FCCA013QualitySystemAuditO.
QSIT FDA官方培训讲义(FDA审核员现场审核技巧)

Management Controls Inspectional Objectives
1. Verify that a quality policy, management review and quality audit procedures, quality plan, and quality system procedures and instructions have been defined & documented.
Satellites
Sterilization Process Controls are inspected as a satellite system to the Production and Process Controls Subsystem Medical Device Reporting, Reports of Corrections and Removals, and Medical Device Tracking are satellites of CAPA
Management Controls Design Controls Corrective & Preventive Action Controls Production & Process Controls
The Three Other Subsystems
Facility & equipment controls Product controls Documents, records & change controls
QSIT
QSIT is a Top down approach
Medical device QMS GMP system and audit

QMS ordinance
MHLW Notifications
Overview of presentation
Guidelines
・ISO Documents ・GHTF SG3 Documents etc..
QMS inspection
PMDA FDA NB
QMS ISO13485 Ordinance
:2003
21CFR Part 820
QMS requirements
Establishment of QMS
Role of GHTF SG3
“SG3 is responsible for the task of examining existing quality system requirements in countries having developed device regulatory systems and identifying areas suitable for harmonization.” /sg3/
/sg3/sg3-final.html
GHTF SG3 Documents GHTF/SG3/N15: 2005 Implementation of Risk Management Principles and Activities within a Quality Management System
Seihin Hyojun Sho and ISO Documents
Seihin Hyojun Sho
QMS Ordinance requires that the manufacturer shall establish and maintain the document defining the product specifications and QMS requirements (Seihin Hyojun Sho), or identifying Seihin Hyojun Sho. (QMS Ordinance Article 6 Para 2) Seihin Hyojun Sho can be substituted by ISO documents if the relationship between Seihin Hyojun Sho and ISO documents is clearly identified.
一阶段审核指南

Guidance for Completing Quality Management SystemAudit Report (Stage 1)质量管理体系第一阶段审核报告编写指南Audit Conclusion and Recommendation:审核结论及推荐意见:Audit Objective:审核目的:1. The objective relating to the type of audit being conducted must be recorded, i.e. select theappropriate objective against the different audit type.必须记录与本次审核类型相关的审核目的。
Audit Summary:审核总结:Information relating to the organisation and its management system:有关被审核方及其管理体系的信息:Customers and markets of the client:产品最终的客户以及市场领域:Critical area, processes, functions to be stressed during stage 2 audit, and suggestions for planning stage 2 audit:第二阶段审核应关注的重点过程、重点区域、重点岗位,以及审核安排建议:Critical product realization processes identified including: 主要的产品实现过程已得到识别,包括: Main Areas/ Dept./ Personnel Involved 涉及的主要区域/或部门/或人员Name of process…过程的名称…Name of process…过程的名称…Name of process…过程的名称…Name of process…过程的名称… Processes outsourced… 要外包过程…Key processes identified including: 特殊/关键生产、服务过程, 包括: Main Areas/ Dept./ Personnel Involved 涉及的主要区域/或部门/或人员Name of key process… 特殊/关键过程的名称… Name of key process… 特殊/关键过程的名称… ……。
Walmart FCCA 质量体系审核验厂要求的详细解释及文件资料范例2020年11月9日更新
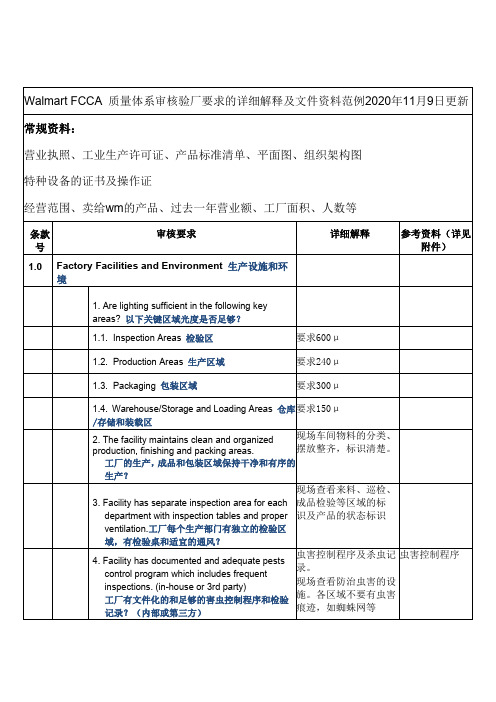
Walmart FCCA质量体系审核验厂要求的详细解释及文件资料范例2020年11月9日更新常规资料:营业执照、工业生产许可证、产品标准清单、平面图、组织架构图特种设备的证书及操作证经营范围、卖给wm的产品、过去一年营业额、工厂面积、人数等条款号审核要求详细解释参考资料(详见附件)1.0Factory Facilities and Environment生产设施和环境1.Are lighting sufficient in the following keyareas?以下关键区域光度是否足够?1.1.Inspection Areas检验区要求600μ1.2.Production Areas生产区域要求240μ1.3.Packaging包装区域要求300μ1.4.Warehouse/Storage and Loading Areas仓库/存储和装载区要求150μ2.The facility maintains clean and organized production,finishing and packing areas.工厂的生产,成品和包装区域保持干净和有序的生产?现场车间物料的分类、摆放整齐,标识清楚。
3.Facility has separate inspection area for eachdepartment with inspection tables and proper ventilation.工厂每个生产部门有独立的检验区域,有检验桌和适宜的通风?现场查看来料、巡检、成品检验等区域的标识及产品的状态标识4.Facility has documented and adequate pestscontrol program which includes frequentinspections.(in-house or3rd party)工厂有文件化的和足够的害虫控制程序和检验记录?(内部或第三方)虫害控制程序及杀虫记录。
FCCA验厂

