VO-Ohpic_trihydrate_DataSheet_MedChemExpress
HPLC测定奥美沙坦酯氢氯噻嗪片含量和有关物质

呵护公众健康合理用药China Licensed Pharmacist Jul.2012,Vol.9No.7奥美沙坦酯氢氯噻嗪片是治疗高血压新药,2010年在我国上市,临床用于治疗高血压、充血性心力衰竭。
奥美沙坦酯与氢氯噻嗪有互补的作用机制,两者合用既能协同降压,又可以减少氢氯噻嗪的应用剂量,具有快速起效、强效降压和持久降压的特点,适于国内众多高血压患者使用。
由于该产品上市时间短,目前文献少见对该产品有关物质测定方法的报道,本研究建立了该产品含量和关物质的HPLC测定方法,简单快速,结果准确。
1仪器与试药安捷伦1200高效液相色谱仪及其色谱工作站(安捷伦公司);奥美沙坦酯对照品(原中国药品HPLC测定奥美沙坦酯氢氯噻嗪片含量和有关物质赵建峰(北京万生药业有限责任公司,北京101113)【摘要】目的:建立高效液相色谱法(HPLC)测定奥美沙坦酯氢氯噻嗪片含量和有关物质的测定方法。
方法:采用HPLC,色谱柱:C18(4.6mm×250mm,Kromasil);流动相A:0.02mol/L磷酸二氢钠溶液(用磷酸调节pH至3.0),流动相B:甲醇-乙腈(100∶900),梯度洗脱。
结果:奥美沙坦酯和氢氯噻嗪均能与其他杂质较好分离;奥美沙坦酯浓度在0.002026~0.04052mg/mL(r= 0.999)范围内线性良好,氢氯噻嗪在0.001265~0.02530mg/mL范围内线性良好(r=1.000)。
结论:本方法灵敏、准确,可作为奥美沙坦酯氢氯噻嗪片含量和有关物质的测定方法。
【关键词】高效液相色谱法;奥美沙坦酯氢氯噻嗪片;含量测定doi:10.3969/j.issn.1672-5433.2012.07.006Determination of the Content and Related Substances of Olmesartan Medoxomil/Hydrochloroth-iazide Tablets by HPLCZhao Jianfeng(Beijing Winsunny Pharmaceutical Co.Ltd.,Beijing101113,China)ABSTRACT Objective:To establish a method by HPLC to determine the content and the related substances of olmesartan medoxomil/hydrochlorothiazide tablets.Methods:The HPLC was performed on a C18column(4.6mm×250mm,Kromasil)with mobile phase A:0.02%sodium dihydrogen phosphate solution(pH3.0adjusted with phosphoric acid)and mobile phase B:methanol-acetonitrile(100∶900). Gradient elution was conducted.Results:Olmesartan medoxomil and hydrochlorothiazide were completely separated from impurities.There was a good linear range from0.002026to0.04052mg/mL(r=0.999)for olmesartan medoxomil and from0.001265to0.02530mg/mL(r=1.000)for hydrochlorothiazide. Conclusion:The method was proved good in sensitivity and accuracy and can be applied to the determination of olmesartan medoxomil/hydrochlorothiazide tablets and its related substances.KEY WORDS HPLC;Olmesartan Medoxomil/Hydrochlorothiazide Tablets;Content Determination作者简介:赵建峰,男,工程师。
CAS号737763-37-0_360A iodide_MedBio技术参数

CAS
包装
纯度
MedBio
MED11537
CCT244747
CCT244747
1404095-34-6
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11470
Monomethyl auristatin E
Monomethyl auristatin E
474645-27-7
10mM (in 1mL DMSO)
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11536
ELR510444
ELR510444
1233948-35-0
25mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11426
Acetyl Angiotensinogen (1-14), porcine
5mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11516
Eribulin mesylate
Eribulin mesylate
441045-17-6
500μg
≥98%Βιβλιοθήκη 品牌货号中文名称英文名称
CAS
包装
纯度
MedBio
MED11540
AZD-5597
AZD-5597
924641-59-8
3、同类产品列表:
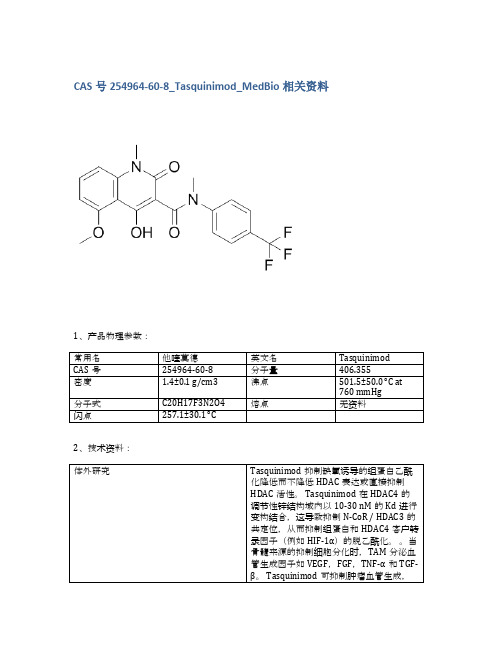
CAS号254964-60-8_Tasquinimod_MedBio相关资料
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11931
Cyclophosphamide
Cyclophosphamide
50-18-0
10mM (in 1mL DMSO)
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED12034
Mupirocin
包装
纯度
MedBio
MED11950
Resminostat hydrochloride
Resminostat hydrochloride
1187075-34-8
100mg
≥98%
品牌
CAS
包装
纯度
MedBio
MED12017
Raltitrexed
Raltitrexed
112887-68-0
10mM (in 1mL DMSO)
≥98%
体内研究
当通过管饲法或饮用水给予成年雄性小鼠(即C57B1 / 6J或无胸腺裸鼠)0.1-30mg / kg(即0.2-74μmoles/ kg)时,Tasquinimod的生物利用度和口服吸收是极好的。Tasquinimod的效力表示为抑制癌症生长50%的Tasquinimod每日口服剂量范围为0.1-1.0 mg / kg / d(即0.24-2.40μmoles/ kg /天),相对于一系列(n> 5)免疫缺陷小鼠中的人前列腺癌异种移植物。通过饮用水以5mg / kg /天的慢性剂量服用的Tasquinimod在免疫活性同系小鼠中产生> 80%的TRAMP-C2小鼠前列腺癌生长抑制(p <0.05)[2]。携带皮下LNCaP肿瘤的裸鼠用Tasquinimod治疗3周。在接种后第7天开始以1mg / kg /天和10mg / kg /天暴露于Tasquinimod。与接种后28天的未处理对照组相比,1 mg / kg /天和10 mg / kg /天的肿瘤重量均有统计学显着的剂量依赖性降低(p <0.001),说明Tasquinimod的抗肿瘤作用[3]。
奥美拉唑-欧洲8.0药典

EUROPEAN PHARMACOPOEIA 8.0Omeprazole04/2013:0942OMEPRAZOLEOmeprazolum C 17H 19N 3O 3S M r 345.4[73590-58-6]DEFINITION 5-Methoxy-2-[(RS )-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-1H -benzimidazole.Content :99.0per cent to 101.0per cent (dried substance).CHARACTERSAppearance :white or almost white powder.Solubility :very slightly soluble in water,soluble in methylene chloride,sparingly soluble in ethanol (96per cent)and in methanol.It dissolves in dilute solutions of alkali hydroxides.It shows polymorphism (5.9).IDENTIFICATIONInfrared absorption spectrophotometry (2.2.24).Comparison :omeprazole CRS .If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in methanol R ,evaporate to dryness and record new spectra using the residues.TESTSSolution S .Dissolve 0.50g in methylene chloride R and dilute to 25mL with the same solvent.Appearance of solution .Solution S is clear (2.2.1).Impurities F and G :maximum 350ppm for the sum of the contents.The absorbance (2.2.25)of solution S determined at 440nm is not greater than 0.10.Related substances .Liquid chromatography (2.2.29).Prepare the solutions immediately before use .Test solution .Dissolve 3mg of the substance to be examined in the mobile phase and dilute to 25.0mL with the mobile phase.Reference solution (a).Dissolve 1mg of omeprazole CRS and 1mg of omeprazole impurity D CRS in the mobile phase and dilute to 10.0mL with the mobile phase.Reference solution (b).Dilute 1.0mL of the test solution to 100.0mL with the mobile phase.Dilute 1.0mL of this solution to 10.0mL with the mobile phase.Reference solution (c).Dissolve 3mg of omeprazole for peak identification CRS (containing impurity E)in the mobile phase and dilute to 20.0mL with the mobile phase.Column :–size :l =0.125m,Ø=4.6mm;–stationary phase :octylsilyl silica gel for chromatography R (5μm).Mobile phase :mix 27volumes of acetonitrile R and 73volumes of a 1.4g/L solution of disodium hydrogen phosphate R previously adjusted to pH 7.6with phosphoric acid R .Flow rate :1mL/min.Detection :spectrophotometer at 280nm.Injection :40μL.Run time :5times the retention time of omeprazole.Identification of impurities :use the chromatogram obtained with reference solution (a)to identify the peak due to impurity D;use the chromatogram supplied with omeprazole for peak identification CRS and the chromatogram obtainedwith reference solution (c)to identify the peak due toimpurity E.Relative retention with reference to omeprazole(retention time =about 9min):impurity E =about 0.6;impurity D =about 0.8.System suitability :reference solution (a):–resolution :minimum 3.0between the peaks due