, a prodrug
which is converted to active metabolite A77 1726 [20],
shows immunosuppressive activity. Many excellent research
publications characterize the exponential growth in therapeu-tic potential of DHODH inhibitors. In 1999 Batt, D.G. [21]
compiled the literature of DHODH inhibitors and described
the earlier attempts to find DHODH inhibitors based on ORO and CoQ binding site. Leban, J. and Vitt, D. [22] pub-lished a review describes DHODH inhibitors as a promising
agent for the treatment of autoimmune and inflammatory
diseases. The scope of this review is to update the key ad-vances in the medicinal chemistry and biology of DHODH inhibitors as a new target for drug discovery. In the later sec-
1040 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
tion, major impetus is given to medicinal chemistry of DHODH inhibitors as therapeutic potential agents.
2. DIHYDROOROTATE DEHYDROGENASE (DHODH)
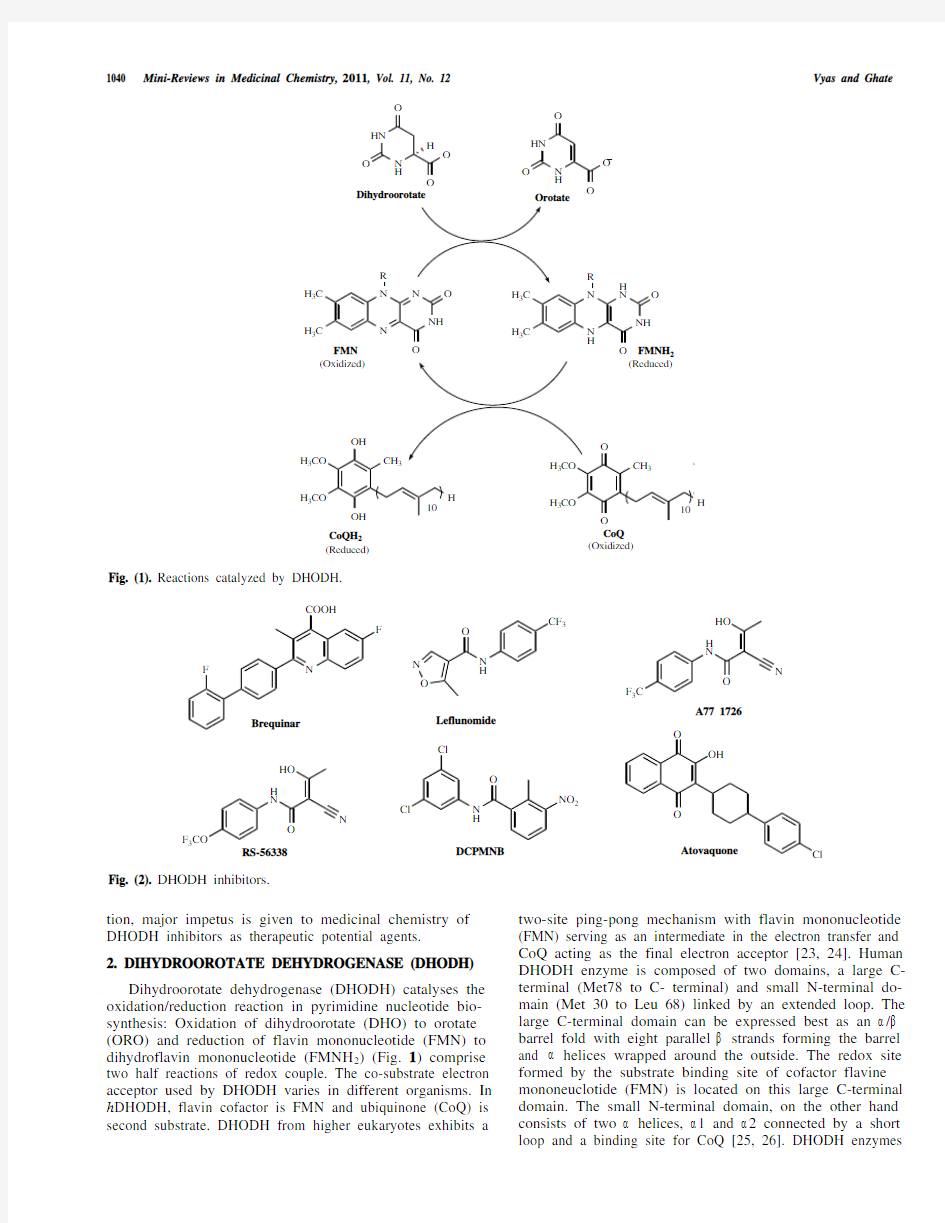
Dihydroorotate dehydrogenase (DHODH) catalyses the oxidation/reduction reaction in pyrimidine nucleotide bio-synthesis: Oxidation of dihydroorotate (DHO) to orotate (ORO) and reduction of flavin mononucleotide (FMN) to dihydroflavin mononucleotide (FMNH2) (Fig. 1) comprise two half reactions of redox couple. The co-substrate electron acceptor used by DHODH varies in different organisms. In h DHODH, flavin cofactor is FMN and ubiquinone (CoQ) is second substrate. DHODH from higher eukaryotes exhibits a two-site ping-pong mechanism with flavin mononucleotide (FMN) serving as an intermediate in the electron transfer and CoQ acting as the final electron acceptor [23, 24]. Human DHODH enzyme is composed of two domains, a large C-terminal (Met78 to C- terminal) and small N-terminal do-main (Met 30 to Leu 68) linked by an extended loop. The large C-terminal domain can be expressed best as an / barrel fold with eight parallel strands forming the barrel and helices wrapped around the outside. The redox site formed by the substrate binding site of cofactor flavine mononeuclotide (FMN) is located on this large C-terminal domain. The small N-terminal domain, on the other hand consists of two helices, 1 and 2 connected by a short loop and a binding site for CoQ [25, 26]. DHODH enzymes
O
Dihydroorotate
O
H3
H3
R
H
H3
H3
R
FMN
H
H3
H3
CoQ
(Oxidized)
CoQH2
(Reduced)
Fig. (1). Reactions catalyzed by DHODH.
3
F3
Leflunomide
A77 1726
F3
RS-56338
2
DCPMNB
Fig. (2). DHODH inhibitors.
Recent Developments in the Medicinal Chemistry Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1041
are divided into two families, based upon their localization, amino acid sequence, substrate/cofactor dependence, and cellular localization [27, 28]. Family-1 enzymes are located in the cytosol, electron acceptors involved in second half reaction of redox process are either fumarate or NAD+ whereas family-2 enzymes transfer electrons to ubiquinone (CoQ), to which h DHODH belongs, is distinguished from family-1 by the use of ubiquinone (CoQ) for reoxidizing FMN. N-terminus is proposed as binding site for electron acceptor, which is responsible for association of enzyme with inner mitochondrial membrane [29]. Family-1 enzymes are further divided into family-1A, homodimeric proteins which contain a cysteine residue as their active site base and use fumarate as electron receptor [30-32] and family-1B (NAD-dependent) enzymes, hetero tetramers, which contain not only FMN but also an iron–sulfur cluster and FAD, where the enzyme is oxidized by NAD+ [33] and a new type 1S identified in Sulfolobus solfataricus [34]. Family-1S DHODH can use CoQ and molecular oxygen as electron acceptors.
3. STRUCTURE OF DHODHS
Clardy, J. et al. [1] solved high-resolution crystal struc-tures of h DHODH in complex with two different inhibitors A77 1726 and brequinar, and refined them to crystallo-graphic R factors of 16.8% and 16.2% at resolutions of 1.6 ? and 1.8 ? respectively. Human DHODH has two domains: / -barrel domain containing the active site and an -helical domain that forms the opening of a tunnel leading to the ac-tive site. Both the inhibitors share a common binding site in this tunnel, and differences in binding region govern drug sensitivity or resistance. Fishwick, C.W.G. and colleagues [35] reported X-ray crystal structure of h DHODH and Pf DHODH with inhibitors and observed different binding modes. In Pf DHODH-inhibitor complex, three direct hydro-gen bonds can be observed (to His185, Arg265 and Tyr528), whereas in h DHODH-inhibitor complex, there are water-mediated hydrogen bonds to Arg265 and Gln47 in addition to direct H-bond to Tyr356. Walse, B. et al. [36] determined the crystal structure of N-terminally truncated DHODH in complex with brequinar analogue (6-chloro brequinar, in which 6-fluoro group of brequinar is replaced with a 6-chloro group) and a novel inhibitor (fenamic acid derivative), and presented a crystal structure of the inhibitor-free N-terminally truncated (Met30-Arg396) h DHODH. In this study extensive network of interactions provides the basis of high affinity of brequinar analogues for h DHODH. Findings of this study suggested a new approach to design DHODH inhibitors. Baumgartner, R. et al. [37] solved five high-resolution X-ray structures in complex with low molecular weight inhibitors. The data provides clear and excellent elec-tron densities for the inhibitors and therefore, enables a de-tailed analysis of prevailing interactions and modes of bind-ing. The structure of Pf DHODH with a bound inhibitor was described by Hurt, D.E. et al. [24] Pf DHODH and h DHODH have different sensitivities to inhibitors. The three-dimensional structure (2.4 ?, R = 20.1%) of Pf DHODH-leflunomide complex was determined by X-ray crystallogra-phy. Comparison of the structures of h DHODH and Pf DHODH reveals structural differences between the CoQ-binding tunnels and an absolutely different binding mode for the same inhibitor is observed. Gojkovic, Z. et al. [38] inves-tigated crystal structure of Candida albicans DHODH and proposed that the enzyme is a member of the DHODH fam-ily-2. In this study full-length DHODH and N-terminally truncated DHODH, were sub-cloned from Candida albicans, recombinantly expressed in Escherichia coli, purified and characterized for their kinetics and substrate specificity. The result of this study provides a background for development of DHODH inhibitors for decreasing pyrimidine nucleotide pools in Candida albicans. T7 RNA polymerase expression system in Escherichia coli was used to produce h DHODH as a fusion protein containing an amino-terminal decahistidine tag [39]. Kinetic analysis of this recombinant h DHODH in-dicates that it has a two site ping-pong mechanism where dihydroorotate (DHO) is oxidized at one site and second substrate CoQ is reduced at other. The functional expression of h DHODH in pyr4 mutants of Ustilago maydis was studied by Bolker M. et al. [40]. Engineered U. maydis strains can be used in in vivo assays for development of novel h DHODH and fungal DHODH inhibitors. Chargas disease is consid-ered as a public health problem in Latin America caused by hemoflagellate protozoan Trypanosoma cruzi. Nonato, M.C. et al. [41] reported the crystal structure of DHODH from Trypanosoma cruzi strain and solved at 2.2? resolution. Crystal structure of T. cruzi DHODH suggested potential sites for the design of highly specific inhibitors.
4. MOLECULAR MECHANISM OF ACTION OF DHODH INHIBITORS
Inhibition of DHODH can cause a lowering of intracellu-lar pyrimidine nucleotides pools in cells, which makes DHODH as an attractive target for drug design in different biological and clinical applications for cancer, arthritis and malaria. The mechanism of action of DHODH inhibitors was studied in several kinetic studies by Bennett, L. and col-leagues at Kettering-Meyer Laboratory, Southern Research Institute, Birmingham, Alabama [42]. They investigated the mechanism of toxicity of dichloroallyl lawsone (DCL) to tumor cells in culture. DCL specifically inhibits biosynthesis of pyrimidine nucleotides by inhibition of DHO and it was confirmed in studies with isolated mitochondria from mouse liver. Cleaveland, E.S. et al. [43] discovered a novel DHODH inhibitor (NSC 665564) and proposed that it has the same mechanism of inhibition as brequinar. Cytotoxicity results were experimentally determined using MOLT-4 lym-phoblast (the site of DHODH) and found the IC50 of brequi-nar (0.5 M) and NSC 665564 (0.3 M) were comparable. The induced cytotoxicity was reversed using either uridine or cytidine. Davis, J.P. et al. [44] investigated the effects of leflunomide and its active metabolites (A77 1726) on the activity of purified recombinant h DHODH. It was found that A77 1726 is a potent inhibitor of DHODH (Ki = 179 +/- 19 nM), while the parent compound leflunomide had no inhibi-tory effect (Concentration >1 M). These result suggested that A77 1726 is a competitive inhibitor of the CoQ binding site and is noncompetitive with respect to DHO.Williamson, R.A. et al. [45]isolated and characterized target protein from mouse spleen, purified and identified as mitochondrial DHODH by a series of structural and functional criteria. In-hibition of the DHODH activity by A77 1726 and its ana-logues clearly showed antiproliferative effect on a wide
1042 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
range of cells due to depletion of intracellular pools of uridyl triphospahtate (UTP) and cytidyl triphospahtate (CTP). 5. MEDICINAL CHEMISTRY OF DHODH INHI-BITORS
DHODH is a central enzyme of pyrimidine biosynthesis that catalyzes oxidation of dihydroorotate to orotate. DHODH inhibitors are considered as promising targets for the development of antiproliferative, antiparasitic and im-munosuppressive drugs since rapid cell proliferation often depends on de novo synthesis of pyrimidine nucleotides. The X-ray structures of Pf DHODH [24] and h DHODH [26] are determined. Orotate and FMN load against each other in the center of the / barrel, and the inhibitor binding site is formed nearby to this site by two -helices, that are posi-tioned between predicted N-terminal transmembrane domain and the canonical / barrel domain. Binding mode studies of DHODH with inhibitors have suggested that inhibitor binding-pocket has extensive variation in amino acid se-quence between Pf DHODH and h DHODH. The intention of the next section is to review all known DHODH inhibitors to cover recent advances in the medicinal chemistry and thera-peutic potential of this class of agents.
