药物分子设计

摘要
本文简单介绍了定量药物关系与药物分子设计的关系、定量药物关系的分类、定量药物关系的研究步骤。主要介绍了二维定量药物关系的研究方法,相关参数及数学模型。研究方法最常用的是HANSCH方程,Free-Wilson方法。构成定量构效关系的两大要素是结构参数和活性参数,常见的结构参数有:疏水参数、电性参数、立体参数、几何参数、拓扑参数、理化性质参数以及纯粹的结构参数等,大部分的参数是有标准表可查到的,少数则需要计算转化才能得到。常见的活性参数有:半数有效量、半数有效浓度、半数抑菌浓度、半数致死量、最小抑菌浓度等。最后介绍了定量构效关系的应用和发展前景。
关键词药物分子设计定量药物关系二维定量药物关系HANSCH方程,Free-Wilson方法结构参数活性参数应用发展前景
Abstract
Omit
1、引言
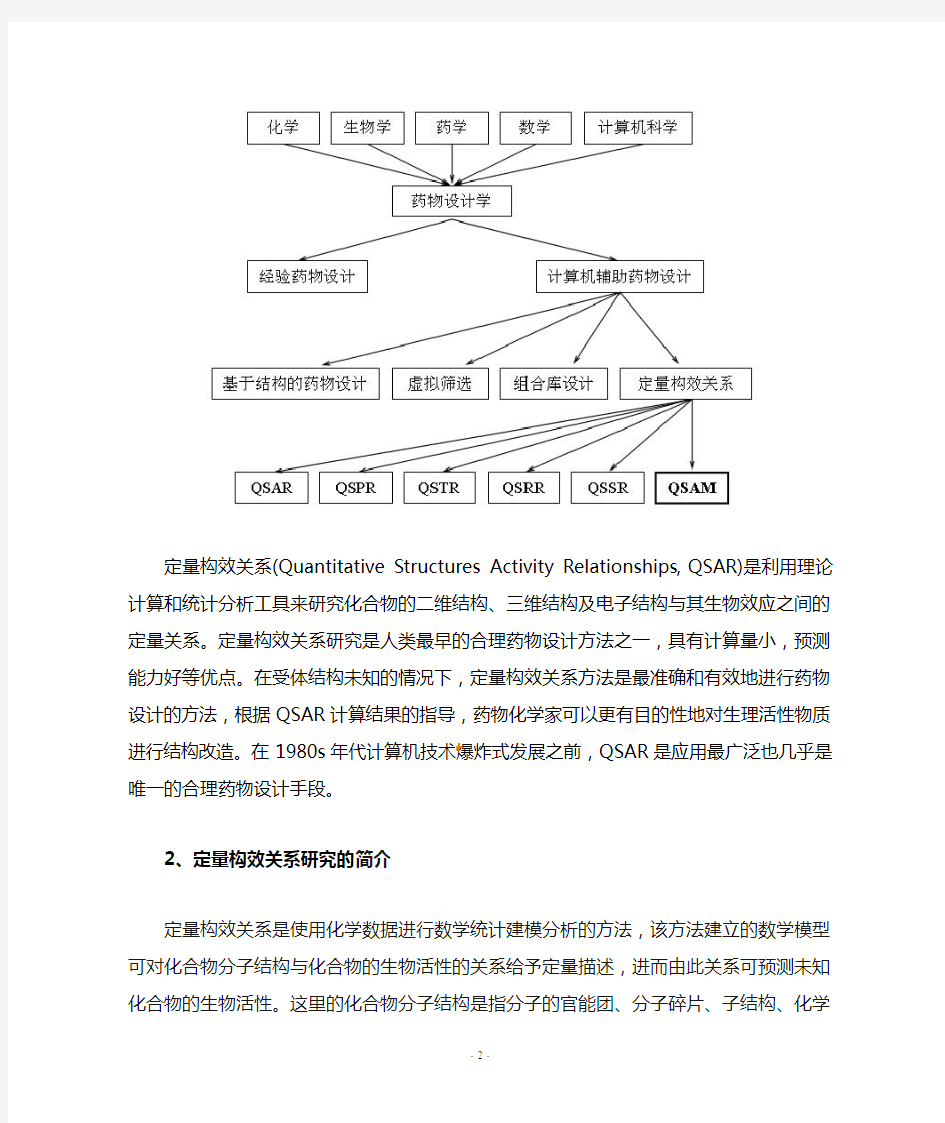
药物设计方法主要分为两种:一种是从小分子结构出发,通过改造、修饰等方法得到活性更好、毒性更低的新化合物,称为间接药物设计;另一种是从生物靶标大分子结构出发,寻找、设计能够与它发生相互作用并调节其功能的小分子,称为直接药物设计。间接药物设计包括定量构效关系(QSAR )和三维药效基团模型方法,直接药物设计分为分子对接和全新药物设计两种方法。
定量构效关系(Quantitative Structures Activity Relationships,QSAR)是利用理论计算和统计分析工具来研究化合物的二维结构、三维结构及电子结构与其生物效应之间的定量关系。定量构效关系研究是人类最早的合理药物设计方法之一,具有计算量小,预测能力好等优点。在受体结构未知的情况下,定量构效关系方法是最准确和有效地进行药物设计的方法,根据QSAR计算结果的指导,药物化学家可以更有目的性地对生理活性物质进行结构改造。在1980s年代计算机技术爆炸式发展之前,QSAR是应用最广泛也几乎是唯一的合理药物设计手段。
2、定量构效关系研究的简介
定量构效关系是使用化学数据进行数学统计建模分析的方法,该方法建立的数学模型可对化合物分子结构与化合物的生物活性的关系给予定量描述,进而由此关系可预测未知化合物的生物活性。这里的化合物分子结构是指分子的官能团、分子碎片、子结构、化学组成,一般可由物理化学、生物化学、量子化学等参数来描述。化合物的生物活性是指化合物的毒性、致癌、致变、致畸及生物降解、生物积累等性质。定量构效关系已经成为研究的一个热点方向,并被广泛应
用于药物设计、化合物毒性分析、生物降解性的预测等领域。定量构效关系的核心问题是采用有效算法构建定量构效关系的数学模型。目前,已有很多应用定量构效关系成功的例子。例如通过定量构效关系的研究,在蛋白质配基特别是酶抑制剂机理方面获得了很大成就,如胰蛋白酶,木瓜蛋白酶等许多酶的定量构效关系。一些定量构效关系新模型,新算法的提出以及新软件的开发,更进一步加快了定量构效关系的快速发展。例如,Accelrys公司开发了一系列计算定量构效关系的软件包,这些软件包内置了多个高质量的定量构效关系模型,利用预测准确性评估验证技术,能够对各类有机化合物的性质进行预测。由于该软件包预测结果可靠,已被包括美国FDA、加拿大环境部、加拿大健康部、美国Abbott实验室和美国LosAlamos国家实验室等单位广泛应用。科学家通过这些软件在计算机上进行基于生物活性小分子的结构的先导化合物寻找,活性构象假说的确定。定量构效关系研究的解决方案,为高质量的化合物库的设计合成提供指导,而且在实际的工作之前,还能够帮助科学家们对要进行设计的化合物进行药物代谢和动力学性质、毒理学性质的预测。定量构效关系帮助研究者辨明方向,提高效率。
3、定量构效关系的分类
定量构效关系研究大致可分为二维定量构效关系(2D-QSAR),三维定量构效关系(3D-QSAR)和多维定量构效关系。早在19世纪,很多研究人员就对化合物活性与其化学性质之间的定量关系进行了探讨。1900年前后,Mcyer和Overton 等提出了麻醉作用的类脂学说。其后几十年,随着Hansch法的提出,多变量解析技术的引入加之计算机技术的进步,QSAR方法才逐渐发展起来。现在QSAR 方法已成为药物设计的必备工具。在此仅介绍2D-QSAR的相关研究。
4、定量构效关系的研究步骤
QSAR的研究程序一般可以归纳为五步:
①获得待测试数据资料;
②选取结构参数及活性参数;
③建立活性参数和结构参数的定量关系模型;
④检验模型;
⑤实际应用和预测
QSAR研究中获得数据的方法主要有两种:一种是实验参数,通过实验测得;另一种理论参数则可通过计算分子的结构信息获取,或由文献资料中获得或者从数据库中下载。研究者认为理想的实验数据是在同样的实验条件下测得的,也可以是从同一个实验室或者同一个课题组的研究者测得的,这一观点是由Cronin 等人提出的。
5、二维定量构效关系方法
二维定量构效关系方法是将分子整体的结构性质作为参数,对分子生理活性进行回归分析,建立化学结构与生理活性相关性模型的一种药物设计方法,常见的二维定量构效关系方法有hansch方法、free-Wilson方法、分子连接性方法等,最为著名和应用最广泛的是hansch方法。
6、二维定量构效关系研究中的参数
1.生物活性的表示方法:活性参数