toimpurity D and omeprazole;if necessary,adjust the pH of the aqueous part of the mobile phase or the concentration of acetonitrile R ;an increase in the pH will improve theresolution.Limits :–impurities D,E :for each impurity,not more than 1.5times the area of the principal peak in the chromatogramobtained with reference solution (b)(0.15per cent);–unspecified impurities :for each impurity,not more than thearea of the principal peak in the chromatogram obtained with reference solution (b)(0.10per cent);–total :not more than 5times the area of the principal peak in the chromatogram obtained with reference solution (b)(0.5per cent);–disregard limit :0.5times the area of the principal peak in the chromatogram obtained with reference solution (b)(0.05per cent).Loss on drying (2.2.32):maximum 0.2per cent,determined on 1.000g by drying under high vacuum at 60°C for 4h.Sulfated ash (2.4.14):maximum 0.1per cent,determined on1.0g.ASSAYDissolve 0.250g in a mixture of 10mL of water R and 40mL of ethanol (96per cent)R .Titrate with 0.1M sodium hydroxide ,determining the end-point potentiometrically (2.2.20).1mL of 0.1M sodium hydroxide is equivalent to 34.54mg of C 17H 19N 3O 3S.STORAGEIn an airtight container,protected from light,at a temperature of 2°C to 8°C.IMPURITIESSpecified impurities:D,E,F,G .Other detectable impurities (the following substances would,if present at a sufficient level,be detected by one or other of the tests in the monograph.They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034).It is therefore not necessary to identify these impurities for demonstration of compliance.See also 5.10.Control of impurities in substances for pharmaceutical use ):A,B,C,H,I.A.5-methoxy-1H-benzimidazole-2-thiol,B.2-[(RS )-[(3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-5-methoxy-1H -benzimidazole,General Notices (1)apply to all monographs and other texts2911Omeprazole magnesium EUROPEAN PHARMACOPOEIA8.0C.5-methoxy-2-[[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfanyl]-1H-benzimidazole(ufiprazole),D.5-methoxy-2-[[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfonyl]-1H-benzimidazole(omeprazolesulfone),E.4-methoxy-2-[[(RS)-(5-methoxy-1H-benzimidazol-2-yl)sulfinyl]methyl]-3,5-dimethylpyridine1-oxide,F.8-methoxy-1,3-dimethyl-12-thioxopyrido[1′,2′:3,4]-imidazo[1,2-a]benzimidazol-2(12H)-one,G.9-methoxy-1,3-dimethyl-12-thioxopyrido[1′,2′:3,4]-imidazo[1,2-a]benzimidazol-2(12H)-one,H.2-[(RS)-[(4-chloro-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-5-methoxy-1H-benzimidazole,I.4-methoxy-2-[[(5-methoxy-1H-benzimidazol-2-yl)sulfonyl]methyl]-3,5-dimethylpyridine1-oxide.01/2009:2374corrected6.7OMEPRAZOLE MAGNESIUMOmeprazolummagnesicumC34H36MgN6O6S2Mr713[95382-33-5]DEFINITIONMagnesium bis[5-methoxy-2-[(RS)-[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl]-1H-benzimidazol-1-ide].It contains a variable quantity of water.Content:97.5per cent to102.0per cent(anhydrous substance).CHARACTERSAppearance:white or almost white,hygroscopic powder.Solubility:very slightly soluble in water,sparingly soluble inmethanol,practically insoluble in heptane.IDENTIFICATIONCarry out either tests A,B,C or tests A,B,D.A.Optical rotation(2.2.7):−0.10°to+0.10°.Dissolve0.250g in methanol R and dilute to25.0mL withthe same solvent.B.Infrared absorption spectrophotometry(2.2.24).Comparison:omeprazole magnesium CRS.C.Atomic absorption spectrometry(2.2.23)as described inthe test for magnesium.The test solution shows the absorption maximum at285.2nm.D.Ignite about0.5g of the substance to be examinedaccording to the procedure for the sulfated ash test(2.4.14).Dissolve the residue in10mL of water R.2mL of thissolution gives the reaction of magnesium(2.3.1).TESTSAbsorbance(2.2.25):maximum0.10at440nm.Dissolve0.500g in methanol R and dilute to25.0mL with thesame solvent.Filter the solution through a membranefilter(nominal pore size0.45μm).Related substances.Liquid chromatography(2.2.29):use thenormalisation procedure.Prepare the solutions immediatelybefore use.Test solution.Dissolve3.5mg of the substance to be examinedin the mobile phase and dilute to25.0mL with the mobilephase.Reference solution(a).Dissolve1mg of omeprazole CRS and1mg of omeprazole impurity D CRS in the mobile phase anddilute to10.0mL with the mobile phase.Reference solution(b).Dissolve3mg of omeprazole for peakidentification CRS(containing impurity E)in the mobile phaseand dilute to20.0mL with the mobile phase.Reference solution(c).Dilute1.0mL of the test solution to100.0mL with the mobile phase.Dilute1.0mL of this solutionto10.0mL with the mobile phase.Column:–size:l=0.125m,Ø=4.6mm;–stationary phase:octylsilyl silica gel for chromatography R(5μm).2912See the information section on general monographs(cover pages)。
200种化学品理化特性表(83页)

200种化学品目录丙烯酸丁酯的理化及危险特性表 (1)丙烯酸的理化及危险特性表 (2)硝酸锌的理化及危险特性表 (3)过氧化二苯甲酰的理化及危险特性表 (4)过硫酸铵的理化及危险特性表 (5)丁酮的理化及危险特性表 (6)苯酚的主要理化和危险特性 (7)甲基叔丁基醚的主要理化及危险特性 (8)三聚氰酸的主要理化和危险特性 (9)季戊四醇的主要理化和危险特性 (10)硬脂醇的主要理化和危险特性 (11)多聚甲醛的主要理化和危险特性 (12)1,2-二甲苯的主要理化及危险特性 (13)1,3-二甲苯的主要理化及危险特性 (14)1,4-二甲苯的主要理化及危险特性 (15)环己烯的主要理化和危险特性 (16)二丁胺的主要理化和危险特性 (17)三乙胺的主要理化和危险特性 (18)甲基二氯硅烷的主要理化和危险特性 (19)异丁烯的主要理化和危险特性 (20)邻叔丁基苯酚的主要理化和危险特性 (21)对叔丁基苯酚的主要理化和危险特性 (22)环己烷的主要理化和危险特性 (23)二氯甲烷的主要理化和危险特性 (24)三氯甲烷的主要理化及危险特性 (25)硫脲的主要理化和危险特性 (26)六亚甲基四胺的主要理化和危险特性 (27)抗氧剂AT-10的主要理化和危险特性 (28)抗氧剂AT-76的主要理化和危险特性 (29)抗氧剂AT-3114的主要理化和危险特性 (30)催化剂JH-CMMS的主要理化和危险特性 (31)抗氧剂AT-168的主要理化和危险特性 (32)甲氧基钠的主要理化和危险特性 (33)氧气的理化及危险特性 (34)硝酸钾的理化及危险特性表 (35)硝酸钙的理化及危险特性表 (36)液氯的理化及危险特性 (37)盐酸的理化及危险特性 (38)丙酮理化及危险特性表 (39)次氯酸钠的理化及危险特性表 (40)二甲氧基甲烷的理化及危险特性表 (41)乙酸的理化及危险特性表 (42)香蕉水的理化及危险特性表 (43)甲醇的理化及危险特性表 (44)氰化亚铜的理化及危险特性 (45)硝酸的理化及危险特性 (46)硫酸的理化及危险特性 (47)铬酸的理化及危险特性 (48)铬酸酐的理化及危险特性 (49)氰化钠的理化及危险特性 (50)丁醇的理化及危险特性 (51)异丙醇的的理化及危险特性 (52)硝酸银的理化及危险特性表 (53)间戊二烯的理化及危险特性 (54)环戊二烯的理化及危险特性 (55)甲苯二异氰酸酯的理化及危险特性表 (56)200#溶剂油的理化及危险特性表 (57)聚氨酯漆固化剂理化及危险特性表 (58)聚氨酯漆稀释剂理化及危险特性表 (59)N,N--二甲基甲酰胺理化及危险特性 (60)丙烯腈理化及危险特性 (61)二氧化硫的特性 (62)异戊二烯的理化及危险特性 (63)醋酸丁酯的理化及危险特性 (64)醋酸乙酯的理化及危险特性 (65)甲苯的理化及危险特性 (66)环己酮的理化及危险特性 (67)三氯化铝(无水)的理化及危险特性 (68)氢氧化钠的理化及危险特性 (69)硝酸铅的理化及危险特性表 (70)电石理化及危险特性表 (71)乙炔理化及危险特性表 (72)氰化亚铜的理化及危险特性 (73)丙烯酸甲酯理化及危险特性................ 错误!未定义书签。