5.1. Triazolopyrimidine-Based DHODH Inhibitors Phillips, M.A. et al . [46] synthesized triazolopyrimidines based inhibitors of Pf DHODH with potent antimalarial activ-ity in whole cell assay. Compound 1 (5-methyl-[1,2,4] tria-zolo[1,5-a ]pyrimidin-7-yl)naphthalen-2-ylamine (Fig. 3) was a representative compound of triazolopyrimidines based in-hibitors and showed potent activity (IC 50 Pf DHODH = 0.047 μM) against P. falciparum . Compound 1 was discov-ered by HTS of a 220,000 compound library of “druglike” molecules using a colorometric enzyme assay. Structure ac-tivity relationship study revealed that substitutions at R and R 1 position of triazolopyrimidine with alkyl group (Fig. 3) 2 (IC 50 Pf DHODH = 0.21 μM), 3 (IC 50 Pf DHODH = 0.19 μM) 4 (IC 50 Pf DHODH = 0.16 μM) resulted in modest de-crease in activity, while the substitutions at R 2 (Fig. 3) com-pound 5 (IC 50 Pf DHODH = 3.0 μM) and 6 (IC 50 Pf DHODH = 93 μM) resulted in a loss of potency. Introduction of het-eroatoms on, or in, the napthyl ring reduce activity 7 (IC 50 Pf DHODH >200 μM), naphthalene ring attached at 1-position showed reduced activity 8 (IC 50 Pf DHODH = 45 μM) and anthracene moiety at 2-position 9 (IC 50 Pf DHODH = 0.056 μM) showed potent inhibitory activity.
In another work the authors [47] studied species differ-ences in inhibitor potency between Pf DHODH and P. ber-ghei dihydroorotate dehydrogenase (Pb DHODH). Com-pound 1 (IC 50 Pb DHODH = 0.23μM) and 9 (IC 50 Pb DHODH = 3.7 μM) do not suppress parasite levels in P. berghei mouse model. Analogs of compound 1 were synthe-sized by replacing the naphthyl moiety with substituted phenyl groups and tested for activity against recombinant enzyme Pf DHODH, Pb DHODH, h DHODH and in whole cell P. falciparum parasite assays. Compound 10 contained para and meta substitutions (Fig. 3) with the best activity against Pf DHODH (IC 50 = 0.077 μM), which has similar potency to compound 1 and showed equal potency against Pf DHODH and Pb DHODH. Binding mode and specific se-lectivity of compound 1 was studied in detail to gain insight into the structural requirement for the selective and potent binding of 1 to Pf DHODH. Steady-state kinetic analysis showed that the Pf DHODH inhibitory activity increased linearly with increasing CoQ D concentration . The authors found that compound 1 inhibited the oxidative half-reaction and prevented transfer of electrons from FMNH 2 to CoQ , without affecting DHO dependent reductive half-reaction. It was suggested that all the inhibitors utilized the same mechanism to inhibit DHODH. Binding mode of 1 and its derivatives in receptor cavity explained the species-selective binding due to variation in amino acid sequence between the human and malarial DHODH.
Phillips, M. A. and Rathod P.K. published a review [48] on identification of several different classes (tria-zolopyrimidine, phenylbenzamides, thiofuran) of compounds by high throughput screening (HTS) as potent inhibitors of Pf DHODH. The most common chemical classes identified by the authors were phenylbenzamides 11, ureas 12 and naphthamides 13 (Fig. 4). These compounds showed strong selectivity for the Pf DHODH against h DHODH. The authors identified a potent and selective triazolopyrimidine-based compound 14, showed potent activity against P. falciparum 3D7 in whole cell assays. The X-ray structures of Pf DHODH bound to triazolopyrimdine-based inhibitors (1, 9 and 10) were solved recently, provided the first structures of Pf DHODH bound to a parasite selective potent inhibitor [49]. Binding-site is composed of two sites for tria-zolopyrimidine based inhibitor: 1) a completely hydrophobic site, which binds naphthyl (1), anthracenyl (9) and phenyl-trifluoromethyl (10), and 2) triazolopyrimidine pocket capa-ble of H-bond interactions. Both the sites provides a signifi-
N N N
R 3
R R 2
1: R = CH 3 R 1 = H R 2 = H R 3 = 2-naphthyl 2: R = CF 3 R 1 = H R 2 = H, R 3 = 2-naphthyl 3: R = C 2H 5 R 1 = H R 2 = H, R 3 = 2-naphthyl 4: R = CH 3 R 1 = CH 3 R 2 = H, R 3 = 2-naphthyl 5: R = CH 3 R 1 = H R 2 = CH 3 R 3 = 2-naphthyl 6: R = CH 3 R 1 = H R 2 = H R 3 = 2-naphthyl 7: R = CH 3 R 1 = H R 2 = H R 3 = 2-benzamide 8: R = CH 3 R 1 = H R 2 = H R 3 = 1-naphthyl 9: R = CH 3 R 1 = H R 2 = H R 3 = 2-anthracenyl 10: R = CH 3 R 1 = H R 2 = H R 3 = 3-F- 4-CF 3-Ph
Fig. (3). Triazolopyrimidine-based derivatives.
Recent Developments in the Medicinal Chemistry
Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1043
cant amount of binding energy to the inhibitor series; -stacking interactions are observed between the naphthyl and two Phe residues (227 and 188), and H-bonds to H185 and R265.
The triazolopyrimidine based inhibitors overlap the bind-ing site of the A77 1726 observed in the previously reported Pf DHODH-A77 1726 crystal structure [25]. The naphthyl binding-site access an earlier unidentified pocket formed by rotation of Phe188, which moves this residue into a position that overlaps the phenyl portion of A77 1726. The authors suggested that these data provides information on structural features as to how Pf DHODH is able to bind different classes of compounds with high affinity.
Recently QSAR and molecular docking studies [50] were performed on triazolopyrimidine based DHODH inhibitors [46, 47] as antimalarial agents. The QSAR studies revealed that spatial, electronic, thermodynamic and structural de-scriptors play an important role in determining DHODH in-hibitory activity of triazolopyrimidine derivatives. Docking studies inferred that hydrophobicity is necessary for better DHODH inhibitory activity. The authors designed some new compounds (15-17) (Fig. 5), performed their docking studies and observed similar binding interaction pattern. Docking study suggested different binding mode of substituted tria-zolopyrimidine derivatives. 2-Methyltriazolopyrimidine ring interacts with some polar (Arg265 and His185) and some nonpolar amino acids (Ileu263, Gly181, Leu176, Cys175,
Cys184, Val532 and Leu172), whereas the substituted phenyl rings interact with a hydrophobic pocket of the DHODH, created by some nonpolar amino acid residues. Ligand-receptor interaction studies inferred that volume and hydrophobicity of the para substituents of phenyl ring should be high but limited, while meta substituents with reasonable volume and moderate hydrophobicity were essential for bet-ter DHODH inhibitory activity. Designed compounds showed good in silico predicted activity as per the developed QSAR models.
5.2. Trifluoromethy Phenyl Butenamide Derivatives Fishwick, C.W.G. et al. [35] applied SPROUT-LeadOpt, a software package for computational structure based lead optimization exercise aiming for improving binding of A77 1726 to CoQ binding cavity in DHODH. The authors synthe-sized and tested analogs of A77 1726 against human and P. falciparum DHODH in inhibition assays. The SAR study of analogs of A77 1726 revealed that substitution of methyl group for a cyclopropyl group as in 18 led to increase in binding affinity for both Plasmodium and human DHODH (IC 50 Pf DHODH = 92.5 μM and IC 50 h DHODH = 0.117 μM). Large hydrophobic substituents at this position (as in com-pounds 19, 20) (Fig. 6) resulted in an adverse effect upon DHODH inhibition. Replacement of the trifluoromethyl group in A77 1726 with phenyl 21 (IC 50 Pf DHODH = 25.7
2
12
13
N
14
Fig. (4). Analogs of triazolopyrimidine identified by HTS.
N H 3
15: R = 3-I, 4-CH(CH 3)2 Dock score = 34.384 16: 3-Br, 4-I Dock score = 32.91617: 3-CH 3, 4-I Dock score = 32.449
Fig. (5). Designed triazolopyrimidine-based DHODH Inhibitors with dock scores.
R 118: R 1 = Cyclopropanyl R 2 = CF 319: R 1 = Cyclobutanyl R 2 = CF 320: R 1 = Cyclopentanyl R 2 = CF 321: R 1 = CH 3 R 2 = Phenyl
22: R 1 = Cyclopropanyl R 2 = Phenyl
Fig. (6). Trifluoromethy phenyl butenamide derivatives.
1044 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
μM and IC50 h DHODH = 0.09 μM) resulted in a significant improvement in binding affinities for both the enzymes. Ar-rangement of these features in compound 22 resulted in good inhibition of h DHODH but an 8-fold reduction in activity against Pf DHODH.
The authors investigated interaction of inhibitors with h DHODH using X-ray crystallography. Co-crystallized structure of h DHODH-inhibitors complex suggested that head moiety of inhibitors occupied same part of the binding cavity as A77 1726. In case of Pf DHODH- A77 1726 com-plex, three direct hydrogen bonds were observed (to His185, Arg265, and Tyr528), whereas in case of h DHODH- A77 1726 complex, there were water-mediated hydrogen bonds to Arg265 and Gln47 as well as the direct hydrogen bond to Tyr356. The biphenyl ring (tail) of each inhibitor was pre-dicted to extend toward the surface of the protein and bind in large hydrophobic region of binding cavity. All the inhibitors made direct H-bonding interaction with binding cavity. The cocrystal structures of 21 involved two water mediated H-bonds to Arg136 and Gln47, and a direct hydrogen bond involving the hydroxyl of Tyr356. The authors predicted binding mode of inhibitors in Pf DHODH and found that in-hibitors binds in a similar fashion to that observed in 21, with head group making direct H-bonds to residues Arg265, His185 and Tyr528. The biphenyl tail was predicted to bind in the large hydrophobic region of the CoQ binding cavities in a manner similar to that found for these inhibitors in h DHODH.All the designed inhibitors showed enhanced levels of inhibition for both Plasmodium and human DHODH enzymes compared to that observed for the existing inhibitor A77 1726.
5.3. Ethoxy Aromatic Amide-Based DHODH Inhibitors
A set of compounds were designed and synthesized [51] as Pf DHODH inhibitors with high binding affinities for ana-lyzing the mode of binding through modeling and mutagene-sis. Two different docking systems Autodock 3.0 and eHITS were used for docking of 23 (IC50 Pf DHODH = 0.16 M) and 24 (IC50 Pf DHODH = 0.44 M) (Fig. 7) into DHODH structures with DHO. Docking studies suggested that inhibi-tors (23, 24) form H-bonds to H185, R265, and Y528 in a similar manner to that observed for A77 1726 binding in the Pf DHODH crystal structure. Mutagenesis studies further emphasized that H185 and R265 residue were involved in binding of these inhibitors. H-bonding and hydrophobic in-teractions were favorable interaction of inhibitors with CoQ binding channel.