FCCA验厂FCCA验厂是沃尔玛(Wal-mart)新推行的全称为:Factory Capability & Capacity Assessment,即工厂产量及能力评估,其目的是审核工厂的产量及生成能力是否符合沃尔玛的产能和质量要求,其主要内容包括以下几个方面:1、Factory Facilities and Environment工厂设施和环境2、Machine Calibration and Maintenance机器校准和维护3、Quality Management System质量管理体系4、Incoming Materials Control来料控制5、Process and Production Control过程和生产控制6、In-House Lab-Testing内部实验室测试7、Final inspection最终检验FCCA验厂文件和内容一、验厂文件的定义验厂文件是指工厂在验厂时为审核员提供工厂的一些基本文件资料及生产记录(包括:营业执照、员工手册、消防演习记录等),验厂文件主要是在人权、环境、安全、卫生和反恐及现场方面的文件资料,根据工厂的客户的要求可能侧重点有所不一样,有的重点是在人权,有的重点是在安全,这个要看工厂的客户是那一家。
专业的验厂文件是保证公司顺利通过验厂的护航使者根据自己的一些经历,总结的验厂文件清单请参考如下(并设有部分问卷):二、最新验厂文件清单文件资料1)品质手册和管理会议记录2)检验程序,检验标准及最近3个月的检验报告。
(来料,制程,包装,成品)3)主要机器设备清单(请准备中英文复印件各一份)4)机器设备保养计划和记录5)机器保养人员的专业证书6)产品规格书7)原料的来料和发料记录8)员工培训计划及培训记录9)产前会议程序及记录和质量会议记录10)供应商管理程序,记录,采购单及物料规格单11)实验室操作手册及测试报告12)缺陷统计报告及出货记录13)工厂组织架构图(请准备复印件一份)14)工厂营业执照(请准备复印件一份)15)当前的品质水准记录16)出货及时率的统计记录17)ISO证书及最近一次的审查报告(如有,请准备正本及复印件一份) 18)纠正预防措施的记录现场布设1)安全通道线2)安全出口、应急灯、火警铃3)化学药品摆放4)仓库特殊要点5)饮水区划分6)意见箱的设立及程序文件三、反恐重点C-TPAT证书保安制度装柜作业程序货柜检查程序及纪录封柜纪录封条管理程序则人权是随机抽查的FCCA验厂审核项目一、童工CL/未成年工U L二、非志愿劳工IL三、胁迫与骚扰CH四、不歧视DI五、最低工资WM六、加班工资OW七、加班时间OT八、社会保险SB九、福利OB十、监测与守法十一、卫生与安全HS&DO十二、宿舍DO十三、环保PE十四、分包Sub十五、其它要求OLFCCA质量验厂文件资料清单1. 营业执照 ;2. 质量体系认证证书 ;3. 组织架构图 ;4. 质量手册 ;5. 程序文件;6. 质量体系内审计划;7. 质量体系内审记录:1)内审员资格证书;2)首末次会议 ;3)检查表 ;4)不符合项报告;5)内审报告8. 质量体系管理评审计划9. 质量体系管理评审记录: 1)管理评审会议记录;2)管理评审报告;3)决议事项的跟进记录10. 主要生产设备清单;11. 设备保养计划 ;12. 设备保养记录;13. 仪器清单 ;14. 仪器校准计划 ;15. 仪器校准记录:1)外校报告 ;2)内校人员资格证书3)内校规程;4)内校报告16. 年度培训计划17. 培训记录 :1)签到表;2)测试卷18. 品管人员岗前资质认定资料(培训及测试记录;19. 新产品设计开发资料 :1)产品规格书;2) BOM表(BOM);3)安规认证证书;4)样品检测报告5)试产记录 ;6)试产评估报告 ;7)作业指导书 ;8)检验标准 ;9) FMEA分析资料 ;10)产品质量控制计划(QC工程图。
FDA行业指南 委托生产质量协议 中英文