EPZ015666_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:EPZ015666 is an orally available inhibitor of PRMT5 enzymatic activity in biochemical assays with IC 50 of 22 nM and broad selectivity against a panel of other histone methyltransferases.IC50 & Target: IC50: 22 nM (PRMT5)[1]In Vitro: Treatment of MCL cell lines with EPZ015666 leads to inhibition of SmD3 methylation and cell death, with IC 50 values in the nanomolar range [1]. EPZ015666, a potent peptide–competitive and SAM–cooperative inhibitor with >10,000–fold specificity againstPRMT5 relative to other methyltransferases [2].In Vivo: EPZ015666 is orally bioavailable and amenable to in vivo studies. We performed 21–d efficacy studies in severe combined immunodeficiency (SCID) mice bearing subcutaneous Z–138 and Maver–1 xenografts, with twice–daily (BID) oral dosing on four dose groups: 25, 50, 100 and 200 mg per kilogram of body weight (mg/kg). After 21 d of continuous dosing, animals areeuthanized, and blood and tissues are analyzed to determine the relationship between methyl–mark pharmacodynamics andtumor–growth inhibition (TGI). EPZ015666 showed dose–dependent exposure and TGI after 21 d in both MCL models. Tumors in all EPZ015666 dose groups measured on day 21 showed statistically significant differences in weight, volume and tumor growth compared to vehicle–treated tumors. Dosing at 200 mg/kg BID induced tumor stasis in Z–138 cells, with >93% TGI after 21 d,whereas Maver–1 cells showed >70% TGI. Additionally, a third MCL xenograft is tested using the Granta–519 cell line, afast–growing model that reached endpoint on day 18 and showed dose–dependent efficacy with 45% TGI in the 200 mg/kg group.EPZ015666 is well tolerated in all three models, with minimal bodyweight loss in the 200 mg/kg dose group and no other clinical observations [1].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]EPZ015666 is serially diluted threefold from 1,000 to 0.051 nM and spotted into a 384–well polypropyleneV–bottom microplate. 3H–SAM is serially diluted twofold in assay buffer for a seven–point dilution series with a top concentration of 700 nM (final assay concentration). Reactions are initiated by the addition of 4 nM enzyme and 40 nM peptide (final assayconcentrations for both). Reactions are incubated for 60 min and quenched by the addition of 10 μL per well of 600 μM unlabeled SAM in assay buffer (final assay concentration). For the peptide competition, EPZ015666 is serially diluted threefold from 1,000 to 0.051 nM and spotted into a 384–well polypropylene V–bottom microplate. Peptide is serially diluted twofold in assay buffer for a seven–point dilution series with a top concentration of 480 nM (final assay concentration). Reactions are initiated by the addition of 4 nM enzyme and 75 nM 3H–SAM (final assay concentrations for both). Reactions are incubated for 60 min, and the reactions are quenched by the addition of 10 μL per well of 600 μM unlabeled SAM in assay buffer (final assay concentration)[1].Cell Assay: EPZ015666 is dissolved in DMSO and stored, and then diluted with appropriate medium (final DMSO 0.2%)before use [1].[1]Cultured cells in linear/log–phase growth are split to a seeding density of 2×105 cells/mL in 2–20 mL of media,depending on the yield required at the end of the growth period. Compound is diluted in DMSO and added to each culture vesselProduct Name:EPZ015666Cat. No.:HY-12727CAS No.:1616391-65-1Molecular Formula:C 20H 25N 5O 3Molecular Weight:383.44Target:Histone Methyltransferase Pathway:Epigenetics Solubility:DMSOwith a final DMSO concentration of 0.2%. Cells are allowed to grow for 96 h undisturbed. At the conclusion of each treatment period, cells are harvested by centrifugation (5 min, 1,200 rpm), and cell pellets are rinsed once with PBS before being frozen on dry ice pending further processing. Long–term proliferation assays are performed on all MCL lines, with slight adjustments to initial seeding densities, depending on growth characteristics for each cell line. All assays are carried out for 12 d[1].Animal Administration: EPZ015666 is formulated in 20% N–N–dimethylacetamide in water (Mice)[1].[1]Mice[1]Male CD–1 mice (25–40 g; n=6, with 3 per time point) are treated with a single dose of EPZ015666 at 2 mg/kg by intravenoustail–vein injection and 10 mg/kg by oral gavage administration, with both doses formulated in 20% N–N–dimethylacetamide in water. Animals are fasted overnight and weighed before dose administration on the day of dosing. Approximately 30 μL ofblood are taken from animals by submandibular or retro–orbital bleeding at pre–specified time intervals (seven time points). For the last time point (24 h), samples are collected via cardiac puncture while the animals are under anesthesia (70% CO2:30% O2). Blood samples are transferred into K2–EDTA tubes and placed on wet ice before centrifugation at 4°C (3,000g, 15 min) to obtain plasma within 30 min after sample collection. Plasma samples are stored at -70±10°C before protein precipitation and LC–MS/MS analysis. We constructed standard calibration curves by analyzing a series of control plasma aliquots containing 100 ng/mL labetalol as an internal standard and 1–3,000 ng/mL EPZ015666. Four levels of quality control are also included in the analysis (3–2,400 ng/mL EPZ015666). Data are analyzed using Phoenix WinNonlin 6.2.1.References:[1]. Chan–Penebre E, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015 Jun;11(6):432–7.[2]. Kryukov GV, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016 Mar 11;351(6278):1214–8.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
CAS号413611-93-5_10074-G5_MedBio_物理性质

1、产品物理参数:
常用名
10074-G5
英文名
10074-G5
CAS号
413611-93-5
分子量
332.313
密度
1.4±0.1 g/cm3
沸点
538.6±60.0 °C at 760 mmHg
分子式
C18H12N4O3
熔点
无资料
闪点
279.5±32.9 °C
2、技术资料:
体外研究
10074-G5抑制Daudi Burkitt淋巴瘤细胞的生长并破坏c-Myc / Max二聚化。针对Daudi和HL-60细胞的IC50值分别为15.6和13.5μM[1]。10074-G5在区域Arg363-Ile381中结合Myc肽Myc353-437,Kd值为2.8μM。10074-G5结合在由诱导螺旋结构域(Leu370-Arg378)的N末端的扭结(Asp379-Ile381)产生的空腔中[3]。
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11457
CDK inhibitor II
CDK inhibitor II
1269815-17-9