Synthesized compounds were tested against a recombi-nant Pf DHODH expressed from a synthetic gene in which codon usage was optimized for expression in E. coli. The tricyclic compounds (23 and 24) were more active than monocyclic or bicyclic compounds 25 (IC50 Pf DHODH = 27.78 M) and 26 (IC50 Pf DHODH = 40.00 M) (Fig. 6). The compounds showed considerably lower affinity for h DHODH, a 1200-fold higher relative affinity for parasite enzyme 23 (IC50h DHODH =29.57 M) and 24 (IC50
h DHODH = 491.4 M).
5.4. Cyclic Aliphatic and Aromatic Carboxylic Acid Am-ide Derivatives
Baumgartner, R. et al. [37] disclosed a novel series of DHODH inhibitors, based on virtual screening methods and presented high-resolution X-ray structures of h DHODH in complex with a novel class of low molecular weight com-pounds. Molecules that displayed structure activity relation-ship in an in vitro assay with IC50 values in nanomolar range were selected and co-crystallized with h DHODH. Selected compounds showed interesting binding modes termed as “nonbrequinar-like” 27 (IC50 = 280 nM) and “brequinar-like”
28 (IC50 = 2 nM) (Fig. 8) and some compounds showed in-terestingly a dual binding mode 29 (IC50 = 7 nM) strongly depending on the nature of chemical substitution. In “bre-quinar-like” binding mode, 6-fluoro-3-methyl-4-quinoline carboxylic acid moiety formed salt bridge to the side chain of Arg136. Additionally H-bonds were formed between car-boxylic acid and the side chain of Gln47. The biphenyl ring system showed several hydrophobic contacts with binding cavity, which mainly, consist of hydrophobic amino acids. In case of compound 27, different binding mode was observed and the carboxylic acid moiety was located in the opposite direction protruding toward the protein’s interior. This bind-ing mode was termed as “nonbrequinar-like”. “Brequinar-like” binding mode was more favorable with smaller meas-ured IC50 values. The authors performed superposition of binding sites, revealed conformational differences for amino acids (Gln47 and Arg136), whereas conformational differ-
H3
2324
2526 Fig. (7). Structures of ethoxy aromatic amide-based DHODH inhibitors.
Recent Developments in the Medicinal Chemistry
Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1045
ences were very minimum for the rest of the binding cavity. The binding site displayed enough volume to hold a variety of ring substitutions and compound conformations, espe-cially for the hydrophobic part of the binding site.
In another work Leban, J. et al. [52] designed via dock-ing, synthesized and improve via QSAR a novel series of DHODH inhibitors. Docking of 30 (Fig. 9) into crystal struc-ture of DHODH resulted in a positioning similar to that of brequinar. Docking study suggested that carboxylic acid group formed an ionic interaction to amino acid residue Arg-136, and the biphenyl ring showed hydrophobic interaction in receptor pocket. Introduction of small side chains into the
biphenyl ring of lead 30 resulted in compound 31, 32 with additional interactions in hydrophobic pocket of CoQ bind-ing cavity. QSAR analysis resulted in identification of im-portant features of a good DHODH inhibitor. In an in vitro enzyme assay, IC 50 values for DHODH Inhibition were de-termined using N-terminally truncated recombinant h DHODH.
5.5. Aromatic Quinoline Carboxamide Derivatives The authors [53] designed and synthesized quinoline-8-carboxamide derivatives as immunosuppressant to prevent xenograft rejection, dependent on inhibition of antibodies (Ab) produced by B-cells, independently of T-cell signals.
F 3C
27
F 3C
28
F 3C
29
Fig. (8). Cyclic aliphatic and aromatic carboxylic acid amide derivatives.
30: IC 50 = 410 nM
31: IC 50 = 280 nM
32: IC 50 = 18 nM
3
Fig. (9). Structures and activity of cyclopentene carboxylic acid amide derivatives.
R _______________________________________Comp. R R 1 R 2 X ________________________________________33 4-CF 3 H H C 34 4-CF3 H H N 35 H H H C 36 2-CF 3 H H C _______________________________________
2
Fig. (10). Aromatic quinolinecarboxamides derivatives.
1046 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
Compounds 33-36 (Fig. 10) were synthesized, via coupling of acid chloride of quinoline-8-carboxylic acid with corre-sponding anilines. Pharmacological screening of compounds based on modulation of Ab production by trinitrophenyl-lipopolysaccharide (TNP-LPS) stimulated murine B-cells. T-cell immunosuppressive activity and cytotoxicity of com-pounds were assessed in mouse mixed lymphocyte reaction (MLR) and Jurkat proliferation assays.
Compound
33 discriminates between B- and T-cells and showed 10-fold B-cell selectivity over T-cell selectivity. Structure activity relationship (SAR) study revealed the presence and influence of trifluoromethyl substituent. In comparison to 33, the corresponding phenyl derivative 35 was a weak B-cell inhibitor, whereas the ortho trifluoro-methyl substituted derivative 36, although equipotent to le-flunomide has a cytostatic character because it was devoid of selectivity toward T-cells and Jurkat cells.
5.6. 2-Phenylquinoline-4-Carboxylic Acid Derivatives
Boa, A.N. et al. [54] synthesized a series of 2-phenylquinoline-4-carboxylic acid derivatives related to bre-quinar and evaluated as inhibitors of Pf DHODH (antimalar-ial agents). Selected compounds (37, 38) (Fig. 11) were evaluated for inhibition of Pf DHODH and they were found more active than brequinar. The active compounds were also screened for inhibition of h DHODH. Synthesized 2-phenylquinoline-4-carboxylic acid derivatives were consid-erably less active against h DHODH than brequinar.
5.7. Cyclopentene Dicarboxylic Acid Amides Derivatives Leban,
J.
et al. [55] synthesized novel DHODH inhibitors by the introduction of heteroatoms into the cyclopentene ring and attached a hydroxyl group to it. Compounds were syn-thesized by reacting dicarboxylic acid anhydrides with ani-lines in an inert solvent.DHODH inhibition was measured in an in vitro enzyme assay using N-terminally truncated re-combinant h DHODH. SAR study revealed that introduction of sulfur atom into the petacyclic ring of lead compound 39 (IC50 = 0.41 M) resulted in equipotent compound 40(IC50 = 0.667 M) (Fig. 12).
Oxidation of sulfide to sulfone led to less active com-pound 41 (IC50 = 3.8 M). Introduction of oxygen atom in pentacyclic ring system resulted in compounds with lower DHODH inhibitory activity.X-ray crystallography showed a brequinar-like binding mode of these compounds with in-creasing number of fluoro substituents in the first aromatic ring.
5.8. Terphenyl Carboxylic Acid Amide Derivatives
Clardy, J. et al. [56] synthesized a series of potentially selective inhibitors of DHODH (42-45) (Fig. 13) via itera-tive, chemoselective Suzuki cross-couplings reaction using biaryl chlorides. The authors [57] disclosed brequinar-derived asymmetric terphenyl compounds. X-ray crystallo-graphic structure of an analog of brequinar bound to h DHODH was determined. Terphenyl compound 42 bound to CoQ binding site in a similar fashion to brequinar, phenyl tail bound in pocket by hydrophobic interactions, and car-boxylic acid moiety formed H-bonds with R136 and amide group forms H-bonds with Y356 near FMN group. Docking study with eHiTS suggested that most of the compounds dock well to human and rat DHODH, confirmed by their docking scores and matched with experimentally determined DHODH inhibition. Synthesized compounds were screened
37: IC50Pf DHODH = 38 M IC50h DHODH = 38 M
38: IC50Pf DHODH = 162 M IC50h DHODH = 80 M
Fig. (11). Structures and activity of 2-phenylquinoline-4-carboxylic acid derivatives.
39
40
41
Fig. (12). Structures of cyclopentene dicarboxylic acid amides derivatives.
Recent Developments in the Medicinal Chemistry
Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1047
via a crude visual enzyme assay based on the color change in the dye 2,6-dichloroindophenol (DCIP) ( 595 = 18.8 mM -1 cm -1), required to established a threshold concentration for DHODH inhibition.
5.9. Cyclopropane Carbonyl Derivatives
Yang, D.Y. et al. [58] synthesized cyclopropane carbonyl analogues as inhibitors of 4-hydroxyphenylpyruvate dioxy-genase (HPPD) and DHODH. The authors reported X-ray crystal structures, and suggested that cyclopropanecarbonyl derivative 46 (IC 50 = 21 nM) was 14 times more potent than the corresponding isopropylcarbonyl derivative 47 (IC 50 = 295 nM) in rat DHODH inhibition due to H-bonding of cy-clopropanecarbonyl oxygen of 46 to hydroxyl group of Tyr356. This specific H-bonding induces cyclopropyl group
in 46 to adopt the fixed bisected conformation, causing 46 to function as a conformation-restricted DHODH inhibitor. 5.10. Biaryl Carboxyamide Derivatives
Fishwick, C.W.G. et al. [59] applied de novo molecular design program SPROUT to design novel inhibitors of Pf DHODH. The authors made a detailed c omparison of the crystal structure of Pf DHODH and h DHODH in the dimen-sions and topography of the hydrophobic CoQ binding chan-nel. The study suggested that the topography of h DHODH is flattened by the projection of a methyl group in the region which was occupied by the aromatic ring of the bound in-hibitor and required inhibitors of this type to be essentially planar. In case of Pf DHODH, same region was much less congested unlike that in h DHODH and could accommodate
3
4243
44
45
Fig. (13). Structures of terphenyl carboxylic acid amide derivatives.
F 3
F 3
4746
Fig. (14).
Structures of cyclopropane carbonyl derivatives.
49: IC 50 Pf DHODH = 153.3 M IC 50 h DHODH = 5.0 M
48: IC 50 Pf DHODH = 142.6 M IC 50 h DHODH = 8.4 M
Fig. (15). Structures and activity of biaryl carboxyamide derivatives .
1048 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
inhibitors that were cylindrical in shape. Two amino acids His185 and Arg265, made direct H-bonding contacts to the designed inhibitors. Pf DHODH and h DHODH inhibition was measured by crude visual assay based on the color change in the dye DCIP, using saturated concentrations of DHO and decylubiquinone (CoQd). Structure based de novo design produced 20 different small molecule templates, bind-ing affinities of some of them were predicted in micromolar range (48, 49) (Fig. 15).
5.11. Biphenyl-4-ylcarbamoyl Thiophene/Cyclopentene Carboxylic Acids Derivatives
Replacement of cyclopentene ring by aromatic heterocy-cles resulted in a series of biphenyl-4-ylcarbamoyl thiophene carboxylic acids derivatives (50, 51) [60] (Fig. 16). SAR study revealed that compounds with thiophene ring (50, 51) were more potent as compared to cyclopentene ring (52, 53). The authors compared X-ray crystal structures of 52 and 55, and found that cyclopentene derivatives (52) showed a single binding mode (non-brequinar like) and thiophene analog (55) showed a dual binding mode which was “brequinar like” and “non-brequinar like”. DHODH inhibition was measured in an in vitro enzyme assay using N-terminally truncated recombinant h DHODH. Compounds 54 (IC50 = 2 M) and 55 (IC50 = 2 M) displayed potent antiproliferatory effects on peripheral blood mononuclear cells (PBMCs), and suggested potential of such compounds for treatment of auto-immune diseases.
5.12. Amino-Benzoic Acid Derivatives
Recently, novel amino-benzoic acid derivatives[61]were disclosed as DHODH inhibitors by virtual screening and X-ray crystallographic studies. The authors determined binding mode of selected hits by X-ray crystallography, suggested the H-bonding interaction to the side-chain of either Arg136 or Tyr365 and a hydrophobic interaction to the side-chain of Val134 and Tyr356. H-bond network was created around the acidic group that made bifurcated H-bonds to the guanidinyl group of Arg136. One carboxylate oxygen atom formed H-bond to a crystallographic water molecule (H-bonds to Gln-47), other carboxylate oxygen formed H- bonds to a second crystallographic water (H-bonds to the NHs of the urea linker of the inhibitor and the backbone carbonyl oxygen of Thr-360). The benzoic acid phenyl ring occupied hydropho-bic pocket formed by the FMN and the side-chains of Val134 and Tyr356 amino acid residue. The software pro-gram GLIDE was used for virtual screening which resulted in 200 hits with IC50 <10 M. The binding modes of the compounds 56, 57 were revealed by co-crystal structures with h DHODH. Selected compounds were screened via en-zymatic assay using recombinant h DHODH.