活性参数是构成二维定量构效关系的要素之一,活性参数必须是定量的描述数据,最好选用化合物对药物靶标的生物活性数据以便能更好地反应药物与受体的相互作用。人们根据研究的体系选择不同的活性参数,常见的活性参数有:半数有效量、半数有效浓度、半数抑菌浓度、半数致死量、最小抑菌浓度等,所有活性参数均必须采用物质的量作为计量单位,以便消除分子量的影响,从而真实地反应分子水平的生理活性。为了获得较好的数学模型,活性参数在二维定量构效关系中一般取负对数后进行统计分析。
2. 结构参数
结构参数是构成定量构效关系的另一大要素,常见的结构参数有:疏水参数、电性参数、立体参数、几何参数、拓扑参数、理化性质参数以及纯粹的结构参数等,大部分的参数是有标准表可查到的,少数则需要计算转化才能得到。
(1)疏水参数:药物在体内吸收和分布的过程与其疏水性密切相关,因而疏水性是影响药物生理活性的一个重要性质,在二维定量构效关系中采用的疏水参数最常见的是脂水分配系数,其定义为分子在正辛醇与水中分配的比例,对于分子母环上的取代基,脂水分配系数的对数值具有加和性,可以通过简单的代数计算获得某一取代结构的疏水参数。主要有脂水分配系数P;疏水常数π;还可用薄层色谱或纸色谱法化合物的Rf值表示疏水性,其中logP和π应用较多。
(2)电性参数:二维定量构效关系中的电性参数直接继承了哈密顿公式和塔夫托公式中的电性参数的定义,用以表征取代基团对分子整体电子分配的影响,其数值对于取代基也具有加和性。如电性常数(σ)是芳香族取代基最常使用的参数;直接共轭时的取代基电性效应常数σp-,σp+;取代基场效应常数F及共轭效应常数R ;Taft取代基常数σ* ;偶极矩(μ)、pKa、量子化学参数及波谱数据。可以使用的电性参数有多种,其中以σ和σ*使用的较多。
(3)立体参数:立体参数可以表征分子内部由于各个基团相互作用对药效构象产生的影响以及对药物和生物大分子结合模式产生的影响,常用的立体参数有塔夫托(Taft)立体参数Es、摩尔折射率MR、摩尔分子体积MV、摩尔分子等张比容Pr和范德华半径等。
(4)几何参数:几何参数是与分子构象相关的立体参数,因为这类参数常常在定量构效关系中占据一定地位,故而将其与立体参数分割考虑,常见的几何参数有分子表面积、溶剂可极化表面积、分子体积、多维立体参数等
(5)拓扑参数:在分子连接性方法中使用的结构参数,拓扑参数根据分子的拓扑结构将各个原子编码,用形成的代码来表征分子结构。
(6)理化性质参数:偶极矩、分子光谱数据、前线轨道能级、酸碱解离常数等理化性质参数有时也用做结构参数参予定量构效关系研究
(7)纯粹的结构参数:在Free-Wilson方法中,使用纯粹的结构参数,这种参数以某一特定结构的分子为参考标准,依照结构母环上功能基团的有无对分子结构进行编码,进行回归分析,为每一个功能基团计算出回归系数,从而获得定量构效关系模型。
7、二维定量构效关系的数学模型
二维定量构效关系中最常见的数学模型是线性回归分析,如Hansch方法和Free-Wilson方法均采用回归分析。
除了回归分析,遗传算法、人工神经网络、偏最小二乘分析、模式识别、单纯形方法等统计分析方法也会应用于二维定量构效关系数学模型的建立。传统的二维定量构效关系使用的结构数据常仅能反应分子整体的性质,通过改良结构参数,使得二维结构参数能够在一定程度上反应分子在三维空间内的伸展状况,成为二维定量构效关系的一个发展方向。扩展二维定量构效关系能够模拟的数据结构的范围,提高QSAR模型的预测能力是2D-QSAR的主要发展方向。目前,二维定量构效关系的研究集中在两个方向:结构数据的改良和统计方法的优化。
7.1 Hansch方程
回归分析中的经典的Hansch方程形式为
logl/C = -Kl(1ogP)2 + K2logP + K3σ+K4Es + Ks
当logP用π代替时,得:
logl/C = -Klπ2+K2π+K3σ+K4Es+Ks
式中:C—化合物产生某种特定生物活性(如ED50,LD50等)的浓度;
P—脂水分配系数;
π—疏水参数;
σ—电子效应参数,即Hammett常数;
Es—Taft立体参数(空间效应参数)。
Kl、K2、K3、K4、Ks均为回归系数
7.2 Free-Wilson模型
1964年,Free与Wilson根据多变量回归分析理论,认为在一组同源化合物中,生物活性为母体结构(基本结构)的活性贡献与取代基的活性贡献之和。特定位置的取代基对生物活性的贡献不受其他位置取代基的影响。不同位置取代基对生物活性的贡献有其相互独立性,从而可以加和。据此,Free与Wilson提出De Novo可加性模型。模型假定分子的活性是母体化合物和取代基的活性贡献之和,不论其他位置取代基变换与否,每一取代基对生物活性的贡献是恒定与可加的。
最初的方程:BR=∑ai + u
式中:ai—取代基的活性贡献; u—母体的活性贡献
修改后的Free-Wilson方程:
log1/C = A + ∑∑Gij Xij + ε
式中:Gij—第j取代位置上,取代基i的基团活性贡献;
Xij—指示变量,表示取代基i在第j位置上的有无,有取代基i则赋值1,否则取0;
P—被取代的位置数;
Si-1—第j位置上取代基个数;
A—母体化合物的理论活性值;
ε—实验误差,衡量基团活性贡献有无统计意义.