50mg
≥98%
品牌
货号
中文名称
英文名称
CAS
包装
纯度
MedBio
MED11591
(S)-CCG-1423
(S)-CCG-1423
None
体内研究
静脉注射20 mg / kg小鼠的血浆半衰期为10074-G5,为37分钟,血药浓度峰值为58μM,比肿瘤峰值浓度高10倍[1]。
3、同类产品列表:
WHO_TRS_937__annex8_eng

© World Health OrganizationWHO Technical Report Series, No. 937, 2006Annex 8Proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release, solid oral dosage forms Introduction1. Background2. WHO revisions to the criteria for Biopharmaceutics Classifi cation Systemclassifi cation3. WHO extensions to the scope of application of the biowaiver4. WHO additional criteria for application of the biowaiver procedure5. Explanation of the tables6. Biowaiver testing procedure according to WHOIntroductionThis proposal is closely linked to the Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchange-ability (WHO Technical Report Series, No. 937, Annex 7). It aims to give national authorities suffi cient background information on the various orally administered active pharmaceutical ingredients (APIs) on the WHO Model List of Essential Medicines (EML), also taking into account local usage of the API, to enable them to make an informed decision as to whether generic formulations should be subjected to in vivo bioequivalence (B E) studies or whether a biowaiver can be granted. In light of scientifi c work and dis-cussion in the last decade, some of the criteria used to evaluate the API in terms of potential for a biowaiver have been revised to allow a broadened scope of application. The result is that many APIs on the EML can now be considered for the biowaiver procedure, subject to the usage and risks in the national setting.1. Background1.1Initiatives to allow biowaivers based on the BiopharmaceuticsClassifi cation SystemIn 1995 the American Department of Health and Human Services, US Food and Drug Administration (HHS-FDA) instigated the B iopharmaceutics391Classifi cation System (BCS), with the aim of granting so-called biowaiv-ers for scale-up and post-approval changes (SUPAC) (/cder/ guidance/cmc5.pdf). A biowaiver means that in vivo bioavailability and/or bioequivalence studies may be waived (i.e. not considered necessary for product approval). Instead of conducting expensive and time-consuming in vivo studies, a dissolution test could be adopted as the surrogate basis for the decision as to whether two pharmaceutical products are equivalent. At that time the biowaiver was only considered for SUPAC to pharmaceutical products.More recently, the application of the biowaiver concept has been extended to approval of certain orally administered generic products (/ cder/guidance/3618fnl.htm).Within the context of the documents cited above, only APIs with high solu-bility and high permeability and which are formulated in solid, immediate-release (IR) oral formulations can be approved on the basis of the biowaiver procedure. A major advantage of the biowaiver procedure is the simplifi ca-tion of the product approval process and the reduction of the time required, thus reducing the cost of bringing new products to market.1.2What is the Biopharmaceutics Classifi cation System?The Biopharmaceutics Classifi cation System (BCS) was proposed in 1995 by Amidon et al.1 It is a scientifi c framework which divides APIs into four groups, according to their solubility and permeability properties.1.3 Classifi cation of active pharmaceutical ingredients accordingto the Biopharmaceutics Classifi cation SystemAccording to the HHS-FDA defi nitions in the documents cited above, the four possible categories for an API according to the BCS are:•BCS class I: “high” solubility – “high” permeability•BCS class II: “low” solubility – “high” permeability•BCS class III: “high” solubility – “low” permeability•BCS class IV: “low” solubility – “low” permeability.Depending on the classifi cation, the oral availability of the API may be expected to range from being heavily dependent on the formulation and manufacturing method (e.g. Class II APIs: poorly soluble yet highly perme-able) to being mostly dependent on the API permeability properties (e.g.Class III APIs: highly soluble yet poorly permeable).1Amidon GL, Lennemas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classifi cation: the correlation of in vitro drug product dissolution and in vivo bioavailability. Phar-maceutics Research, 1995, 12:413–420.3921.4How is high or low solubility currently defi ned by the Departmentof Health and Human Services, US Food and Drug Administration?The aqueous solubility of a drug substance is considered as high according to the HHS-FDA BCS criteria when:• the ratio of the highest orally administered dose (in mg) to the solubility (mg/ml) is 250 ml or lower.—This criterion is met over the pH range 1–7.5 at 37 °C.According to HHS-FDA guidances, the determination of the equilibrium solubility should be carried out with the shake-fl ask method (other methods such as acid or base titration are permitted when their ability to predict the equilibrium solubility is justifi ed). The experiments should be carried out at a temperature of 37 ± 1°C. Further, a suffi cient number of pH conditions should be chosen to cover the pH range of 1–7.5 and each determination should be carried out at least in triplicate. The buffer solutions given in the United States Pharmacopeia (USP) are appropriate for the tests, but other buffers are also allowed for these experiments. The pH value of each buffer solution should be checked before and after each experiment. Degradation of the API due to pH or buffer composition should be reported together with other stability data.The reason for the 250-ml cut-off criterion for the dose:solubility ratio is that in pharmacokinetic bioequivalence studies, the API formulation is to be ingested with a large glass of water (8 ounces corresponds to about 250 ml). If the highest orally administered dose can be completely dissolved in this amount of water, independent of the physiological pH value (hence the determination over the pH range 1–7.5), solubility problems are not expected to hinder the uptake of the API in the small intestine.The other important parameter for the BCS is the