5.13. N-Arylaminomethylene Malonate
Johnson, A.P. et al. [62] studied the effects of substitu-ents on the selectivity of binding of N-arylaminomethylene malonate derivatives to DHODH. Molecular modeling study suggested that substitution at aryl portion with H-bond ac-ceptors (58-60) (Fig. 18) increased selectivity of these in-hibitors for Pf DHODH. The authors described the synthesis of novel series of DHODH inhibitors containing either car-boxylic acid or ester functionality. Synthesized compound were screened for binding affinity against Pf DHODH and h DHODH at 50 M using the DCIP dye. X-ray crystal struc-tures of selective inhibitors bound to Pf DHODH showed
50: IC50 = 44 nM51: IC50 = 3 nM
52: IC50
= 134 nM
53: IC50 = 410 nM
O
54 :IC50 = 2 M55: IC50 = 2 M
Fig. (16). Structures and activity of biphenyl-4-ylcarbamoyl thiophene/cyclopentene carboxylic acids derivatives.
Recent Developments in the Medicinal Chemistry Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1049
mutation of His185 to Ala causing a large reduction in bind-ing affinity. N-acyl-2-aminomethylene malonate inhibitors bound to DHODH in a similar fashion to that of A77 1726. Examination of binding cavities for both Pf DHODH and h DHODH revealed that, in addition to a large hydrophobic cavity, a smaller hydrophobic cavity was also located near to Arg265 in Pf DHODH. The docking studies indicated that, volume of the small hydrophobic cavity accommodated one of the two ester-derived ethyl groups.
5.14. 4-Hydroxycoumarins, Fenamic Acids and N-(alkylcarbonyl) Anthranilic Acids Derivatives
Fritzson,
I.
et al. [63] identified three different classes of h DHODH inhibitor based upon virtual screening and struc-ture-guided selection of fragments. The authors created pharmacophores by using structure based virtual screening method. This allowed the identification of some structural features like hydrophobic, donor, acceptor and ionizable groups for interaction. On the basis of structure-guided fragment selection method, it was suggested that substitution at third position of benzoic acid caused polar interaction with Try356 amino acid in inner subside of CoQ biding site. Three different class of compounds (4-hydroxycoumarins, fenamic acids, and N-(alkylcarbonyl)anthranilic acids) were designed, synthesized and evaluated for inhibition of h DHODH. Derivatives of N-(alkylcarbonyl) anthranilic acid (61, 62) showed potent h DHODH inhibitory activity than 4-hydroxycoumarins (63, 64) and fenamic acids derivatives (65, 66) (Fig. 19).
5.15. Inhibitors of Trypanosoma cruzi DHODH
Nonato,
M.C.
et al. [64] identified a novel series of Try-panosoma cruzi DHODH inhibitors by a combination of virtual screening (structure-based and ligand-based) and iso-thermal titration calorimetry (ITC) methods. Trypanosoma cruzi DHODH (Tc DHODH) is a member of family-1A DHODH and h DHODH is a member of family-2 DHODHs. Members of family-1A are homodimeric proteins whereas family-2 enzymes are monomeric. The authors have ex-plored active site of DHODHs, to map structural differences between h DHODH, Trypanosoma cruzi(Tc DHODH) and Leishmania major(Lm DHODH) enzymes. The topographi-cal analysis identified four new regions (S1, S2, S3 and S4) which were able to accommodate inhibitors. The results from ITC assays provided insights into the role of different substituents in determining binding affinity of compounds tested against Tc DHODH. The authors proposed SAR study based on the calorimetric assays, together with structural information provided by X-ray diffraction analysis of 3D structures. In compounds 67-72 (Fig. 20) substitution at C5 was more favorable than C6 (carboxylic oxygen), and rigidi-fication of the carboxyl group resulted in a complete loss of molecular interaction. The authors determined the apparent inhibition constant (Ki app) and elucidated the mechanism of inhibition of compounds.
Schematic diagram of important interactions of inhibitor with DHODH is given in Fig. (21).
5.1
6.Alkyl-5-Benzimidazole Thiophene-2-Carboxamide Derivatives
Booker, M.L.
et al. [65] recently identified novel inhibi-tors of DHODH. Compounds 73-75 were identified form HTS screening of 215,000 diverse compounds [66]. The authors evaluated i n vitro and in vivo drug absorption, distri-bution, metabolism, and excretion properties of inhibitors
56: IC50 = 0.7 M57: IC50 = 0.5 M
Fig. (17). Structures and activity of amino-benzoic acid derivatives.
1
_________________________________________________________________
Compound R1 R2 Pf DHODH(50 M) h DHODH(50 M)
_________________________________________________________________
58 CO2Et m-CO2Et 0 ± 4 5 ± 2
59CO2Et p-CO2Et 15 ± 4 4 ± 2
60 CN p-CO2Et 2 ± 4 2 ± 2
_________________________________________________________________
Fig. (18). Structures and activity of N-arylaminomethylene malonate derivatives.
1050 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
62:
IC50 = 0.015 M
63: IC50 = 0.23
M
64: IC50 = 11
M
3
65: IC50 = 0.24
M
66: IC50 = 0.26 M
H3
Fig. (19). Structures and activity of 4-hydroxycoumarins, fenamic acids and N-(alkylcarbonyl) anthranilic acids derivatives.
H
H
H
2
H
2
H
67:Ki app = 19.28 ± 4.01 ( M)68:Ki app = 21.70 ± 4.36( M)
69:Ki app = 72.21 ± 7.41( M)
70:Ki app = 94.79 ± 14.98 ( M)
71:Ki app = 139.24 ± 26.04( M)72:Ki app = 300 ± 20.08( M)
Fig. (20). Structures of Inhibitors of Trypanosoma cruzi DHODH.
Recent Developments in the Medicinal Chemistry Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1051
(B)
Head
Group
Pf DHODH)
H-bonds to Gln47/Arg136
and to Tyr356(h DHODH)
Hydrophobic interactions
(hydrophobic region)
(C)
(D)(E)
H-bonds to Gln-47, Thr-360
Val-134,Tyr-356
(F)
Large
Hydrophobic
Cavity
(G)
Fig. (21) Schematic diagram of important interactions of inhibitors with DHODH A: Aromatic carboxylic acid amide derivatives [37], B: Triazolopyrimidine-based derivatives[50], C: Trifluoromethy phenyl butenamide derivatives [35], D: Terphenyl carboxylic acid amide de-rivatives [56, 57], E: Cyclopropane carbonyl derivatives [58], F:Amino-benzoic acid derivatives [61], G:N-arylaminomethylene malonate [62].
and found double-digit nanomolar potency against
Pf DHODH, Pb DHODH and Pv DHODH, while lacking ac-
tivity against h DHODH (Fig. 22). Molecular modeling stud-
ies suggested that the structure of Pf DHODH complexed
with 73 was similar to the structures of Pf DHODH bound to
the triazolopyrimidine based inhibitors and A77 1726. X-ray
structure determination of 73 in complex with Pf DHODH
reveled that cyclopropyl ring system binds a largely hydro-
phobic pocket formed by Val532, Ile272, and Ile263. Ion
pair H-bonds between His-185 and the nitrogen of methyl-
formamide, and between Arg265 and oxygen of methylfor-
mamide (non-hydrophobic contacts) were observed near to
hydrophobic pocket. The benzimidazole ring bound in a hy-
drophobic pocket formed by Tyr168, Cys175, Phe171,
Leu172, Phe188, Leu191, and Leu31 suggested that the se-
lective binding of 73 to Pf DHODH was due to amino acid
substitutions in the benzimidazole ring.
5.17.Amino Nicotinic Acid and Isonicotinic Acid Deriva-
tives
Castro, P.L.J.C. et al. [67] granted a U.S. patent on
discloser of a series of amino nicotinic acid and isonicotinic
3
S
N
O
Fig. (22). Structures and activity of Alkyl-5- benzimidazole thiophene-2-carboxamide derivatives.
1052 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
acid derivatives as h DHODH inhibitors. The authors de-scribed the method of preparation of h DHODH inhibitors, biological evaluation and use of the compounds in the manu-facturing of medicine for treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondilytis, multiple sclerosis, Wegener’s granulomatosis, systematic lupus erythematosus and psoriasis and method of treatment of pathological condi-tions or diseases susceptible to amelioration by inhibition of DHODH. Some of the compounds (76-79) with h DHODH inhibitory activity are shown in Fig. (23) [67]. 6. CONCLUSION AND PERSPECTIVE
Purine and pyrimidine bases are essential for cellular metabolism and cell growth and are considered as important precursors in DNA and RNA synthesis. DHODH catalyzes the conversion of dihydroorotate to orotate, which is the pre-cursor for uridine and cytidine. This is a rate limiting step in the pyrimidine biosynthesis pathway, necessary for cell growth and proliferation. Rapidly proliferating cells (cancer cells, T-lymphocytes) do not acquire sufficient nucleosides from the salvage pathway for their survival. DHODH inhibi-tors block the growth of fast proliferating cell (de novo syn-thesis). The cells which grow at normal speed can meet the requirement of pyrimidines base (uracil, cytosine and thymine) from normal metabolic cycle (salvage pathways). Lymphocytes, the primary mediators of the immune re-sponse, use pyrimidines exclusively, for their growth and rely mainly on de novo synthesis to meet their requirement for activation and proliferation. Compounds that reduce the availability or utilization of pyrimidines are important agents for the treatment of auto immune diseases. A large number of DHODH inhibitors are emerging as potential intervention points for developing therapeutic agents for treatment of malaria, cancer therapy and as an immunosuppressive drug for the treatment of rheumatoid arthritis. The X-ray structure of h DHODH in complex with inhibitors is also resolved. All the h DHODH inhibitors, reported to date in the literatures, bind to the CoQ binding channel and display favorable ac-tivities. The CoQ binding site of all DHODH enzymes con-tains two highly conserved residues, an arginine and a tyro-sine. Binding mode analysis of A77 1726 with co-crystallized DHODH structures reveals the important of car-bonyl group in making H-bond with water, which bounds to Arg136. The variable nature of the CoQ binding site in all the DHODHs is considered to be responsible for species-related inhibition for compounds that bind in the tunnel. The inhibitor-binding pocket can be divided into site A (H-bonding pocket), which forms H-bonding interactions with the bound inhibitors, and site B, which is completely hydro-phobic. Knowledge of DHODH structures can help in de novo design and discovery of DHODH inhibitors by compet-ing for the CoQ binding site. Binding profile studies con-firmed that differences in substituents and their pattern of substitution were largely dictated by different levels of inhi-bition of Pf DHODH and h DHODH. Based upon various experimental finding discussed above the important pharma-cophoric groups are identified. DHODH is capped by a hy-drophobic cavity and inhibitors have to diffuse through a very hydrophobic environment. DHODH inhibitors often have a heterocyclic rings in their structure. Lone pair of elec-tron on N-atom in triazolopyrimidine, qunoline, isoxazol and imidazole ring system act as hydrogen acceptor and respon-sible for H-bonding with CoQ binding site. Secondly, the biphenyl or aryl ring system showed several hydrophobic interactions in the enzyme binding pocket. The substituted biphenyl (biaryl tail) ring especially with fluoro, trifluoro, methoxy and ethoxy in first aromatic ring led toward better inhibitory activity. Carboxy group attached to five-membered ring (cyclopentene, thiophene) can form a dual binding mode. Compounds containing a carbonyl group at either the meta- or para-positions of the aryl ring, showed H-bonding. Beyond these findings, several other important chemical scaffolds are identified. Compounds containing cyclopropane, cyclopentene, naphthyl, anthracenyl and other pentacyclic aromatic heterocycles showed good binding af-finities for both Plasmodium and human DHODH. HTS [68] and virtual screening [69] methods have proved a successful strategy to identify potent and highly species selective in-hibitors of DHODH. The most common structural features across the different classes of inhibitors are 1) 5 or 6 mem-
76: IC 50 = 3 m
O
CF 3
77: IC 78: IC 50 = 14 m
O
CH 3
O
Li CF 3
79: IC 50 = 5 m
Fig. (23). Structures and activity of amino nicotinic acid and isonicotinic acid derivatives.