近年来,有人将Hansch分析与Free-Wilson模型联合应用,对药物的系列设计有较大的价值。
8、QSAR的应用
QSAR广泛应用于化学毒剂、农药、医药等领域,在农药方面用于研制高效、高选择性的除草剂,在医药方面研制开发高效的药物分子等。QSAR参与生物活性分子的合理设计过程,借助分子的结构参数或理化性质参数,以数学和统计学手段定量地研究诸如蛋白质分子等生物大分子与有机小分子化合物相互作用、有机小分子在生物体内吸收、分布、代谢、排泄等生理相关性质。QSAR研究是通过分析现有的具有ADME活性物质(例如一组具有相同药理作用及相近的化合物),以化合物的物理化学参数或者结构参数等作为自变量,生物活性(例如半数抑制浓度IC50和半数效应浓度EC50)为分子对接及定量构效关系研究。因变量,运用包括数理统计方法在内的一系列方法建立起化合物的化学结构与生物活性之间的定量关系,推测小分子和受体蛋白质之间的相互作用机理,最后可以根据新化合物的结构数据改变现有小分子的结构从而提高其生物活性或者对新合成的小分子进行活性预测。
9、定量构效关系的发展前景
定量构效关系的应用已经取得了相当大的进步,这一点虽然勿庸置疑,但是定量构效关系依然存在着许多不尽人意的地方,需要提出新模型,新算法进一步
改进定量构效关系的模型,拓展定量构效关系的应用范围。
经典的定量构效关系及药物分子设计已取得了较好的成效和重要进展。但是其严重不足之处在于所采用的参数均是基于化合物的二维结构或是由实际测量得到的。然而受体如蛋白质和DNA 均处于三维结构关系的空间,因此为了更准确地表达药物-受体的超分子(间)相互作用, 有必要进行三维构效关系(3D-QSAR) 或多维构效关系(MD-QSAR)的研究,建立合理的模型或获得更强的预测。
早期的药物设计,占据主导地位的是定量构效关系方法,但是随着近年来生物大分子三维结构的准确测定和计算机计算能力的显著提高,20世纪90年代以来基于结构修饰的药物设计逐渐取代QSAR在药物设计领域的主导地位,例如分子对接等。但是定量构效关系方法在药学研究以及药物设计中仍然发挥着非常重要的作用。
10、参考文献:
药物分子设计的策略_药理活性与成药性
药物分子设计的策略: 药理活性与成药性 郭宗儒* (中国医学科学院药物研究所, 北京 100050) 摘要: 化合物的内在活性和成药性是创新药物的两个基本要素, 活性是药物的基础和核心, 成药性是辅佐 活性发挥药效的必要条件, 两者互为依存。药物在体内的药剂相、药代动力相和药效相可概括为活性和成药性 的展示过程。成药性是药物除活性外的其他所有性质, 包括物理化学性质、生物化学性质、药代动力学性质和 毒副作用, 这是在不同层次上表征药物的性质和行为, 但又相互关联与制约。活性与成药性由化学结构所决定, 体现在微观结构与宏观性质的结合上, 寓于分子的结构之中。先导物的优化是对活性、物化、生化、药代和安 全性等性质的多维空间的分子操作, 因而具有丰富的药物化学内涵。 关键词: 分子设计; 内在活性; 成药性; 先导物优化 中图分类号: R916 文献标识码:A 文章编号: 0513-4870 (2010) 05-0539-09 Strategy of molecular drug design: activity and druggability GUO Zong-ru* (Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing 100050, China) Abstract: Intrinsic activity and druggability represent two essences of innovative drugs. Activity is the fundamental and core virtue of a drug, whereas druggability is essential to translate activity to therapeutic usefulness. Activity and druggability are interconnected natures residing in molecular structure. The pharma-ceutical, pharmacokinetic and pharmacodynamic phases in vivo can be conceived as an overall exhibition of activity and druggability. Druggability actually involves all properties, except for intrinsic activity, of a drug. It embraces physico-chemical, bio-chemical, pharmacokinetic and toxicological characteristics, which are inter-twined properties determining the attributes and behaviors of a drug in different aspects. Activity and drugga-bility of a drug are endowed in the chemical structure and reflected in the microscopic structure and macroscopic property of a drug molecule. The lead optimization implicates molecular manipulation in multidimensional space covering activity, physicochemistry, biochemistry, pharmacokinetics and safety, and embodies abundant contents of medicinal chemistry. Key words: drug design; intrinsic activity; druggability; lead optimization 研发有机小分子药物的药物化学模式, 大都是针对某药物靶标发现苗头化合物 (hit), 将苗头物演化成先导物 (hit-to-lead) 以确定先导物 (lead discovery), 经优化 (lead optimization) 确定候选药物 (drug candidate), 最终达到临床应用的目的。这个全过程是通过结构变换和改造将活性化合物发展成患者可使用的药物, 从分子水平由非药向成药的演 收稿日期: 2009-11-30. *通讯作者Tel / Fax: 86-10-83155752, E-mail: zrguo@https://www.360docs.net/doc/c611231147.html, 化。笔者已从不同侧面阐述了药物分子设计的策略内涵[1?3], 本文以药物的活性和成药性的视角, 讨论构建药物化学结构应注重的问题。 1 类药性和成药性 类药性 (drug-like) 是对苗头物和先导物结构的基本要求, 是Lipinski分析了临床大量口服药物的分子结构, 归纳和提炼出的经验性特征, 类药5原则(Rule of five) 成为筛选苗头和先导物、构建化合物库的重要标准[4], 开阔了人们研发新药的理念。然而, ·综述·