intestinal permeability of the API.1.5How is high or low permeability currently defi ned by the Departmentof Health and Human Services, US Food and Drug Administration?According to HHS-FDA a drug is considered highly permeable, when 90 % or more of the orally administered dose is absorbed in the small intestine.Permeability can be assessed by pharmacokinetic studies (for example, mass balance studies), or intestinal permeability methods, e.g. intestinal perfusion in humans, animal models, Caco 2 cell lines or other suitable, validated cell lines. In vivo or in situ animal models or in vitro models (cell lines) are only considered appropriate by HHS-FDA for passively trans-ported drugs. It should be noted that all of these measurements assess the fraction absorbed (as opposed to the bioavailability, which can be reduced substantially by fi rst-pass metabolism).393HHS-FDA suggests use of two different methods for determining the per-meability classifi cation if results with one method are inconclusive.1.6Which pharmaceutical formulations can currently be consideredfor a biowaiver according to the Department of Health andHuman Services, US Food and Drug Administration?To be considered bioequivalent according to the HHS-FDA biowaiver pro-cedure, a pharmaceutical product:• should contain a Class I API;• should be rapidly dissolving, meaning it should release at least 85% of its content in 30 minutes in three different media (pH 1.2, pH 4.5 and pH6.8, composition see “Multisource document”)1 in a paddle (50 rpm) orbasket (100 rpm) apparatus at 37 °C and a volume of 900 ml;• should not contain excipients which could infl uence the absorption of the API;• should not contain an API with a narrow therapeutic index; and• should not be designed to be absorbed from the oral cavity.The reasoning for the above-mentioned dissolution restrictions is that whena highly soluble, highly permeable API dissolves rapidly, it behaves like asolution in the gastrointestinal tract. If this is the case, the pharmaceutical composition of the product is insignifi cant, provided that excipients which infl uence the uptake across the gut wall are excluded from the formulation.The API is not prone to precipitation after its dissolution due to its good solu-bility under all pH conditions likely to be found in the upper gastrointestinal tract. The high permeability ensures the complete uptake (> 90%) of the API during its passage through the small intestine. The rapid dissolution of the product guarantees that the API is available long enough for the uptake in the small intestine (the passage time in the small intestine is approximately four hours) and negates any slight differences between the formulations.Pharmaceutical products containing an API with a narrow therapeutic index should always be tested with in vivo methods, because the risk to the patient resulting from a possible incorrect bioequivalence decision using the bio-waiver procedure is considered too high with these kinds of APIs.As the BCS is only applicable to APIs which are absorbed from the small intestine; drugs absorbed from other sites (e.g. from the oral cavity) are not eligible for a biowaiver.It is clear that the HHS-FDA requirements for the classifi cation of APIs and eligibility criteria for the biowaiver are very strict. During the last decade,1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).394several publications and continuing scientifi c discussions have suggested that the original HHS-FDA criteria for application of the biowaiver pro-cedure could be relaxed without substantially increasing the risk to public health or to the individual patient. On the basis of these publications and dialogue, WHO has proposed revised BCS criteria and additional consid-erations for the eligibility of a pharmaceutical product for the biowaiver procedure in the “Multisource document”.12.WHO revisions to the criteria for BCS classifi cationWHO revisions to the BCS criteria are as follows:•WHO high-solubility defi nitionWhen an API shows a dose:solubility ratio of 250 ml or lower at 37 °C over a pH range of 1.2–6.8, it can be classifi ed as “highly soluble”. The decrease in pH from 7.5 in the FDA guidances to 6.8 refl ects the need to dissolve the drug before it reaches the mid-jejunum to ensure absorption from the gastrointestinal tract.• Furthermore, the dose that is to be used for the calculation is the highestdose indicated in the Model List of Essential Medicines (EML). In some countries, products may be available at doses exceeding the highest dose on the EML. In such cases, the classifi cation given in the tables at the end of this Annex may no longer be appropriate and the dose:solubil-ity ratio and the permeability will have to be reassessed at the product dose.•WHO permeability defi nitionWhen an API is absorbed to an extent of 85% or more, it is considered to be “highly permeable”. The permeability criterion was relaxed from 90% in the FDA guidance to 85% in the WHO “Multisource document”.Some examples of APIs now included in BCS Class I that were previ-ously considered to be in Class III are paracetamol, acetylsalicylic acid, allopurinol, lamivudine and promethazine.Application of these revised criteria has changed the classifi cation of some APIs in the list. Thus, the classifi cations in the tables attached to this docu-ment supersede those in previous publications. As new APIs appear on the EML, it will be necessary to classify them according to the revised BCS;so it is therefore anticipated that the tables will be revised regularly. In addition, some APIs have not yet been suffi ciently characterized to assign them a BCS classifi cation. As the tables evolve, it is anticipated that more concrete information will be generated for these APIs as well.1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).395the basket apparatus (applies to pharmaceutical products containingClass III APIs);—rapidly dissolving (release of > 85% of the labelled amount of drug in 30 minutes) in standard media at pH 1.2, 4.5 and 6.8, at a rota-tional speed of 75 rpm in the paddle apparatus or 100 rpm in the bas-ket apparatus (applies to pharmaceutical products containing Class IAPIs and/or Class II APIs which are weak acids and meet the 250 mldose:solubility requirement at pH 6.8).