Recent Developments in the Medicinal Chemistry Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1053
bered aromatic or non-aromatic ring system with acidic (car-boxylic acid) head group, which interacts (H-bonding) with the Arg136 (CoQ binding cavity), 2) a planar functional group, and 3) a bulky hydrophobic substituent (biphenyl ring system) which fulfills shape requirement in the hydrophobic binding pocket of the CoQ binding cavity. The most signifi-cant feature for good DHODH inhibitory activity is the sub-stitution pattern on one of the biphenyl rings with fluoro or trifluoromethyl group. Several structural features necessary for DHODH inhibition are reviewed in present communica-tion. These recent advances in design, synthesis and thera-peutic potential of different classes of DHODH inhibitors will aid the structural insights into the development of potent DHODH inhibitors.
ACKNOWLEDGEMENT
The authors are grateful to Director Institute of Phar-macy, Nirma University, Ahmedabad for providing facilities to complete this work.
REFERENCES
[1]Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J.
Structures of human dihydroorotate dehydrogenase in complex
with antiproliferative agents. Struct. Fold Des., 2000, 8, 25-33. [2]Loeffler, M.; Zameitat, E. Pyrimidine biosynthesis. Encyc. Biol.
Chem.,2004, 3, 600–605.
[3]Mascia, L.; Turchi, G.; Bemi, V.; Ipata, P. L. Uracil salvage path-
way in PC12 cells. Biochem. Biophys. Acta,2000, 1524, 45–50. [4]Fairbanks, L.D.; Bofill, M.; Ruckemann, K.; Simmonds, H.A.
Importance of ribonucleotide availability to proliferating T-
lymphocytes from healthy humans. J. Biol. Chem., 1995, 270,
29682-29689.
[5]Nara, T.; Hshimoto, T.; Aoki, T. Evolutionary implications of the
mosaic pyrimidine-biosynthetic pathway in eukaryotes. Gene,
2000, 257, 209-222.
[6]Loeffler, M.; Fairbanks, L.D.; Zameitat, E.; Marinaki, A.M.; Sim-
monds H. A. Pyrimidine pathways in health and disease. Trends in
Molecular Medicine, 2005, 11, 330-437.
[7]Shawver, L.K.; Schwartz, D.P.; Mann, E.; Chen, H.; Tsai, J.; Chu,
L.; Taylorson, L.; Longhi, M.; Meredith, S.; Germain, L.; Jacobs,
J.S.; Tang, C.; Ullrich, A.; Berens, M.E.; Hersh, E.; McMahon, G.;
Hirth, K.P.; Powell, T.J. Inhibition of platelet-derived growth fac-
tor-mediated signal transduction and tumor growth by N-[4-
(trifluoromethyl)-phenyl]-5-methylisoxazole- 4-carboxamide. Clin.
Cancer Res., 1997, 3, 1167–1177.
[8]Baumann, P.; Mandl, W.S.; Volkl, A.; Adam, C.; Bumeder, I.;
Oduncu, F.; Schmidmaier, R. A prospective, randomised, con-
trolled, double-blind phase I-II clinical trial on the safety of A-
Part(R) Gel as adhesion prophylaxis after major abdominal surgery
versus non-treated group. Mol. Cancer Ther., 2009, 8, 366-375. [9]Chen, S.F.; Ruben, R.L.; Dexter, D.L. Mechanism of action of the
novel anticancer agent 6-fluoro-2-(2'-fluoro-1,1'-biphenyl-4-yl)-3-
methyl-4-quinolinecarboxylic acid sodium salt (NSC 368390): in-
hibition of de novo pyrimidine nucleotide biosynthesis. Cancer
Res., 198646, 5014-5019.
[10]Herrmann, M.L.; Schleyerbach, R.; Kirschbaum, B.J. Leflunomide:
an immunomodulatory drug for the treatment of rheumatoid arthri-
tis and other autoimmune diseases. Immunopharmacology, 2004,
47, 273-289
[11]Merrill, J.E.; Hanak, S.; Pu, S.F.; Liang, J.; Dang, C.; Iglesias,
B.D.; Harvey, B.; Zhu, B.; McMonagle, S.K. Teriflunomide re-
duces behavioral, electrophysiological, and histopathological defi-
cits in the Dark Agouti rat model of experimental autoimmune en-
cephalomyelitis. J. Neurol., 2009, 256, 89-103
[12]Copeland, R. A.; Marcinkeviciene, J.; Haque, T. S.; Kopcho, L. M.;
Jiang, W.; Wang, K.; Ecret, L. D.; Sizemore, C.; Amsler, K. A.;
Foster, L.; Tadesse, S.; Combs, A. P.; Stern, A. M.; Trainor, G. L.;
Slee, A.; Rogers, M. J.; Hobbs, F. Helicobacter pylori-selective an-
tibacterials based on inhibition of pyrimidine biosynthesis. J. Biol.
Chem., 2000, 275, 33373-33378. [13]Marcinkeviciene, J.; Rogers, M. J.; Kopcho, L.; Jiang, W.; Wang,
K.; Murphy, D. J.; Lippy, J.; Link, S.; Chung, T. D.; Hobbs, F.;
Haque, T.; Trainor, G. L.; Slee, A.; Stern, A. M.; Copeland, R. A.
Selective inhibition of bacterial dihydroorotate dehydrogenases by
thiadiazolidinediones. Biochem. Pharmacol., 2000, 60, 339-342. [14]Tamta, H.; Mukhopadhyay, A. K. Biochemical targets for malaria
chemotherapy. Current Research & Information on Pharmaceuti-
cal Science (CRIPS), 2003, 4, 6–9.
[15]Morand, P.; Courtin, O.; Sautes, C.; Westwood, R.; Hercend, T.;
Kuo, E. A.; Ruuth, E. Dihydroorotate dehydrogenase is a target for
the biological effects of leflunomide. Transplant. Proc., 1996, 28,
3088– 3091.
[16]Gustafson, G.; Davis, G.; Waldron, C.; Smith, A.; Henry, M. Iden-
tification of a new antifungal target site through a dual biochemical
and molecular-genetics approach. Curr. Genet., 1996, 30, 159–165.
[17]Shannon, P.V.R.; Eichholtz, T.; Linstead, D.; Masdin, P.; Skinner,
R. Condensed heterocyclic compounds as anti-inflammatory and
immunomodulatory agents. International patent number WO 99/
45926, 1999.
[18]Fox, R.I.; Herrmann, M.L.; Frangou, C.G.; Wahl, G.M.; Morris,
R.E.; Strand, V.; Kirschbaum, B.J. Mechanism of action for le-
flunomide in rheumatoid arthritis. Clin. Immunol., 1999, 93, 198-
208.
[19]Rozman, B. Clinical experience with leflunomide in rheumatoid
arthritis. J. Rheumatol Suppl., 1998, 53, 27-31.
[20]Williamson, R.A.; Yea, C.M.; Robson, P.A.; Curnock, A.P.; Gad-
her, S.; Hambleton, A.B.; Woodward, K.; Bruneau, J.M.; Hamble-
ton, P.; Spinella-Jaegle, S.; Morand, P.; Courtin, O.; Sautes, C.;
Westwood, R.; Hercend, T.; Kuo, E. A.; Ruuth, E. Dihydroorotate
dehydrogenase is a target for the biological effects of leflunomide.
Transplant. Proc., 1996, 28, 3088-3091.
[21]Batt, D.G. Inhibitors of dihydroorotate dehydrogenase. Expert
Opin. Ther. Pat.,1999, 9(1), 41-54.
[22]Leban, J.; Vitt D.l Human dihydroorotate dehydrogenase inhibitors,
a novel approach for the treatment of autoimmune and
inflammatory diseases. Arzneimittelforschung, 20116(1), 66-72. [23]Hansen, M.; Le Nours, J.; Johansson, E.; Antal, T.; Ullrich, A.;
Loffler, M.; Larsen, S. Inhibitor binding in a class 2 dihydroorotate
dehydrogenase causes variations in the membrane-associated
Nterminal domain. Protein Sci., 2004, 13, 1031-1042.
[24]Hurt, D.E.; Widom, J.; Clardy, J. Structure of Plasmodium falcipa-
rum dihydroorotate dehydrogenase with a bound inhibitor. Acta
Crystallogr., 2006, 62, 312-323.
[25]Nagy, M.; Lacroute, F.; Thomas, D. Divergent evolution of
pyrimidine biosynthesis between anaerobic and aerobic yeasts.
Proc. Natl. Acad. Sci. USA, 1992, 89, 8966–8970
[26]Liu, S.; Neidhardt, E.A.; Grossman, T.H.; Ocain, T.; Clardy, J.
Structures of human dihydroorotate dehydrogenase in complex
with antiproliferative agents. Structure, 2000, 8(1), 25-33.
[27]Norager, S.; Jensen, K.F.; Bjornberg, O.; Larsen, S. E. coli dihy-
droorotate dehydrogenase reveals structural and functional distinc-
tions between different classes of dihydroorotate dehydrogenase.
Structure, 2002, 10, 1211–1223.
[28]Bjornberg, O.; Gruner, A.C.; Roepstorff, P.; Jensen, K.F. The ac-
tivity of Escherichia coli dihydroorotate dehydrogenase is depend-
ent on a conserved loop identified by sequence homology, mutage-
nesis, and limited proteolysis. Biochemistry, 1999, 38, 2899–2908 [29]Bjornberg, O.; Rowland, P.; Larsen, S.; Jensen, K.F. Active site of
dihydroorotate dehydrogenase A from Lactococcus lactis investi-
gated by chemical modification and mutagenesis. Biochemistry,
1997, 36, 16197–1620.
[30]Feliciano, P.R.; Cordeiro, A.T.; Costa, A.J.; Nonato, M.C. Cloning,
expression, purification, and characterization of Leishmania major
dihydroorotate dehydrogenase. Protein Expr. Purif., 2006, 48, 98–
103.
[31]Cordeiro, A.T.; Feliciano, P.R.; Nonato, M.C. Crystallization and
preliminary X-ray diffraction analysis of Leishmania major Dihy-
droorotate dehydrogenase. Acta Cryst. Section F, 2006, 62, 1049–
1051.
[32]Rowland, P.; Norager, S.; Jensen, K.F.; Larsen, S. Structure of
roorotate dehydrogenase B: electron transfer between two flavin
groups bridged by a iron-sulphur cluster. Structure, 2000, 8(12),
1227-1238
[33]Nielsen, F.S.; Andersen, P.S.; Jensen, K.F. The B form of dihydro-
orotate dehydrogenase from Lactococcus lactis consists of two dif-
1054 Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 Vyas and Ghate
ferent subunits, encoded by the pyrDb and pyrK genes, and con-
tains FMN, FAD, and [FeS] redox centers. J. Biol. Chem., 1996,
271, 29359–29365.
[34]Sorensen, P.G.; Dandanell, G. A new type of dihydroorotate dehy-
drogenase, type 1S, from the thermoacidophilic archaeon Sul-
folobus solfataricus. Extremophiles, 2002, 6, 245–251.
[35]Davies, M.; Heikkila, T.; McConkey, G.A.; Fishwick, C.W.G.;
Parsons, M.R.; Johnson, A.P. Structure-based design, synthesis,
and characterization of inhibitors of human and Plasmodium falci-
parum dihydroorotate dehydrogenases. J. Med. Chem., 2009, 52(9),
2683-2693.
[36]Walse, B.; Dufe, V.T.; Svensson, B.; Fritzson, I.; Dahlberg, L.;
Khairoullina, A.; Wellmar, U.; Al-Karadaghi, S. The structures of
human dihydroorotate dehydrogenase with and without inhibitor
reveal conformational flexibility in the inhibitor and substrate bind-
ing sites. Biochemistry, 2008, 47(34), 8929-8936.
[37]Baumgartner, R.; Walloschek, M.; Kralik, M.; Gotschlich, A.;
Tasler, S.; Mies, J.; Leban, J. Dual binding mode of a novel series
of DHODH inhibitors. J. Med. Chem., 2006, 49(4), 1239-1247. [38]Zameitat, E.; Gojkovic, Z.; Knecht, W.; Piskur J.; Loffler M. Bio-
chemical characterization of recombinant dihydroorotate dehydro-
genase from the opportunistic pathogenic yeast Candida albicans.