分子对接在药物研发中的应用
分子对接在药物研发中的应用 药物发现(drug discovery)就是新药的研究与开发,其过程涉及化学、药学、医学、生命科学和计算机科学。药物研发的理想策略是能够同时直接针对药物研究的两个对象:受体和药物。药物分子必须分布到受体生物大分子部位并与受体结合,才有可能发挥药理作用。药物设计的目的是预测小分子与受体大分子的结合位点,最终目标是发现全新的先导化合物。新药的研究与开发是一个周期长、投入高、创新性强的多学科交叉与渗透的系统工程。利用计算机模拟可以探测受体大分子的性质,解释实验结果以及预测由于配体和受体结构变化而引起的新性质;通过改变配体和受体分子形状和方向的操作,非常直观地看到小分子与受体大分子在靶点的相互作用,由此确定受体和药物的结合位点,还能对配体小分子结构进行修饰,提出改善药物的药效学和动力学性质的改良方案。 标签:计算;模拟;药物研发;分子对接 Abstract:Drug discovery is the research and development of new drugs. The process involves chemistry,pharmacy,medicine,life science and computer science. The ideal strategy for drug development is to be able to target both the receptor and the drug directly at the same time. In order to play a pharmacological role,drug molecules must be distributed to the receptor biomolecules and bind to the receptor. Drug design is designed to predict the binding sites of small molecules to receptor macromolecules with the ultimate goal of discovering new lead compounds. The research and development of new drugs is a system engineering with long cycle,high investment and strong innovation. Computer simulation can be used to detect the properties of the receptor macromolecules,to explain the experimental results and to predict the new properties caused by the changes of ligand and receptor structures. By changing the shape and direction of the ligand and receptor molecules,the interaction between small molecules and receptor macromolecules at the target sites can be seen intuitively,and the binding sites between the receptors and the drugs can be determined. It can also modify the structure of ligands and improve the pharmacodynamics and kinetic properties of drugs. Keywords:calculation;simulation;drug development;molecular docking 1 分子對接 作为药物设计的核心技术,分子对接(docking)是利用计算机模拟配体和受体分子之间通过匹配原则相互识别的过程。在配体小分子发生药物作用过程中,配体小分子与生物大分子相互接近对方,双方采取合适的取向,使配体小分子与生物大分子在活性位点达到契合,并相互作用,再不断调整构象,形成稳定的复合物构象。通过计算机模拟软件确定复合物中的配体小分子与生物大分子的相对位置和取向,再对两个分子的构象以及底物构象在形成复合物过程中的变化进行研究、判断、计算,最终确定药物作用机制。
虚拟药物筛选与药物分子设计教程与实战
药物分子设计前沿 摘要:近些年来,各种各样的新型疾病依次出现。因此,寻找可以治愈这些疾病的药物对人们来说至关重要。随着计算机技术的高速发展,运用计算机进行新药的模拟实验已经成为一种新的方法。本文就对这些方法做一个总的综述来介绍这些方法在新药设计过程中的应用过程。计算机辅助药物设计方法(CADD)是药物分子设计的基础。从2O世纪6O年代构效关系方法(QSAR)提出以后.经过40多年的努力和探索,CADD方法已经发展成为一门完善和新兴的研究领域。计算机辅助药物设计是应用量子力学、分子动力学、构效关系等基础理论数据研究药物对酶、受体等作用的药效模型,从而达到药物设计之目的。计算机辅助药物设计方法(CADD)大体可以分为三类:基于小分子的药物分子设计方法、基于受体结构的药物分子设计方法、计算组合方法。计算机辅助药物设计是研究与开发新药的一种崭新技术,它大大加快了新药设计的速度,节省了创创新药工作的人力和物力,使药物学家能够以理论作指导,有目的地开发新药。 关键词:药物分子设计计算机模拟分子模拟活性位点分析法 ABSTRACT:In those past years, a variety of new diseases were appeared. So, it’s vary essential for us to find the drugs that can cure these diseases. And with the fast development of computer technology, the applying of computer in the simulations of these new drugs has become a new method. In this paper, I will draw a general overview of those methods to introduce the applications in the design process of the new drugs. The method of Computer Aided Drug Design(℃ADD)was the basis 0f drugs molecule design which was proposed in 1960.During the last 40 years,the CADD method has been widely applied as a burgeoning and potential research area.The aim of CADD is to design drug according