(4)Considerations relating to excipientsThe national authority should be aware that some excipients can infl uencemotility and/or permeability in the gastrointestinal tract. Therefore, the ex-cipients used in the multisource product formulation should be scrutinized.In this regard, the national authority can draw on the experience relat-ing to formulations which have been approved on the basis of humanbioequivalence studies in their own or in other jurisdictions.If the multisource product under consideration contains excipients thathave been used before in similar amounts in other formulations of thesame API, it can be reasonably concluded that these excipients will haveno unexpected consequences for the bioavailability of the product. If,however, the formulation contains different excipients, or amounts ofthe same excipients that are very different from usual, the national au-thority may choose to declare the biowaiver procedure inapplicable.A list of usual and acceptable excipients can be found at the following website: /cder/iig/iigfaqWEB.htm; formulations of some productscan be found on the web sites of some national drug regulatory authorities.5.Explanation of the tablesThe decision of a national authority to allow a biowaiver based on the BCS should take into consideration the solubility and permeability char-acteristics as well as the therapeutic use and therapeutic index of the API, its pharmacokinetic properties, the similarity of the dissolution profi les of the multisource and the comparator products in standard buffers with a pH of 1.2, pH 4.5 and pH 6.8 at 37 °C. Data related to the excipients compo-sition in the multisource product are also required. A systematic approach to the biowaiver decision has been established by the International Pharma-ceutical Federation (FIP) and published in the Journal of Pharmaceutical Sciences (/cgi-bin/jhome/68503813).The relevant documents can also be downloaded from the FIP web site at: http://www.fi/. These monographs provide detailed information which should be taken into account whenever available in the biowaiver consideration.3985.1Which active pharmaceutical ingredients are included in thetables?The substances listed in the 14th WHO Model List of Essential Medicines (EML) of March 2005 have been evaluated and classifi ed according to the revised criteria given above.5.2Where do the data come from?The solubility and permeability values were found in the publicly available literature, such as Martindale’s, the Merck Index and scientifi c journals.Please note that the doses used for the calculation of the dose:solubility ratio are those stated in the EML.The indications given in the tables are reproduced directly from the EML. If the EML specifi es the dosage form (e.g. sublingual tablet) this is indicated under “comments”.5.3“Worst case” approach to the Biopharmaceutics Classifi cationSystemThe drugs listed in the EML were classifi ed according to the criteria explained above. Where no clear classifi cation could be made, the “worst case” was as-sumed. For example if a substance is highly soluble, but absolute bioavailability data were not available, the test conditions for BCS Class III substances have been proposed. The same procedure was adopted for fi xed combinations, for example amoxicillin and clavulanic acid, the testing procedure was always fi xed according to the “worst” BCS classifi cation, in this example clavulanic acid (BCS Class III/1), because amoxicillin is a BCS Class I drug. This com-bination would therefore be tested according to BCS Class III requirements.The results of the revised classifi cation can be found in Tables 1–3.5.4Why are there three Tables?Table 1 lists all APIs on the EML that are administered orally, with the excep-tion of the APIs listed as complementary. Table 2 summarizes the APIs listed as complementary in the EML and Table 3 lists the APIs for which no classifi cation had previously been assigned, or that had been introduced with the 14th EML (March 2005), together with a more detailed explanation of their classifi cation.5.5 Risk assessmentTo minimize the risks of an incorrect biowaiver decision in terms of public health and risks to individual patients, the therapeutic indications of the API, known pharmacokinetic variations, food effects, etc. should be evalu-ated based on local clinical experience, taking into account the indications399for which the API is prescribed in that country as well as specifi c pharmaco-kinetic population variations (for example CYP polymorphisms). Known potential risks are listed under “potential risks” in the tables. The absence of an entry under “potential risks” should not, however, be misconstrued as meaning that there are no risks associated with the use of the medicine. 6.Biowaiver testing procedure according to WHODepending on the BCS classifi cation of the API, based on solubility and permeability characteristics listed in the accompanying tables, the testing procedure is defi ned in section 9.2.1 of the “Multisource document”1:6.1For pharmaceutical products containing BiopharmaceuticsClassifi cation System Class I (highly soluble, highlypermeable) APIsFor rapidly dissolving (as defi ned above) pharmaceutical products contain-ing BCS Class I APIs, more than 85% dissolution of the labelled amount is required within 30 minutes in standard media at pH 1.2, 4.5 and 6.8 using the paddle apparatus at 75 rpm or the basket apparatus at 100 rpm. The dis-solution profi les of the comparator and the multisource products should be compared by an f> 50 or an equivalent statistical criterion.2If within 15 minutes more than 85% of the API are released from the compar-ator and the multisource formulation under the above-mentioned conditions the products will be considered very rapidly dissolving. In this case the prod-ucts are deemed to be equivalent and a profi le comparison is not required.6.2For pharmaceutical products containing