FEBS Journal, 2006, 273, 3183–3191.
[39]Neidhardt, E.A.; Punreddy, S.R.; McLean, J.E.; Hedstrom, L.;
Grossman, T.H. Expression and Characterization of E. coli-
produced Soluble, Functional Human Dihydroorotate Dehydro-
genase: a Potential Target for Immunosuppression. J. Molec. Mi-
crobiol. Biotechnol., 1999, 1(1), 183-188.
[40]Zameitat, E.; Freymark, G.; Dietz, C.D.; Loffler, M.; Bolker, M.
Functional Expression of Human Dihydroorotate Dehydrogenase
(DHODH) in pyr4 Mutants of Ustilago maydis Allows Target
Validation of DHODH Inhibitors In Vivo. Appl. Environ. Micro-
biol., 2007, 73(10), 3371–3379.
[41]Pinheiro, M.P.; Iulek, J.; M. Nonato C. Crystal structure of Try-
panosoma cruzi Dihydroorotate dehydrogenase from Y strain. Bio-
chem. Biophys. Res. Commun., 2008, 369, 812–817.
[42]Bennett, L. L Jr.; Smithers, D.; Rose, L.M.; Adamson, D.J.; Tho-
mas. H.J. Inhibition of Synthesis of Pyrimidine Nucleotides by 2-
Hydroxy-3-(3,3-dichloroallyl) -1 ,4-naphthoquinone. Cancer Res.,
1979, 39, 4868-4874.
[43]Cleaveland, E.S.; Zaharevitz, D.W.; Kelley, J.A.; Paull, K.;
Cooney, D.A.; Ford, H. Jr. Identification of a novel inhibitor (NSC
665564) of dihydroorotate dehydrogenase with a potency equiva-
lent to brequinar. Biochem. Biophys. Res. Commun., 1996, 223(3),
654-659.
[44]Davis, J.P.; Cain, G.A.; Pitts, W.J.; Magolda, R.L.; Copeland, R.A.
The immunosuppressive metabolite of leflunomide is a potent in-
hibitor of human dihydroorotate dehydrogenase. Biochemistry,
1996, 35(4), 1270-1273.
[45]Williamson, R.A.; Yea, C.M.; Robson, P.A.; Curnock, A.P.; Gad-
her, S.; Hambleton, A.B.; Woodward, K.; Bruneau, J.M., Hamble-
ton, P.; Moss, D., Thomson, T.A.; Spinella-Jaegle, S.; Morand, P.;
Courtin, O.; Sautes, C.; Westwood, R.; Hercend, T.; Kuo, E. A.;
Ruuth, E. Dihydroorotate dehydrogenase is a high affinity binding
protein for A77 1726 and mediator of a range of biological effects
of the immunomodulatory compound. J. Biol. Chem., 1995, 270,
22467–22472.
[46]Phillips, M. A.; Gujjar R.; Malmquist, N.A.; White, J.; Mazouni,
F.E.; Baldwin, J.; Rathod, P.K. Triazolopyrimidine-based dihydro-
orotate dehydrogenase inhibitors with potent and selective activity
against the malaria parasite Plasmodium falciparum. J. Med.
Chem., 2008, 51, 3649–3653.
[47]Gujjar, R.; Marwaha, A.; Mazouni, F.E.; White, J.; White, K.L.;
Creason, S.; Shackleford, D.M.; Baldwin, J.; Charman, W.N.;
Buckner, F.S.; Charman, S.; Rathod, P.K.; Phillips, M.A. Identifi-
cation of a metabolically stable triazolopyrimidine-based dihydro-
orotate dehydrogenase inhibitor with antimalarial activity in mice.
J. Med. Chem.2009,52, 1864–1872.
[48]Phillips, M. A Rathod, P.K.Plasmodium dihydroorotate dehydro-
genase: a promising target for novel anti-malarial chemotherapy.
Infectious Disorders - Drug Targets, 2010, 10, 226-239.
[49]Deng, X.; Gujjar, R.; El Mazouni, F.; Kaminsky, W.; Malmquist,
N.A.; Goldsmith, E.J.; Rathod, P.K.; Phillips, M.A. Structural plas-
ticity of malaria dihydroorotate dehydrogenase allows selective
binding of diverse chemical scaffolds. J. Biol. Chem., 2009,
284(39), 26999-27009.
[50]Ojha, P.K.; Roy K.Chemometric modeling, docking and in silico
design of triazolopyrimidine-based dihydroorotate dehydrogenase
inhibitors as antimalarials. Eu. J. Med. Chem., 2010, 45, 4645-
4656.
[51]Heikkila T.; Ramsey, C.; Davies, M.; Galtier, C.; Stead, A.M.W.,
Johnson, A. P.; Fishwick, C.W. G. Boa, A.N.; McConke G.A. De-
sign and Synthesis of Potent Inhibitors of the Malaria Parasite Di-
hydroorotate Dehydrogenase J. Med. Chem., 2007, 50, 186-191. [52]Leban, J.; Saeb, W.; Garcia, G.; Baumgartner R.; Kramer, B.Dis-
covery of a novel series of DHODH inhibitors by a docking proce-
dure and QSAR refinement. Bioorg. Med. Chem. Lett., 2004, 14,
55–58.
[53]Papageorgiou, C.; Matt, A.V.; Joergensen, J.; Andersen, E.; Wag-
ner, K.; Beerli, C.; Than, T.; Borer, X.; Florineth, A.; Rihs, G.;
Schreier, M.H.; Weckbecker, G.; Heusser C. Aromatic quinoline-
carboxamides as selective, orally active antibody production inhibi-
tors for prevention of acute xenograft rejection. J. Med. Chem.
2001,44, 1986-1992.
[54]Boa, A. N.; Canavan, S.P.; Hirst, P.R.; Ramsey, C.; Stead, A. M.
W.; McConkey, G.A. Synthesis of brequinar analogue inhibitors of
malaria parasite dihydroorotate dehydrogenase. Bioorg. Med.
Chem.,2005, 13, 1945–1967.
[55]Leban, J.; Kralik, M.; Mies, J.; Gassen, M.; Tentschert K.; Baum-
gartner R. SAR, species specificity, and cellular activity of cy-
clopentene dicarboxylic acid amides as DHODH inhibitors. Bioorg.
Med. Chem. Lett., 2005, 15, 4854–4857.
[56]Sutton, A.E.; Clardy, J. The synthesis of potentially selective in-
hibitors of dihydroorotate dehydrogenase. The utilization of che-
moselective Suzuki cross-coupling reactions in a parallel synthesis.
Tet. Lett.,2001, 42, 547–551.
[57]Hurt, D.E.; Suttona, A.E.; Clardy J. Brequinar derivatives and
species-specific drug design for dihydroorotate dehydrogenase.
Bioorg. Med. Chem. Lett., 2006, 16, 1610–1615.
[58]Kuo, P.Y.; Shie, T. L.; Chen, Y.S. Lai J.T.; Yang, D.Y. Enzyme
inhibition potency enhancement by active site metal chelating and
hydrogen bonding induced conformation restricted cyclopropane-
carbonyl derivatives. Bioorg. Med. Chem. Lett., 2006, 16, 6024–
6027.
[59]Heikkila, T.; Thirumalairajan, S.; Davies, M.; Parsons, M.R.;
McConkey A.G.; Fishwick, C.W. G.; Johnson, A.P. The first de
novo designed inhibitors of Plasmodium falciparum dihydroorotate
dehydrogenase. Bioorg. Med. Chem. Lett., 2006, 16, 88–92.
[60]Leban, J.; Kralik, M.; Mies, J.; Baumgartner, R.; Gassen M.; Tasler
S. Biphenyl-4-ylcarbamoyl thiophene carboxylic acids as potent
DHODH inhibitors. Bioorg. Med. Chem. Lett., 2006, 16, 267–270.
[61]McLean L. R.; Zhang, Y.; Degnen W.; Peppard, J.; Cabel, D.; Zou,
C.; Tsay, J. T.; Subramaniam, A.; Vaz, R. J.; Li Y. Discovery of
novel inhibitors for DHODH via virtual screening and X-ray crys-
tallographic structures. Bioorg. Med. Chem. Lett., 2010, 20, 1981–
1984.
[62]Cowen, D.; Bedingfield, P.; McConkey G.A.; Fishwick, C.W.G.;
Johnson, A. P. A study of the effects of substituents on the selectiv-
ity of the binding of N-arylaminomethylene malonate inhibitors to
DHODH. Bioorg. Med. Chem. Lett., 2010, 20, 1284–1287.
[63]Fritzson, I.; Svensson, B.; Al-Karadaghi, S.; Walse, B.; Wellmar,
U.; Nilsson, U.; Thrige, J. D.G. Jonsson, S. Inhibition of human
DHODH by 4-hydroxycoumarins, fenamic acids, and N-
(alkylcarbonyl)anthranilic acids identified by structure-guided
fragment selection. Chem. Med. Chem., 2010, 5(4), 608–617.
[64]Cheleski, J.; Rocha J.R.; Pinheiro, M. P.; Wiggers, H. J.; Da Silva
A.B.F.; Nonato M.C.; Montanari C.A. Novel insights for dihydro-
orotate dehydrogenase class 1A inhibitors discovery. Eu. J. Med.
Chem. 2010, 45, 5899-5909.
[65]Booker, M.L.; Bastos C.M.; Kramer, M.L.;Barker, R.H.; Skerlj,
R.; Sidhu, A.B.; Deng, X.; Celatka C.;Cortese, J.F.;Bravo, J.E.G.;
Llado, K. N.C.; Serrano, A..E.;Barturen, I.A.;Jimenez-Díaz, M.B.;
Viera, S.;Garutn, H.; Wittlin, S.; Papastogiannidis, P.; Lin, J.W.;
Janse, C.J.; Khan, S.M.; Duraisingh, M.; Coleman, B.; Goldsmith,
E.J.; Phillips, M.A.; Munoz, B.; Wirth, D.
F.; Klinger J.D.;Wie-
gand, R.; Sybertz, E. Novel inhibitors of Plasmodium falciparum
dihydroorotate dehydrogenase with anti-malarial activity in the
mouse model. J. Biol. Chem., 2010 285(43), 33054–33064.
Recent Developments in the Medicinal Chemistry Mini-Reviews in Medicinal Chemistry, 2011, Vol. 11, No. 12 1055
[66]Patel, V.; Booker, M.; Kramer, M.; Ross, L.; Celatka, C.A.; Ken-
nedy, L.M.; Dvorin, J.D.; Duraisingh, M.T.; Sliz, P.; Wirth, D.F.;
Clardy, J. Identification and characterization of small molecule in-
hibitors of Plasmodium falciparum dihydroorotate dehydrogenase.
J. Biol. Chem., 2008, 283(50), 35078-35085.
[67]Castro, P.L.J.C.; Erra, S.M.; Lozoya, T.M.E.; Navarro, R.E.;
Amino nicotinic acid and isonicotinic acid derivative as DHODH
inhibitor., U S patent 0074898, March 25, 2010. [68]Baldwin, J.; Michnoff, C. H.; Malmquist, N. A.; White, J.; Roth
M.G.; Rathod, P.K. Phillips M. A. High-throughput screening for
potent and selective inhibitors of plasmodium falciparum dihydro-
orotate dehydrogenase. J. Biol. Chem., 2005, 280(23), 21847–
21853.
[69]Seifert, M.H.J. Assessing the discriminatory power of scoring
functions for virtual screening. J. Chem. Inf. Model.,2006, 46,
1456-1465.