to the pharmacodynamic model between the drugs and the enzyme or receptor which is applied the quantum mechanics.molecular dynamics,and quantitative structure—activity relationship.The CADD includes three methods:method basing on the ligand,method basing on the receptor,and combinatorial chemistry method.The CADD is a new technology to research drug which can accelerate the speed of drug design,economize the manpower and material resources. KEY WORDS:Drug molecular design;computer simulation; molecular simulation;active site analysis 引言 传统药物设计从总体上来讲,缺乏成熟完善的发现途径,具有很大的盲目性,一般平均要筛选10000种以上的化合物才能得到一种新药,因此开发效率很低。随着计算机技术及计算化学、分子生物学和药物化学的发展,药物设计进入了理性阶段,其中药物分子设计是目前新药发现的主要方向。它是依据生物化学、酶学、分子生物学以及遗传学等生命科学的研究成果,针对这些基础研究中所揭示的包括酶、受体、离子通道及核酸等潜在的药物设计靶点,并参考其它类源性配体或天然产物的化学结构特征,设计出合理的药物分子。运用计算机模拟来进行新药的分子结构设计主要有三种方法:分子对接设计、遗传算法以及计算机辅助
多靶点药物分子设计
多靶点药物分子设计 郭彦伸, 郭宗儒* (中国医学科学院、北京协和医学院药物研究所, 北京100050) 摘要: 作用于单一分子靶标的药物治愈多基因相关疾病如癌症、或影响多个组织或细胞类型的疾病如糖尿病等存在的问题逐渐被人们所认识。与选择性药物的治疗作用相比,几个靶标间的平衡调节能够提供较好的疗效和较低的副作用,同时作用于多个靶标的多靶点药物能够较好地控制复杂的疾病。本文详细比较分析了单靶点药物的不足和多靶点药物的优势,介绍了多靶点配体药物分子设计的方法及需要优化的参数。对于多靶点药物设计,关键的挑战是如何保证获得平衡的活性同时又能够实现选择性以及适当的药代动力学性质。到目前为止, 多靶点药物分子设计的方法对于药物化学家、药理学家、毒理学家以及生物化学家仍然是一项新的挑战。 关键词: 多靶点配体; 药效团组合; 药物分子设计 中图分类号: R916.1 文献标识码:A 文章编号: 0513-4870 (2009) 03-0276-06 Design of multiple targeted drugs GUO Y an-shen, GUO Zong-ru* (Institute Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China) Abstract: Drugs designed to act on individual molecular targets usually can not combat multigenic diseases such as cancer, or diseases that affect multiple tissues or cell types such as diabetes. Increasingly, it is being recognised that a balanced modulation of several targets can provide a superior therapeutic effect and side effect profile compared to the action of a selective ligand. The multi-target drugs which impact multiple targets simultaneously are better at controlling complex disease systems and are less prone to drug resistance. Here, we compare the disadvantage of the selective ligands and the predominance of multi-targets drugs in detail and introduce the approaches of designing multiple ligands and the procedure of optimization particularly. A key challenge in the design of multiple ligands is attaining a balanced activity at each target of interest while simul-taneously achieving a wider selectivity and a suitable pharmacokinetic profile. On this point, the multi-target approach represents a new challenge for medicinal chemists, pharmacologists, toxicologists, and biochemists. Key words: multiple targeted ligands; pharmacophore combination; design of drug 药物靶标是指与疾病的发生有因果关系或者参与疾病的发展过程, 并通过药物对其进行调节而实现治疗目的的生物分子。自30多年前引入离体筛选(in vitro) 的概念以来, 基因组学和高通量筛选技术的进步,使药物发现从依赖动物筛选逐渐转变到“一病一靶”。现代药理学研究已深入到细胞和分子水平, 更加强调药物作用的靶标, 发现了许多单一靶点选 收稿日期: 2009-02-11. *通讯作者Tel: 86-10-83155752, E-mail: zrguo@https://www.360docs.net/doc/c611231147.html, 择性的药物, 在临床上表现出显著的疗效, 如选择性的HMG辅酶A还原酶抑制剂[1]。随着进一步的深入研究, 发现单一靶点药物也存在着明显的不足。单一靶点抗肿瘤药物单独用药对于晚期患者的化疗效率不高, 人们逐渐认识到单一靶点药物之间的联合应用或作用于多个分子靶标的“多靶点”药物在治疗功能失调类疾病时将起到重要作用[2,3]。目前人们有意识地、理性地设计作用于特定的多个靶点的配体成为研究趋势, “多靶点”药物研发将成为研究的热点。
(完整版)10分钟教你掌握分子对接模拟软件(医药向)