BiopharmaceuticsClassifi cation System Class III (highly soluble, lowpermeability) APIsA biowaiver can be considered only if both the multisource and the com-parator product are very rapidly dissolving. Eighty-fi ve per cent or more dissolution of the labelled amount of the API should be achieved within15 minutes in standard media at pH 1.2, 4.5 and 6.8 using the paddle ap-paratus at 75 rpm or the basket apparatus at 100 rpm.Generally, the risks of an inappropriate biowaiver decision should be more critically reviewed (e.g. site-specifi c absorption, induction/competition at the absorption site, excipient composition and therapeutic risks) for prod-ucts containing BCS Class III APIs than for BCS Class I drugs.1Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).4006.3For pharmaceutical products containing APIs with highsolubility at pH 6.8 but not at pH 1.2 or 4.5 and with highpermeability (by defi nition, BCS Class II compoundswith weak acidic properties)These are eligible for a biowaiver provided that the multisource product:• is rapidly dissolving, i.e. 85% or more dissolution of the labelled amount of the API should be achieved within 30 minutes in standard media at pH 6.8 using the paddle apparatus at 75 rpm or the basket apparatus at 100 rpm; and• the multisource product exhibits similar dissolution profiles, as deter-mined with the f2 value or equivalent statistical evaluation, to those ofthe comparator product in buffers at all three pH values (pH 1.2, 4.5 and6.8).For multisource products containing BCS Class II APIs with dose:solubility ratios of 250 ml or less, at pH 6.8, the excipients should also be critically evaluated in terms of type and amounts of surfactants in the formulation.Further details of eligibility for the biowaiver and appropriate test proce-dures can be found in sections 5 and 9 of the “Multisource document”.11Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability (WHO Technical Report Series, No. 937, Annex 7).401405c h l o r p h e n a -m i n e h yd r o ge n m a l e a t e 4 m g h i g hB A 25-59%, fi r s t p a s s 3/19.2.1.2C Y P 2D 6 p o l y -m o r p h i s m a n t i a l l e r g i ce x t e n t of fi r s t -p a s s m e t a b o l i s m u n c e r t a i nc h l o r p r o m a z i n e h yd r o c h l o r i de 100 m gh i g hl o w39.2.1.2 p s y c h o t h e r a p e u -t i c m e d i c i n e c i p r o fl o x a c i nh y d r o c h l o r i d e 250 m g h i g hB A 70–82%, p o s s i b l e fi r s t p a s s , h i g h i nC a c o -2 c e l l s3/19.2.1.2 a n t i b a c t e r i a le x t e n t of fi r s t - p a s s m e t a b o l i s m u n c e r t a i nc l o f a z i m i n e100 m gi n s u f fi c i e n t l i t e r a t u r e l o w 4/3N o t e l i g i b l e f o r b i o w a i v e r a t p r e s e n t a n t i l e p r o s y m e d i c i n ec l o m i f e n e c i t r a t e50 m g h i g h i n s u f fi c i e n t l i t e r a t u r e 3/19.2.1.2o v u l a t i o n i n d u c e rc l o m i p r a m i n e h yd r o c h l o r i de 25 m g h i g h66% e x c r e t e d i n t h e u r i n e , t h e r e m a i n d e r b e i n g e l i m i -n a t e d i n t h e f a e c e s 3/19.2.1.2p s y c h o t h e r a p e u -t i c m e d i c i n el a c k o f a b s o l u t e b i o a v a i l a b i l i t y d a t ac l o x a c i l l i n (a s s od i u m s a l t )1000 m g h i g hl o w 39.2.1.2a n t i b a c t e r i a lc ode i n e p h o s p h a t e 30 m g h i g h l o w39.2.1.2r i s k o f a b u s eo p i o i d a n a l g e s i c ,d i a r r h oe a i n a d u l t sd a p s o n e100 m gl o w (w e a k b a s e ) h i g h 2N o t e l i g i b l e f o r b i o w a i v e rG 6P D d e fi -c i e n c ya n t i l e p r o s y m e d i c i n ed i a ze p a m 5 m g h i g hh i g h19.2.1.1p s y c h o t h e r a p e u -t i c m e d i c i n e s c o r e d t a b l e tB A , B i o a v a i l a b i l i t y ; G 6P D , g l u c o s e -6-p h o s p h a t e d e h y d r o g e n a s e .409g l y c e r y l t r i n i t r a t e 500 μgh i g hs u b l i n g u a l a p p l i c a t i o n ,p e r m e a b i l -i t y i n t h e o r a l c a v i t y m o r e i m p o r t a n t t h a n G I p e r m e a b i l i t y3/1N A hl o c a l a b s o r p t i o n a n t i a n g i n a l m e d i c i n e s u b l i n g u a l a p p l i c a t i o ng r i s e o f u l v i n 250 m gl o w (n e u t r a l ) h i g h2N o t e l i g i b l e f o r b i o w a i v e r a n t i f u n g a lh a l o p e r i d o l2 m gb o r d e r l i n e < 0.01 m g /m l 2l o w 4/3N o t e l i g i b l e f o r b i o w a i v e rp s y c h o t h e r a p e u -t i c m e d i c i n eh y d r a l a z i n e h y d r o c h l o r i d e50 m g h i g hl o w 39.2.1.2a n t i h y p e r t e n s i v e m e d i c i n eh y d r o c h l o r o -t h i a z i d e 25 m g h i g h l o w 39.2.1.2a n t i h y p e r t e n s i v e m e d i c i n e , d i u r e t i c a n d u s e d i n h e a r t f a i l u r es c o r e d t a b l e ti b u p r o f e n 400 m gl o w , w e a k a c i d (p K a 4.4,5.2)h i g h 29.2.1.3N S A I D , a n t i m i -g r a i n e m e d i c i n ei n d i n a v i r s u l f a t e 400 m g l o w l o w (?)4/2N o t e l i g i b l e f o r b i o w a i v e r C Y P 450 3A 4, f o o d e f f e c t (–)a n t i r e t r o v i r a lu n k n o w n w h e t h e r p o o r B A i s d u e t o p o o r s o l u b i l i t y o r p o o r s o l u b i l i t y a n d p o o r p e r m e a b i l i t yD :S , D o s e :s o l u b i l i t y r a t i o ; B A , b i o a v a i l a b i l i t y .426T a b l e 3C o m p o u n d s i n t r o d u c e d t o t h e W H O M o d e l L i s t o f E s s e n t i a l M e d i c i n e s s i n c e M a r c h 2005 f o r w h i c h n o c e r t a i n c l a s s i fi c a t i o n h a d b e e n p r e v i o u s l y r e p o r t e d (t h e s e c o m p o u n d s a l s o a p p e a r i n T a b l e 1 a n d T a b l e 2)M e d i c i n e aH i g h e s t o r a l s t r e n g t h a c c o r d i n g t o W H O E s s e n t i a l M e d i c i n e s L i s t a S o l u b i l i t y bP e r m e a b i l i t y c B C S c l a s s dD i s s o l u t i o n t e s t (f o r b i o w a i v e r )e P o t e n t i a l r i s k s fI n d i c a t i o n (s )a c c o r d i n g t o W H O E s s e n t i a l M e d i c i n e s L i s t (E M L )aC o m m e n t s a n d s p e c i a l d o s a g e f o r m i n d i c a t