Received: February 20, 2011 Revised: April 15, 2011 Accepted: June 05, 2011
The way常见用法
The way 的用法 Ⅰ常见用法: 1)the way+ that 2)the way + in which(最为正式的用法) 3)the way + 省略(最为自然的用法) 举例:I like the way in which he talks. I like the way that he talks. I like the way he talks. Ⅱ习惯用法: 在当代美国英语中,the way用作为副词的对格,“the way+ 从句”实际上相当于一个状语从句来修饰整个句子。 1)The way =as I am talking to you just the way I’d talk to my own child. He did not do it the way his friends did. Most fruits are naturally sweet and we can eat them just the way they are—all we have to do is to clean and peel them. 2)The way= according to the way/ judging from the way The way you answer the question, you are an excellent student. The way most people look at you, you’d think trash man is a monster. 3)The way =how/ how much No one can imagine the way he missed her. 4)The way =because
The way的用法及其含义(二)
The way的用法及其含义(二) 二、the way在句中的语法作用 the way在句中可以作主语、宾语或表语: 1.作主语 The way you are doing it is completely crazy.你这个干法简直发疯。 The way she puts on that accent really irritates me. 她故意操那种口音的样子实在令我恼火。The way she behaved towards him was utterly ruthless. 她对待他真是无情至极。 Words are important, but the way a person stands, folds his or her arms or moves his or her hands can also give us information about his or her feelings. 言语固然重要,但人的站姿,抱臂的方式和手势也回告诉我们他(她)的情感。 2.作宾语 I hate the way she stared at me.我讨厌她盯我看的样子。 We like the way that her hair hangs down.我们喜欢她的头发笔直地垂下来。 You could tell she was foreign by the way she was dressed. 从她的穿著就可以看出她是外国人。 She could not hide her amusement at the way he was dancing. 她见他跳舞的姿势,忍俊不禁。 3.作表语 This is the way the accident happened.这就是事故如何发生的。 Believe it or not, that's the way it is. 信不信由你, 反正事情就是这样。 That's the way I look at it, too. 我也是这么想。 That was the way minority nationalities were treated in old China. 那就是少数民族在旧中
(完整版)the的用法
定冠词the的用法: 定冠词the与指示代词this ,that同源,有“那(这)个”的意思,但较弱,可以和一个名词连用,来表示某个或某些特定的人或东西. (1)特指双方都明白的人或物 Take the medicine.把药吃了. (2)上文提到过的人或事 He bought a house.他买了幢房子. I've been to the house.我去过那幢房子. (3)指世界上独一无二的事物 the sun ,the sky ,the moon, the earth (4)单数名词连用表示一类事物 the dollar 美元 the fox 狐狸 或与形容词或分词连用,表示一类人 the rich 富人 the living 生者 (5)用在序数词和形容词最高级,及形容词等前面 Where do you live?你住在哪? I live on the second floor.我住在二楼. That's the very thing I've been looking for.那正是我要找的东西. (6)与复数名词连用,指整个群体 They are the teachers of this school.(指全体教师) They are teachers of this school.(指部分教师) (7)表示所有,相当于物主代词,用在表示身体部位的名词前 She caught me by the arm.她抓住了我的手臂. (8)用在某些有普通名词构成的国家名称,机关团体,阶级等专有名词前 the People's Republic of China 中华人民共和国 the United States 美国 (9)用在表示乐器的名词前 She plays the piano.她会弹钢琴. (10)用在姓氏的复数名词之前,表示一家人 the Greens 格林一家人(或格林夫妇) (11)用在惯用语中 in the day, in the morning... the day before yesterday, the next morning... in the sky... in the dark... in the end... on the whole, by the way...
“the way+从句”结构的意义及用法
“theway+从句”结构的意义及用法 首先让我们来看下面这个句子: Read the followingpassageand talkabout it wi th your classmates.Try totell whatyou think of Tom and ofthe way the childrentreated him. 在这个句子中,the way是先行词,后面是省略了关系副词that或in which的定语从句。 下面我们将叙述“the way+从句”结构的用法。 1.the way之后,引导定语从句的关系词是that而不是how,因此,<<现代英语惯用法词典>>中所给出的下面两个句子是错误的:This is thewayhowithappened. This is the way how he always treats me. 2.在正式语体中,that可被in which所代替;在非正式语体中,that则往往省略。由此我们得到theway后接定语从句时的三种模式:1) the way+that-从句2)the way +in which-从句3) the way +从句 例如:The way(in which ,that) thesecomrade slookatproblems is wrong.这些同志看问题的方法
不对。 Theway(that ,in which)you’re doingit is comple tely crazy.你这么个干法,简直发疯。 Weadmired him for theway inwhich he facesdifficulties. Wallace and Darwingreed on the way inwhi ch different forms of life had begun.华莱士和达尔文对不同类型的生物是如何起源的持相同的观点。 This is the way(that) hedid it. I likedthe way(that) sheorganized the meeting. 3.theway(that)有时可以与how(作“如何”解)通用。例如: That’s the way(that) shespoke. = That’s how shespoke.
way 用法
表示“方式”、“方法”,注意以下用法: 1.表示用某种方法或按某种方式,通常用介词in(此介词有时可省略)。如: Do it (in) your own way. 按你自己的方法做吧。 Please do not talk (in) that way. 请不要那样说。 2.表示做某事的方式或方法,其后可接不定式或of doing sth。 如: It’s the best way of studying [to study] English. 这是学习英语的最好方法。 There are different ways to do [of doing] it. 做这事有不同的办法。 3.其后通常可直接跟一个定语从句(不用任何引导词),也可跟由that 或in which 引导的定语从句,但是其后的从句不能由how 来引导。如: 我不喜欢他说话的态度。 正:I don’t like the way he spoke. 正:I don’t like the way that he spoke. 正:I don’t like the way in which he spoke. 误:I don’t like the way how he spoke. 4.注意以下各句the way 的用法: That’s the way (=how) he spoke. 那就是他说话的方式。 Nobody else loves you the way(=as) I do. 没有人像我这样爱你。 The way (=According as) you are studying now, you won’tmake much progress. 根据你现在学习情况来看,你不会有多大的进步。 2007年陕西省高考英语中有这样一道单项填空题: ——I think he is taking an active part insocial work. ——I agree with you_____. A、in a way B、on the way C、by the way D、in the way 此题答案选A。要想弄清为什么选A,而不选其他几项,则要弄清选项中含way的四个短语的不同意义和用法,下面我们就对此作一归纳和小结。 一、in a way的用法 表示:在一定程度上,从某方面说。如: In a way he was right.在某种程度上他是对的。注:in a way也可说成in one way。 二、on the way的用法 1、表示:即将来(去),就要来(去)。如: Spring is on the way.春天快到了。 I'd better be on my way soon.我最好还是快点儿走。 Radio forecasts said a sixth-grade wind was on the way.无线电预报说将有六级大风。 2、表示:在路上,在行进中。如: He stopped for breakfast on the way.他中途停下吃早点。 We had some good laughs on the way.我们在路上好好笑了一阵子。 3、表示:(婴儿)尚未出生。如: She has two children with another one on the way.她有两个孩子,现在还怀着一个。 She's got five children,and another one is on the way.她已经有5个孩子了,另一个又快生了。 三、by the way的用法
The way的用法及其含义(一)
The way的用法及其含义(一) 有这样一个句子:In 1770 the room was completed the way she wanted. 1770年,这间琥珀屋按照她的要求完成了。 the way在句中的语法作用是什么?其意义如何?在阅读时,学生经常会碰到一些含有the way 的句子,如:No one knows the way he invented the machine. He did not do the experiment the way his teacher told him.等等。他们对the way 的用法和含义比较模糊。在这几个句子中,the way之后的部分都是定语从句。第一句的意思是,“没人知道他是怎样发明这台机器的。”the way的意思相当于how;第二句的意思是,“他没有按照老师说的那样做实验。”the way 的意思相当于as。在In 1770 the room was completed the way she wanted.这句话中,the way也是as的含义。随着现代英语的发展,the way的用法已越来越普遍了。下面,我们从the way的语法作用和意义等方面做一考查和分析: 一、the way作先行词,后接定语从句 以下3种表达都是正确的。例如:“我喜欢她笑的样子。” 1. the way+ in which +从句 I like the way in which she smiles. 2. the way+ that +从句 I like the way that she smiles. 3. the way + 从句(省略了in which或that) I like the way she smiles. 又如:“火灾如何发生的,有好几种说法。” 1. There were several theories about the way in which the fire started. 2. There were several theories about the way that the fire started.
way 的用法
way 的用法 【语境展示】 1. Now I’ll show you how to do the experiment in a different way. 下面我来演示如何用一种不同的方法做这个实验。 2. The teacher had a strange way to make his classes lively and interesting. 这位老师有种奇怪的办法让他的课生动有趣。 3. Can you tell me the best way of working out this problem? 你能告诉我算出这道题的最好方法吗? 4. I don’t know the way (that / in which) he helped her out. 我不知道他用什么方法帮助她摆脱困境的。 5. The way (that / which) he talked about to solve the problem was difficult to understand. 他所谈到的解决这个问题的方法难以理解。 6. I don’t like the way that / which is being widely used for saving water. 我不喜欢这种正在被广泛使用的节水方法。 7. They did not do it the way we do now. 他们以前的做法和我们现在不一样。 【归纳总结】 ●way作“方法,方式”讲时,如表示“以……方式”,前面常加介词in。如例1; ●way作“方法,方式”讲时,其后可接不定式to do sth.,也可接of doing sth. 作定语,表示做某事的方法。如例2,例3;
the-way-的用法讲解学习
t h e-w a y-的用法
The way 的用法 "the way+从句"结构在英语教科书中出现的频率较高, the way 是先行词, 其后是定语从句.它有三种表达形式:1) the way+that 2)the way+ in which 3)the way + 从句(省略了that或in which),在通常情况下, 用in which 引导的定语从句最为正式,用that的次之,而省略了关系代词that 或 in which 的, 反而显得更自然,最为常用.如下面三句话所示,其意义相同. I like the way in which he talks. I like the way that he talks. I like the way he talks. 一.在当代美国英语中,the way用作为副词的对格,"the way+从句"实际上相当于一个状语从句来修饰全句. the way=as 1)I'm talking to you just the way I'd talk to a boy of my own. 我和你说话就象和自己孩子说话一样. 2)He did not do it the way his friend did. 他没有象他朋友那样去做此事. 3)Most fruits are naturally sweet and we can eat them just the way they are ----all we have to do is clean or peel them . 大部分水果天然甜润,可以直接食用,我们只需要把他们清洗一下或去皮.
way的用法总结大全
way的用法总结大全 way的用法你知道多少,今天给大家带来way的用法,希望能够帮助到大家,下面就和大家分享,来欣赏一下吧。 way的用法总结大全 way的意思 n. 道路,方法,方向,某方面 adv. 远远地,大大地 way用法 way可以用作名词 way的基本意思是“路,道,街,径”,一般用来指具体的“路,道路”,也可指通向某地的“方向”“路线”或做某事所采用的手段,即“方式,方法”。way还可指“习俗,作风”“距离”“附近,周围”“某方面”等。 way作“方法,方式,手段”解时,前面常加介词in。如果way前有this, that等限定词,介词可省略,但如果放在句首,介词则不可省略。
way作“方式,方法”解时,其后可接of v -ing或to- v 作定语,也可接定语从句,引导从句的关系代词或关系副词常可省略。 way用作名词的用法例句 I am on my way to the grocery store.我正在去杂货店的路上。 We lost the way in the dark.我们在黑夜中迷路了。 He asked me the way to London.他问我去伦敦的路。 way可以用作副词 way用作副词时意思是“远远地,大大地”,通常指在程度或距离上有一定的差距。 way back表示“很久以前”。 way用作副词的用法例句 It seems like Im always way too busy with work.我工作总是太忙了。 His ideas were way ahead of his time.他的思想远远超越了他那个时代。 She finished the race way ahead of the other runners.她第一个跑到终点,远远领先于其他选手。 way用法例句
the_way的用法大全教案资料
t h e_w a y的用法大全
The way 在the way+从句中, the way 是先行词, 其后是定语从句.它有三种表达形式:1) the way+that 2)the way+ in which 3)the way + 从句(省略了that或in which),在通常情况下, 用in which 引导的定语从句最为正式,用that的次之,而省略了关系代词that 或 in which 的, 反而显得更自然,最为常用.如下面三句话所示,其意义相同. I like the way in which he talks. I like the way that he talks. I like the way he talks. 如果怕弄混淆,下面的可以不看了 另外,在当代美国英语中,the way用作为副词的对格,"the way+从句"实际上相当于一个状语从句来修饰全句. the way=as 1)I'm talking to you just the way I'd talk to a boy of my own. 我和你说话就象和自己孩子说话一样. 2)He did not do it the way his friend did. 他没有象他朋友那样去做此事. 3)Most fruits are naturally sweet and we can eat them just the way they are ----all we have to do is clean or peel them . 大部分水果天然甜润,可以直接食用,我们只需要把他们清洗一下或去皮. the way=according to the way/judging from the way 4)The way you answer the qquestions, you must be an excellent student. 从你回答就知道,你是一个优秀的学生. 5)The way most people look at you, you'd think a trashman was a monster. 从大多数人看你的目光中,你就知道垃圾工在他们眼里是怪物. the way=how/how much 6)I know where you are from by the way you pronounce my name. 从你叫我名字的音调中,我知道你哪里人. 7)No one can imaine the way he misses her. 人们很想想象他是多么想念她. the way=because 8) No wonder that girls looks down upon me, the way you encourage her. 难怪那姑娘看不起我, 原来是你怂恿的
the way 的用法
The way 的用法 "the way+从句"结构在英语教科书中出现的频率较高, the way 是先行词, 其后是定语从句.它有三种表达形式:1) the way+that 2)the way+ in which 3)the way + 从句(省略了that或in which),在通常情况下, 用in which 引导的定语从句最为正式,用that的次之,而省略了关系代词that 或in which 的, 反而显得更自然,最为常用.如下面三句话所示,其意义相同. I like the way in which he talks. I like the way that he talks. I like the way he talks. 一.在当代美国英语中,the way用作为副词的对格,"the way+从句"实际上相当于一个状语从句来修饰全句. the way=as 1)I'm talking to you just the way I'd talk to a boy of my own. 我和你说话就象和自己孩子说话一样. 2)He did not do it the way his friend did. 他没有象他朋友那样去做此事. 3)Most fruits are naturally sweet and we can eat them just the way they are ----all we have to do is clean or peel them . 大部分水果天然甜润,可以直接食用,我们只需要把他们清洗一下或去皮.