首先介绍一下自己吧,本人毕业于南方某知名211大学药学系,目前于澳门科技大学攻读硕士研究生。从本科开始自己就在接触CADD(计算机辅助药物设计)方面的软件知识,在此将分享一些自己的纯干货!下面将以一个实例操作带大家迅速认识和掌握分子模拟对接,希望给各位从事医药行业和药物化学合成的同学带来帮助。 话不多说,下面进入正题。 首先我们搞清楚一个概念:什么是分子模拟对接。分子模拟对接简单来说就是利用电脑软件将受体蛋白与配体分子进行模拟对接,计算它们的结合能(KJ/MOL)大小来判断结合是否紧密,若结合效果比较理想,那么该蛋白受体或配体则是我们理想的分子,可以进一步进行实验室操作,避免盲目实验带来的人力经济损失。 接下来我将介绍一下本篇文章的主角,也是我们所要用到的软件PyRx、Chemdraw、AutodockTools以及PyMol。为了便于理解,简要概括之:Chemdraw为化合物分子绘图软件;PyRx为Autodock Vina算法搭载软件,能够调用其算法直接进行模拟对接;AutodockTools是PyMol为对接结果成像软件,可以进一步分析其结构。 下面正式进入正题,我将大致分为三个板块来进行推进:受体配体的准备;分子对接;结果分析。研究类型为:已知若干配体分子结构,通过受体蛋白测试配体分子活性。 本次筛选意在以COMT酶为受体,从20种与常见氨基酸形成环二肽的目标化合物中筛选出与COMT酶受体结合最为紧密的一种环二肽结构,大大减少了随机筛选的盲目性,有利于进一步研究该类化合物分子的生物学活性与改造成抗帕金森疾病前药的可能。图1展示了20种不同环二肽结构物质的统一结构,随着R基团的不同,所对应的氨基酸也不同。而表1则展示了20种不同环二肽的分子式。 图1 Cycol[DOPA(6-NO2)-AA]
药物分子设计简介
药物分子设计简介 传统药物设计从总体上来讲,缺乏成熟完善的发现途径,具有很大的盲目性,一般平均要筛选10000种化合物以上才能得到一种新药,因此开发效率很低,很难迅速得到合适的新药来治疗越来越多的疑难杂症。 随着计算机技术及计算化学、分子生物学和药物化学的发展,药物设计进入了理性阶段,其中药物分子设计是目前新药发现的主要方向。它是依据生物化学、酶学、分子生物学以及遗传学等生命科学的研究成果,针对这些基础研究中所揭示的包括酶、受体、离子通道及核酸等潜在的药物设计靶点,并参考其它类源性配体或天然产物的化学结构特征,设计出合理的药物分子。计算机辅助药物设计方法(cadd)是药物分子设计的基础。从20世纪60年代构效关系方法(qsar)提出以后,经过40多年的努力和探索,尤其是20世纪90年代以后,随着多种新的方法的出现,cadd方法已经发展成为一门完善和新兴的研究领域,它大大提高了药物开发的效率,为人们攻克一些顽症提供了崭新的思路和成功的希望。 计算机辅助药物设计方法(cadd)大体可以分为三类: 1.基于小分子的药物分子设计方法,这类方法主要是针对受体结构未知的药物分子,主要包括定量构效方法和药效团模型方法; 2.基于受体结构的药物分子设计方法。随着分子生物学和结构生物学的发展,越来越多的生物大分子结构被解析。因此对于一些未知三维结构的受体大分子,它们的结构常常也可以通过同源蛋白质建模的方法得到。在这种情况下,就可以采用基于受体结构的药物分子设计方法来寻找新的先导化合物。基于受体结构的药物分子设计方法的思路是通过研究受体结构的特征以及受体和药物分子之间的相互作用方式来进行药物设计。常用的方法是分子对接方法和从头设计方法; 3.计算组合方法。主要包括两部分的内容,一方面是采用计算机技术设计合成组合库的构造块,通过计算机生成包含足够分子多样性的虚拟组合库;另一方面则是把得到的虚拟组合库和其它分子设计方法结合起来进行药物分子设计。当然上面介绍的这些方法之间也并不是完全独立的。对于某个体系常常会采用多种药物设计方法进行先导化合物的设计。如知道了蛋白质和药物分子相互结合的复合物结构时,可以从蛋白质结构出发用基于分子对接的数据库搜索方法进行先导化合物的寻找;也可以从复合物的相互作用信息,得到药效团模型,然后从药效团模型出发进行药物设计;同时还可以从蛋白质结构出发,用片断生长的方法进行全新药物分子的设计[也就是从头设计方法]。 计算机辅助药物设计方法(cadd)中目前还有几个难题: 1.受体和配体之间自由能的评估,因为分子活性的大小常常是由受体和药物分子之间的结合自由能来决定的,所以体系的自由能得计算非常重要。虽然目前已有很多方法,如线性相互作用能方法等,但总的来看结合自由能的评估还需要做大量的工作; 2.大分子的构象问题。确定构象的方法有两种:实验方法和理论计算方法。用实验方法解析的大分子结构的数目有限,而常用的构象分析方法,如系统搜索方法、距离几何方法、
-合理药物设计
合理药物设计 合理药物设计(rational drug design)是依据与药物作用的靶点即广义上的受体,如酶、受体、离子通道、抗原、病毒、核酸、多糖等,寻找和设计合理的药物分子。主要通过对药物和受体的结构在分子水平甚至电子水平上全面准确地了解,进行基于结构的药物设计和通过对靶点的结构功能与药物作用方式及产生生理活性的机理的认识进行基于机理的药物设计。合理药物设计是化学、生物学、数学、物理学以及计算机科学交叉的产物,是在社会对医药需求的强大推动下逐步发展起来的,主要应用各种理论计算方法和分子图形模拟技术来进行合理药物设计。合理药物设计方法包括3类:①基于配体的药物设计②基于受体结构的药物设计③基于药物作用机理的药物设计。 1.基于配体的药物设计方法 合理药物分子设计必须在已知受体结构模型的条件下才能进行但到目前为止许多已知药物作用的受体结构是未知的在未知受体结构时应用合理药物设计的原理和概念开始药物设计也有了不少的尝试,这方面的研究大致可分为两类;探索系列小分子药物三维结构与活性的关系---主要有3D-QSAR;根据已知药物结构反推受体结构模型,再行合理药物设计,如药效团模型(Pharmacophore Modeling)方法。 1.1定量构效关系(3D-QSAR) 从对药物与受体相互作用的研究可以知道药物的作用是依赖自身空间形状的,其与受体的作用一般为非共价性质虽然在未知受体结
构时无法进行常规意义上的合理药物设计,但可以在对已知药物研究的基础上进行受体形状推测(receptor-mapping),将与药物本身形状有关的参数引入到定量构效关系中,称之为3D-QSAR。该方法是基于被研究的分子结合在同一个靶标生物大分子的相同部位的基本假定,将药物的结构信息、理化参数与生物活性进行拟合计算,建立合理的定量关系的数学模型,再以此关系设计新的化合物。不同方法采用不同的结构性质来确定构效关系。 利用小分子三维结构作为参数的三维定量构效关系方法在预测小分子与生物大分子的相互作用时非常有用,各种在化合物三维结构基础上进行三维定量构效关系研究的方法(3D-QSAR),在药物研究中己经越来越广泛地应用。主要方法为距离几何(Distance Geometry, DG)、分子形状分析(Molecular Shape Analysis, MSA)、比较分子场分析(Comparative Molecular Field Analysis, CoMFA)以及虚拟受体(Pseudo Receptor)方法。 在3D-QSAR中,CoMFA是目前应用最为广泛的方法,它采用化合物周围的静电场、范德华力场等的空间分布作为化合物结构描述变量,通过最小二乘法建立化合物的生物活性与化台物周围各种力场空间分布之间关系的模型。CoMFA是在不了解受体结构的情况下,通过将分子势场图示到网格点上来表示分子的周围环境,比较它们与药物分子的生物活性定量关系,用以推测受体的某些性质,并可依次建立起作用模型来设计新的化合物,定量地预测其活性强度。 1.2药效基团模型方法