i o n s aa m l o d i p i n e 5 m gs l i g h t l y s o l u b l e (1),D :S 5 m lB A a b s60–65%,e x c r e t i o n o f d r u g m e t a b o -l i t e s i n u r i n e 90–95% (2)19.2.1.1a n t i h y p e r t e n s i v e m e d i c i n eB A a b s < 85% a s c r i b e d t o fi r s t -p a s s m e t a b o l i s ma m o d i a q u i n e(b a s e )200 m g45 m g /m l 2,D :S 4.4 m lB A > 75% (3)3/19.2.1.2C Y P 2C 8p o l y m o r p h i s m ,i n c r e a s e d r i s k f o r a g r a n u l o c y -t o s i s a n d h e p a -t o t o x i c i t y (4)a n t i m a l a r i a la m o x i c i l l i n + c l a v u l a n i c a c i d 500 m g + 125 m gf r e e l y s o l u b l e i n w a t e r (1),D :S 1.25 m la b s o r p t i o n > 73% (5)1 + 3/19.2.1.2a n t ib ac t e r i a lt e s t s b a s e d o n c l a v u l a n i c a c i d c l a s s i fi c a t i o na r t e s u n a t e 50 m gv e r ys l i g h t l y s o l u b l e (6),D :S 500 m l ;(w e a k a c i d ,p K a ~ 6.4)B A a b s 82% (1),B A a b s 88% (7),B A a b s 61% (8)4/2N o t e l i g i b l e f o r b i o w a i v e ra n t i m a l a r i a lp e r m e a b i l i t y d e p e n d s o n s e v e r i t y o f d i s e a s eD :S , D o s e : s o l u b i l i t y ; B A , B i o a v a i l a b i l i t y .427a z i t h r o m y c i n 500 m gp r a c t i c a l l y i n s o l u b l e i n w a t e r (1)< 0.01m g /m l , D :S 50 000 m lB A a b s 16% (9);B A 37%(10, 11); 4/2N o t e l i g i b l e f o r b i o w a i v e ra n t ib ac t e r i a l u n k n o w n w h e t h e r p o o r B A i sd ue t o p o o r s o l u b i l i t y o r p o o r s o l u b i l i t y a n d p o o r p e r m e a b i l i t yc a l c i u m f o l i n a t e 15 m gs p a r i n g l y s o l u b l e i n w a t e r (P h . E u r . 5.2); v e r y s o l u b l e (U S P 28); D :S 15 m l a n d 0.015 m l , r e s p e c -t i v e l yB A a b s 92% 25 m g (12, 13);B A a b s 73.4%(15 m g ) (14);f u l l y a b s o r b e d ;A UC a n d t 1/2s i m i l a r a f t e r i.v . & p .o (15)19.2.1.1 a n t i c y t o t o x i c m e d i c i n el e v o d o p a (l ) + c a r b i d o p a (c )(l ) 250 m g + (c ) 25 m g(l ) h i g h +(c ) s o l u b l e 1 i n 500 o f w a t e r , f r e e l y s o l u b l e i n 3 M H C l (1)(l ) h i g h +(c ) B A 58% (16); B A a b s88% (d o g s ) (17)(l ) 1 +(c ) 3/19.2.1.2n a r r o w t h e r a p e u t i c i n d e xa n t i p a r k i n s o n m e d i c i n et e s t s b a s e d o n c a r b i d o p a c l a s s i fi c a t i o nc e fi x i m e 400 m gs l i g h t l ys o l u b l e (2),D :S 400 m l22–54% (2)4N o t e l i g i b l e f o r b i o w a i v e ra n t ib ac t e r i a lD :S , D o s e : s o l u b i l i t y ; B A : B i o a v a i l a b i l i t y ; P h .E u r ., E u r o p e a n P h a r m a c o p o e i a ; U S P , U n i t e d S t a t e s P h a r m a c o p o e i a ; A U C , a r e a u n d e r t h e c u r v e ; i.v ., i n t r a v e n o u s .。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Inhibitors, Agonists, Screening Libraries
Data Sheet
BIOLOGICAL ACTIVITY:
VO–Ohpic trihydrate is an extremely potent inhibitor of PTEN with IC 50 of 46±10 nM.
IC50 & Target: IC50: 46±10 nM (PTEN)[1]
In Vitro: VO–OHpic with two OHpic ligands and an oxo ligand is a sterically demanding molecule, and one will therefore expect that binds substrate will affect the subsequent binding of the inhibitor due to steric hindrance. VO–OHpic significantly inhibits PTEN activity in low nanomolar concentrations (IC 50, 46±10 nM), which is in agreement with the previously determined potency (IC 50,35±2 nM) in a PIP 3–based assay. The inhibition constants K ic and K iu are determined to be 27±6 and 45±11 nM, respectively [1].VO–OHpic is an encouragingly specific and potent PTEN inhibitor. VO–OHpic is the most potent inhibitor (IC 50=35 nM) of the PTEN lipid phosphatase activity [2].In Vivo: PTEN is inhibited in mice by intra–peritoneal injection of VO–OHpic (10 μg/kg) 30 min before ischemia and then exposed them to 30 min of ischemia and 120 min of reperfusion. At the end of the experiment, myocardial infarct size is measured by triphenyltetrazolium chloride (TTC). Myocardial infarct size is significantly decreased in VO–treated mice (25±6 vs. 56±5 %, n=7,P<0.01). There is no difference in the area at risk between these two groups (46±3 vs. 57±3 %, n=7, P>0.05)[3].
PROTOCOL (Extracted from published papers and Only for reference)
Kinase Assay:[1]VO–OHpic is dissolved in DMSO (100 μM) and diluted further to the required concentration with 1% DMSO. For inhibition studies, PTEN is preincubated with VO–OHpic at RT for 10 min before substrate is added to initialise the reaction.
Background absorbance (malachite green assay) and fluorescence (OMFP assay) are determined with VO–OHpic in assay buffer and corrected in the data analysis [1].
Animal Administration: VO–OHpic (VO) is prepared in saline (Mice)[3].[3]Mice [3]
The experiment is performed with male C57BL6 mice. Briefly, mice are anesthetized with pentobarbital (70 mg/kg). The left coronary artery is occluded about 1–2 mm below the left auricle. Reperfusion is accomplished by loosening the ligature. The PTEN inhibitor VO–OHpic is administered by intra–peritoneal injection at the dosage of 10 μg/kg once 30 min before ischemia. Saline is used as control. At the end of the experiment, the animals are euthanized by transecting the aorta and removing the heart for infarct size determination.
References:
[1]. Mak LH, et al. Characterisation of the PTEN inhibitor VO–OHpic. J Chem Biol. 2010 Oct;3(4):157–63.
[2]. Rosivatz, E, et al. A small molecule inhibitor for phosphatase and tensin homologue deleted on chromosome 10 (PTEN). ACS Chem Biol. 2006 Dec 15;1(12):780–90.
[3]. Zu L, et al. PTEN inhibitors cause a negative inotropic and chronotropic effect in mice. Eur J Pharmacol. 2011 Jan 10;650(1):298–302.
Product Name:
VO–Ohpic (trihydrate)Cat. No.:
HY-13074CAS No.:
476310-60-8Molecular Formula:
C 12H 16N 2O 11V+Molecular Weight:
415.20Target:
PTEN Pathway:
PI3K/Akt/mTOR Solubility:10 mM in DMSO; H 2
O: < 0.1 mg/mL
Caution: Product has not been fully validated for medical applications. For research use only.
Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@
Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。