the way=according to the way/judging from the way 4)The way you answer the qquestions, you must be an excellent student. 从你回答就知道,你是一个优秀的学生. 5)The way most people look at you, you'd think a trashman was a monster. 从大多数人看你的目光中,你就知道垃圾工在他们眼里是怪物. the way=how/how much 6)I know where you are from by the way you pronounce my name. 从你叫我名字的音调中,我知道你哪里人. 7)No one can imaine the way he misses her. 人们很想想象他是多么想念她. the way=because 8) No wonder that girls looks down upon me, the way you encourage her. 难怪那姑娘看不起我, 原来是你怂恿的 the way =while/when(表示对比) 9)From that day on, they walked into the classroom carrying defeat on their shoulders the way other students carried textbooks under their arms. 从那天起,其他同学是夹着书本来上课,而他们却带着"失败"的思想负担来上课.
The way的用法及其含义(三)
The way的用法及其含义(三) 三、the way的语义 1. the way=as(像) Please do it the way I’ve told you.请按照我告诉你的那样做。 I'm talking to you just the way I'd talk to a boy of my own.我和你说话就像和自己孩子说话一样。 Plant need water the way they need sun light. 植物需要水就像它们需要阳光一样。 2. the way=how(怎样,多么) No one can imagine the way he misses her.没人能够想象出他是多么想念她! I want to find out the way a volcano has formed.我想弄清楚火山是怎样形成的。 He was filled with anger at the way he had been treated.他因遭受如此待遇而怒火满腔。That’s the way she speaks.她就是那样讲话的。 3. the way=according as (根据) The way you answer the questions, you must be an excellent student.从你回答问题来看,你一定是名优秀的学生。 The way most people look at you, you'd think a trash man was a monster.从大多数人看你的目光中,你就知道垃圾工在他们眼里是怪物。 The way I look at it, it’s not what you do that matters so much.依我看,重要的并不是你做什么。 I might have been his son the way he talked.根据他说话的样子,好像我是他的儿子一样。One would think these men owned the earth the way they behave.他们这样行动,人家竟会以为他们是地球的主人。
way的用法
一.Way:“方式”、“方法” 1.表示用某种方法或按某种方式 Do it (in) your own way. Please do not talk (in) that way. 2.表示做某事的方式或方法 It’s the best way of studying [to study] English.。 There are different ways to do [of doing] it. 3.其后通常可直接跟一个定语从句(不用任何引导词),也可跟由that 或in which 引导的定语从句 正:I don’t like the way he spoke. I don’t like the way that he spoke. I don’t like the way in which he spoke.误:I don’t like the way how he spoke. 4. the way 的从句 That’s the way (=how) he spoke. I know where you are from by the way you pronounce my name. That was the way minority nationalities were treated in old China. Nobody else loves you the way(=as) I do. He did not do it the way his friend did. 二.固定搭配 1. In a/one way:In a way he was right. 2. In the way /get in one’s way I'm afraid your car is in the way, If you are not going to help,at least don't get in the way. You'll have to move-you're in my way. 3. in no way Theory can in no way be separated from practice. 4. On the way (to……) Let’s wait a few moments. He is on the way Spring is on the way. Radio forecasts said a sixth-grade wind was on the way. She has two children with another one on the way. 5. By the way By the way,do you know where Mary lives? 6. By way of Learn English by way of watching US TV series. 8. under way 1. Elbow one’s way He elbowed his way to the front of the queue. 2. shoulder one’s way 3. feel one‘s way 摸索着向前走;We couldn’t see anything in the cave, so we had to feel our way out 4. fight/force one’s way 突破。。。而前进The surrounded soldiers fought their way out. 5.. push/thrust one‘s way(在人群中)挤出一条路He pushed his way through the crowd. 6. wind one’s way 蜿蜒前进 7. lead the way 带路,领路;示范 8. lose one‘s way 迷失方向 9. clear the way 排除障碍,开路迷路 10. make one’s way 前进,行进The team slowly made their way through the jungle.
the way的用法大全
在the way+从句中, the way 是先行词, 其后是定语从句.它有三种表达形式:1) the way+that 2)the way+ in which 3)the way + 从句(省略了that或in which),在通常情况下, 用in which 引导的定语从句最为正式,用that的次之,而省略了关系代词that 或in which 的, 反而显得更自然,最为常用.如下面三句话所示,其意义相同. I like the way in which he talks. I like the way that he talks. I like the way he talks. 如果怕弄混淆,下面的可以不看了 另外,在当代美国英语中,the way用作为副词的对格,"the way+从句"实际上相当于一个状语从句来修饰全句. the way=as 1)I'm talking to you just the way I'd talk to a boy of my own. 我和你说话就象和自己孩子说话一样. 2)He did not do it the way his friend did. 他没有象他朋友那样去做此事. 3)Most fruits are naturally sweet and we can eat them just the way they are ----all we have to do is clean or peel them . 大部分水果天然甜润,可以直接食用,我们只需要把他们清洗一下或去皮. the way=according to the way/judging from the way 4)The way you answer the qquestions, you must be an excellent student. 从你回答就知道,你是一个优秀的学生. 5)The way most people look at you, you'd think a trashman was a monster. 从大多数人看你的目光中,你就知道垃圾工在他们眼里是怪物. the way=how/how much 6)I know where you are from by the way you pronounce my name. 从你叫我名字的音调中,我知道你哪里人. 7)No one can imaine the way he misses her. 人们很想想象他是多么想念她. the way=because 8) No wonder that girls looks down upon me, the way you encourage her. 难怪那姑娘看不起我, 原来是你怂恿的 the way =while/when(表示对比) 9)From that day on, they walked into the classroom carrying defeat on their shoulders the way other students carried textbooks under their arms.
“the-way+从句”结构的意义及用法知识讲解
“the way+从句”结构的意义及用法 首先让我们来看下面这个句子: Read the following passage and talk about it with your classmates. Try to tell what you think of Tom and of the way the children treated him. 在这个句子中,the way是先行词,后面是省略了关系副词that 或in which的定语从句。 下面我们将叙述“the way+从句”结构的用法。 1.the way之后,引导定语从句的关系词是that而不是how,因此,<<现代英语惯用法词典>>中所给出的下面两个句子是错误的:This is the way how it happened. This is the way how he always treats me. 2. 在正式语体中,that可被in which所代替;在非正式语体中,that则往往省略。由此我们得到the way后接定语从句时的三种模式:1) the way +that-从句2) the way +in which-从句3) the way +从句 例如:The way(in which ,that) these comrades look at problems is wrong.这些同志看问题的方法不对。
The way(that ,in which)you’re doing it is completely crazy.你这么个干法,简直发疯。 We admired him for the way in which he faces difficulties. Wallace and Darwin greed on the way in which different forms of life had begun.华莱士和达尔文对不同类型的生物是如何起源的持相同的观点。 This is the way (that) he did it. I liked the way (that) she organized the meeting. 3.the way(that)有时可以与how(作“如何”解)通用。例如: That’s the way (that) she spoke. = That’s how she spoke. I should like to know the way/how you learned to master the fundamental technique within so short a time. 4.the way的其它用法:以上我们讲的都是用作先行词的the way,下面我们将叙述它的一些用法。
定冠词the的12种用法
定冠词the的12种用法 定冠词the 的12 种用法,全知道?快来一起学习吧。下面就和大家分享,来欣赏一下吧。 定冠词the 的12 种用法,全知道? 定冠词the用在各种名词前面,目的是对这个名词做个记号,表示它的特指属性。所以在词汇表中,定冠词the 的词义是“这个,那个,这些,那些”,可见,the 即可以放在可数名词前,也可以修饰不可数名词,the 后面的名词可以是单数,也可以是复数。 定冠词的基本用法: (1) 表示对某人、某物进行特指,所谓的特指就是“不是别的,就是那个!”如: The girl with a red cap is Susan. 戴了个红帽子的女孩是苏珊。 (2) 一旦用到the,表示谈话的俩人都知道说的谁、说的啥。如:
The dog is sick. 狗狗病了。(双方都知道是哪一只狗) (3) 前面提到过的,后文又提到。如: There is a cat in the tree.Thecat is black. 树上有一只猫,猫是黑色的。 (4) 表示世界上唯一的事物。如: The Great Wall is a wonder.万里长城是个奇迹。(5) 方位名词前。如: thenorth of the Yangtze River 长江以北地区 (6) 在序数词和形容词最高级的前面。如: Who is the first?谁第一个? Sam is the tallest.山姆最高。 但是不能认为,最高级前必须加the,如: My best friend. 我最好的朋友。 (7) 在乐器前。如: play the flute 吹笛子
Way的用法
Way用法 A:I think you should phone Jenny and say sorry to her. B:_______. It was her fault. A. No way B. Not possible C. No chance D. Not at all 说明:正确答案是A. No way,意思是“别想!没门!决不!” 我认为你应该打电话给珍妮并向她道歉。 没门!这是她的错。 再看两个关于no way的例句: (1)Give up our tea break? NO way! 让我们放弃喝茶的休息时间?没门儿! (2)No way will I go on working for that boss. 我决不再给那个老板干了。 way一词含义丰富,由它构成的短语用法也很灵活。为了便于同学们掌握和用好它,现结合实例将其用法归纳如下: 一、way的含义 1. 路线
He asked me the way to London. 他问我去伦敦的路。 We had to pick our way along the muddy track. 我们不得不在泥泞的小道上择路而行。 2. (沿某)方向 Look this way, please. 请往这边看。 Kindly step this way, ladies and gentlemen. 女士们、先生们,请这边走。 Look both ways before crossing the road. 过马路前向两边看一看。 Make sure that the sign is right way up. 一定要把符号的上下弄对。 3. 道、路、街,常用以构成复合词 a highway(公路),a waterway(水路),a railway(铁路),wayside(路边)
way与time的特殊用法
way/time的特殊用法 1、当先行词是way意思为”方式.方法”的时候,引导定语从句的关系词有下列3种形式: Way在从句中做宾语 The way that / which he explained to us is quite simple. Way在从句中做状语 The way t hat /in which he explained the sentence to us is quite simple. 2、当先行词是time时,若time表示次数时,应用关系代词that引导定语从句,that可以省略; 若time表示”一段时间”讲时,应用关系副词when或介词at/during + which引导定语从句 1.Is this factory _______ we visited last year? 2.Is this the factory-------we visited last year? A. where B in which C the one D which 3. This is the last time _________ I shall give you a lesson. A. when B that C which D in which 4.I don’t like the way ________ you laugh at her. A . that B on which C which D as 5.He didn’t understand the wa y ________ I worked out the problem. A which B in which C where D what 6.I could hardly remember how many times----I’ve failed. A that B which C in which D when 7.This is the second time--------the president has visited the country. A which B where C that D in which 8.This was at a time------there were no televisions, no computers or radios. A what B when C which D that