分子对接的原理,方法及应用
分子对接的原理,方法及应用 (PPT里弄一些分子对接的照片,照片素材文件里有) 分子对接 是将已知三维结构数据库中的分子逐一放在靶标分子的活性位点处。通过不断优化受体化合物的位置、构象、分子内部可旋转键的二面角和受体的氨基酸残基侧链和骨架,寻找受体小分子化合物与靶标大分子作用的最佳构象,并预测其结合模式、亲和力和通过打分函数挑选出接近天然构象的与受体亲和力最佳的配体的一种理论模拟分子间作用的方法。 通过研究配体小分子和受体生物大分子的相互作用,预测其亲和力,实现基于结构的药物设计的一种重要方法。 原理: 按照受体与配体的形状互补,性质互补原则,对于相关的受体按其三维结构在小分子数据库直接搜索可能的配体,并将它放置在受体的活性位点处,寻找其合理的放置取向和构象,使得配体与受体形状互补,性质互补为最佳匹配 (配体与受体结合时,彼此存在静电相互作用,氢键相互作用,范德华相互作用和疏水相互作用,配体与受体结合必须满足互相匹配原则,即配体与受体几何形状互补匹配,静电相互作用互补匹配,氢键相互作用互补匹配,疏水相互作用互补匹配) 目的: 找到底物分子和受体分子的最佳结合位置 问题: 如何找到最佳的结合位置以及如何评价对接分子之间的结合强度 方法: 1、首先建立大量化合物的三维结构数据库 2、将库中的分子逐一与靶分子进行“对接” 3、通过不断优化小分子化合物的位置以及分子内部柔性键的二面角,寻找小分子化合物与靶标大分子作用的最佳构象,计算其相互作用及结合能 4、在库中所有分子均完成了对接计算之后,即可从中找出与靶标分子结合的最佳分子 应用: 1)直接揭示药物分子和靶点之间的相互作用方式 2)预测小分子与靶点蛋白结合时的构象 3)基于分子对接方法对化合物数据库进行虚拟筛选,用于先导化合物的发现
药物分子设计基础论文
药物分子设计的基本学识论文 摘要 近些年来,各种各样的新型疾病依次出现。因此,寻找可以治愈这些疾病的药物对人们来说至关重要。随着分子生物学和药物化学的发展,药物设计进入了理性阶段,其中药物分子设计是目前新药发现的主要方向。它是依据生物化学、酶学、分子生物学以及遗传学等生命科学的研究成果,针对这些基础研究中所揭示的包括酶、受体、离子通道及核酸等潜在的药物设计靶点,并参考其它类源性配体或天然产物的化学结构特征,设计出合理的药物分子。本文介绍了几种药物设计的方法。关键词:药物;分子设计;靶点。 ABSTRACT In recent years a variety of new disease appeared in turn. Therefore, looking for drugs that c an cure the disease to people is very important. With the development of molecular biology and pharmaceutical chemistry, entered the stage of rational drug design of drug molecular design is t he main direction of drug discovery. It is on the basis of biochemistry, enzymology, molecular biol ogy and genetics biological scientific research achievements, such as iron to these basic research reveals the including enzyme, receptors, ion channels and nucleic acids such as potential targets f or drug design, and refer to other types of source sex ligand or chemical structure characteristics of natural products, design a reasonable drug molecules. Original meaning, this paper introduces several kinds of drug design methods. Keywords: drugs; Molecular design; Targets. 一.药物分子生物学重点。 1.分子生物学:是在分子水平研究生命现象的科学,是现代生命科学的共同语言。核心内容是通过生物的物质基础——核酸、蛋白质、酶等生物大分子的结构、功能及其相互作用等运动规律的研究来阐明生命分子基础,从而探讨生命的奥秘。 2.药学分子生物学:由于分子生物学的新理论、新技术渗入到药学研究领域,从而使药物学研究以化学、药学的培养模式转化为以生命科学、药学和化学相结合的新药模式。 3.分子生物学的主要研究对象:核酸、蛋白质、酶等生物大分子的结构、功能及相互作用。 4.分子生物学在医药工业中的应用: ①DNA重组技术与新药研究 ②药物基因组学、药物蛋白质组学与现代药物研究 ③药物蛋白质组学是基因、蛋白质、疾病三者相连的桥 二.药物设计的发展 1.药物设计是随着药物化学学科的诞生相应出现的。早在20世纪20年代以前,就开始进行天然有效成分的结构改造。直到1932年,欧兰梅耶发表了将有机化学的电子等排原理和环状结构等价概念用于药物设计,首次出现具有理论性的药物分子结构的修饰工作。随后,药物作用的受体理论、生化机制、药物在体内转运等药物设计的理论不断出现。在60年代初出现了构效关系的定量研究,1964年汉希和藤田稔夫提出定量构效关系的汉希分析。药物设计开始由定性进入定量研究阶段,为定量药物设计奠定理论和实践基础。药物设计逐渐形成一门独立的分支学科。70年代以后药物设计开始综合运用药物化学、分子生物学、量子化学、统计数学基础理论和当代科学技术以及电子计算机等手段,开辟了药物设计新局面。随着分子生物学的进展,对酶与受体的理解更趋深入,对有些酶的性质、酶反应历程、药物
药物分子设计的发展
药物分子设计的发展 陈凯先Ξ 罗小民 蒋华良 (上海药物研究所 上海 200031) 摘要 简要回顾了药物分子设计的发展,重点评述了间接药物设计方法和直接药物 设计方法,并列举了若干药物分子设计的成功例子,最后提出了该领域在21世纪的 发展前景。 关键词 药物分子设计,发展,展望 新药的寻找迄今为止仍是一件耗资巨大而效率很低的工作,迫切需要应用新的理论方法和技术加以改进。药物分子设计就是在这种社会需求的强大推动下逐步发展起来的。 药物分子设计作为一个独立和明确的研究领域,始于20世纪60年代,迄今已有40年。值此世纪交替之际,谨就药物分子设计的发展,作一简单的回顾和展望。 很久以前,药物分子设计就已成为人们的美好梦想。但是,直到物理学、化学、计算机科学和现代生物学有了充分的发展之后,药物分子设计才具有现实的可能性。 1 药物分子设计的发展 1894年,Emil Fischer提出了药物作用的“锁钥原理”,即药物作用于体内特定部位,有如钥匙与锁的关系。这一思想虽然过于简单粗糙,但是其基本思路至今仍然富有活力和价值。 从20世纪60年代以来,经过40年的不断探索和努力,现代药物设计的策略和方法已经大为丰富,基本可以分成两大类:间接药物设计和直接药物设计。 1.1 间接药物设计 这类方法是从一组小分子(例如几十个)化合物的结构和生物活性数据出发,研究其结构2活性关系的规律,在此基础上预测新化合物的生物活性(药效)和进行高活性分子的结构设计。在药物设计研究的早期(60—80年代),人们对于药物作用的靶标分子大多缺乏了解,只能从药物小分子化合物的结构和活性出发,去归纳和认识药物分子的构2效关系,因此,间接药物设计成为这一时期药物设计研究的主要方法。 定量构效关系(Quantitative Structure-Activity Relationship,QS AR)是一种重要的间接药物设计方法。最早的QS AR方法由Hansch于1962年提出。它对一组小分子化合物的理化参数2000年 中国科学院 院 刊 第4期Ξ中国科学院院士,上海药物研究所所长 收稿日期:2000年6月20日
合理药物设计药物开发策略
合理药物设计(rational drug design) 依据与药物作用的靶点即广义上的受体,如酶、受体、离子通道、抗原、病毒、核酸、多糖等,寻找和设计合理的药物分子。主要通过对药物和受体的结构在分子水平甚至电子水平上全面准确地了解,进行基于结构的药物设计和通过对靶点的结构功能与药物作用方式及产生生理活性的机理的认识进行基于机理的药物设计。合理药物设计是化学、生物学、数学、物理学以及计算机科学交叉的产物,是在社会对医药需求的强大推动下逐步发展起来的,主要应用各种理论计算方法和分子图形模拟技术来进行合理药物设计。合理药物设计方法包括3 类:①基于配体的药物设计②基于受体结构的药物设计③基于药物作用机理的药物设计。 1. 基于配体的药物设计方法 合理药物分子设计必须在已知受体结构模型的条件下才能进行但到目前为止许多已知药物作用的受体结构是未知的在未知受体结构时应用合理药物设计的原理和概念开始药物设计也有了不少的尝试, 这方面的研究大致可分为两类;探索系列小分子药物三维结构与活性的关系---主要有3D-QSAR;根据已知药物结构反推受体结构模型, 再行合理药物设计,如药效团模型(Pharmacophore Modeling)方法。 1.1 定量构效关系(3D-QSAR) 从对药物与受体相互作用的研究可以知道药物的作用是依赖自身空间形状的, 其与受体的作用一般为非共价性质虽然在未知受体结构时无法进行常规意义上的合理药物设计, 但可以在对已知药物研究的基础上进行受体形状推测(receptor-mapping), 将与药物本身形状有关的参数引入到定量构效关系中, 称之为3D-QSAR。该方法是基于被研究的分子结合在同一个靶标生物大分子的相同部位的基本假定,将药物的结构信息、理化参数与生物活性进行拟合计算,建立合理的定量关系的数学模型,再以此关系设计新的化合物。不同方法采用不同的结构性质来确定构效关系。
计算机辅助药物分子设计
计算机辅助药物分子设计 计算机辅助药物分子设计的方法开始于20世纪80年代早期。当今,随着人类基因组计划的完成、蛋白组学的迅猛发展,以及大量与人类疾病相关基因的发现,药物作用的靶标分子急剧增加;同时,在计算机技术推动下,计算机药物辅助设计在近几年取得了巨大的进展。 计算机辅助药物设计(computer aideddrug design)是以计算机化学为基础,通过计算机的模拟、计算和预算药物与受体生物大分子之间的关系,设计和优化先导化合物的方法。计算机辅助药物设计实际上就是通过模拟和计算受体与配体的这种相互作用,进行先导化合物的优化与设计,从而达到防治疾病、纠正失调的机体内环境的目的。 那么,要做好药物分子设计,需要掌握哪些知识呢? (1)理论基础的学习,主要来自书籍和文献。 (2)软件的学习和使用,主要来自各个软件的说明书或者使用手册。 (3)来自和相关方向研究者的交流,另外还需要掌握一些生物学方面的知识。 原理: 计算机辅助药物设计的一般原理是,首先通过X-单晶衍射等技术获得受体大分子结合部位的结构,并且采用分子模拟软件分析结合部位的结构性质,如静电场、疏水场、氢键作用位点分
布等信息。然后再运用数据库搜寻或者全新药物分子设计技术,识别得到分子形状和理化性质与受体作用位点相匹配的分子,合成并测试这些分子的生物活性,经过几轮循环,即可以发现新的先导化合物。因此,计算机辅助药物分子设计大致包括活性位点分析法、数据库搜寻、全新药物设计。 1.活性位点分析法 该方法可以用来探测与生物大分子的活性位点较好地相互作用的原子或者基团。用于分析的探针可以是一些简单的分子或者碎片,例如水或者苯环,通过分析探针与活性位点的相互作用情况,最终可以找到这些分子或碎片在活性部位中的可能结合位置。由活性位点分析得到的有关受体结合的信息对于全新药物的设计具有指导性。目前,活性位点分析软件有DRID、GREEN、HSITE 等。另外还有一些基于蒙特卡罗、模拟退火技术的软件如MCSS、HINT、BUCKETS等。 2. 数据库搜寻 目前数据库搜寻方法分为两类。一类是基于配体的,即根据药效基团模型进行三维结构数据库搜寻。该类方法一般需先建立一系列活性分子的药效构象,抽提出共有的药效基团,进而在现有的数据库中寻找符合药效基团模型的化合物。该类方法中比较著名的软件有Catalyst和Unity,而以前者应用更普遍。另一类方法是基于受体的,也称为分子对接法,即将小分子配体对接到受体的活性位点,并搜寻其合理的取向和构象,使得配体与受体的形
药物发展史
药物发展史 作者:刘华富专业:药学学号:201343855237 摘要 药最先是从人类社会初期开始的。人类在与大自然作斗争中创造了原始的医药,医药学同其它科学一样,来源于人类的社会实践和物质生活的需要。药学是历代人民大众智慧的结晶,经历了从天然药物,到化学制药再到生物制药三个阶段。它对全人类的健康发展,种族繁衍与发展,有着巨大贡献。 正文 远古时代人们为了生存从生活经验中得知某些天然物质可以治疗疾病与伤痛,这是药物的源始。这些实践经验有不少流传至今,例如饮酒止痛、大黄导泻、楝实祛虫、柳皮退热等。以后在宗教迷信与邪恶斗争及封建君王寻求享乐与长寿中药物也有所发展。但更多的是将民间医药实践经验的累积和流传集成本草,这在我国及埃及、希腊、印度等均有记载,例如在公元一世纪前后我国的《神农本草经》及埃及的《埃伯斯医药籍》等。明朝李时珍的《本草纲目》(1596)在药物发展史上有巨大贡献,是我国传统医学的经典著作,全书共52卷,约190万字,收载药物1892种,插图1160帧,药方11000余条,是现今研究中药的必读书籍,在国际上有七种文字译本流传。在西欧文艺复兴时期(十四世纪开始)后,人们的思维开始摆脱宗教束缚,认为事各有因,只要客观观察都可以认识。瑞士医生Paracelsus(1493-1541)批判了古希腊医生Galen恶液质唯心学说,结束了医学史上1500余年的黑暗时代。后来英国解剖学家W h a r v e s(1578-1657)发现了血液循环,开创了实验药理学新纪元。意大利生理学家F.F o n t a n a(1720-1805)通过动物实验对千余种药物进行了毒性测试,得出了天然药物都有其活性成分,选择作用于机体某个部位而引起典型反应的客观结论。这一结论以后为德国化学家F.W.S e r t u r n e r(1783-1841)首先从罂粟中分离提纯吗啡所证实。18世纪后期英国工业革命开始,不仅促进了工业生产也带动了自然科学的发展。其中有机化学的发展为药理学提供了物质基础,从植物药中不断提纯其活性成分,得到纯度高的药物,如依米丁、奎宁、士的宁、可卡因等。 l9世纪末,化学工业的兴起,Ehrlich化学治疗概念的建立,为20世纪初