FDA Guidance for Industry: ContractManufacturing ArrangementsforDrugs: QualityAgreements Page 1 / 21 Contract M anufacturing Arrangements for Drugs: Quality AgreementsGuidance for Industry行业指南:药品委托生产安排:质量协议U.S. Department of Health and Human Services Food and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Pharmaceutical Quality/Manufacturing Standards (CGMP)FDA 行业指南:药品委托生产安排:质量协议翻译:JuliaGuidance for IndustryAdditional copies are available from:Office of Communications, Division of Drug Information Center for Drug Evaluation and ResearchFood and Drug Administration10001 New Hampshire Ave., Hillandale Bldg., 4th Floor Silver Spring, MD 20993-0002Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353Email:druginfo@/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/orOffice of Communication, Outreach and Development Center for Biologics Evaluation and ResearchFood and Drug Administration10903 New Hampshire Ave., Bldg. 71, Room 3128 Silver Spring, MD 20993-0002Phone: 800-835-4709 or 240-402-8010Email:ocod@/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/orPolicy and Regulations Staff, HFV-6 Center for Veterinary Medicine Food and Drug Administration7519 Standish Place, Rockville, MD 20855/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/default.htmU.S. Department of Health and Human Services Food and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Biologics Evaluation and Research (CBER)Center for Veterinary Medicine (CVM)November 2016Pharmaceutical Quality/Manufacturing Standards (CGMP)TABLE OF CONTENTS 目录TABLE OF CONTENTS 目录 (3)I. INTRODUCTION 前言 (4)II. DEFINING THE WHO AND WHAT OF CONTRACT MANUFACTURING 指定委托生产的人和事 (6)III. RESPONSIBILITIES OF PARTIES INVOLVED IN CONTRACT MANUFACTURING 委托生产所涉及各方的职责7 IV. DOCUMENTING CGMP ACTIVITIES IN QUALITY AGREEMENTS 在质量协议中记录CGMP 活动 (10)A. What Is a Quality Agreement? 什么是质量协议? (10)B. Elements of a Quality Agreement 质量协议的要素 (11)V. ILLUSTRATIVE SCENARIOS 案例 (17)A. Owners and Contract Facilities Are Both Responsible for CGMP 所有者和受托场所是否都对CGMP 负责?18B. CGMPs Apply to all Contract Facilities, Including Analytical Testing Laboratories 适用于所有合同场所,包括分析化验室的CGMP (19)C. Owners and Contract Facilities Perform Change Control Activities 所有者和受托方实施变更控制活动20VI. RECOMMENDATIONS 建议 (21)Contract Manufacturing Arrangements for Drugs: QualityAgreementsGuidance for Industry1行业指南:药品委托生产安排:质量协议This guidance represents the current thinking of the Food and Drug Administration (FDA or Agency) onthis topic. It does not establish any rights for any person and is not binding on FDA or the public. You canuse an alternative approach if it satisfies the requirements of the applicable statutes and regulations. Todiscuss an alternative approach, contact the FDA office responsible for this guidance as listed on the titlepage.本指南代表FDA 当前对此问题的看法。
FDA最新工艺验证指南(2011.1版)北大中英对译-已打印

中文译稿:北京大学药物信息与工程研究中心 info@
另外的副本可从以下部门得到: 马里兰州银泉市新罕布什尔大道10193号2201室 药品信息处,对外信息办公室, 邮政编码:20993 电话:301-796-3400; 传真:301-847-8714
Guidance for Industry
行业指南 Process Validation: General Principles and Practices 工艺验证:一般原则与规范
U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Center for Veterinary Medicine (CVM) January 2011 Current Good Manufacturing Practices (CGMP) Revision 1 美国卫生与人类服务部 食品药品管理局 药物评价和研究中心(CDER) 生物制品评价和研究中心(CBER) 兽药中心(CVM) 2011年1月 现行药品质量生产管理规范(CGMP) 修订版 1
/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
and/or Communications Staff, HFV-12 Center for Veterinary Medicine Food and Drug Administration 7519 Standish Place, Rockville, MD 20855 (Tel) 240-276-9300
FDA 质量体系手册 第一章 质量体系法规

知识。
This manual covers the Quality System regulation and the basic Good Manufacturing Practices (GMP) requirements that all manufacturers and distributors must consider when they plan to manufacture medical devices, including medical device kits, trays or packs, for distribution in the United States. Model procedures and sample forms are also included in the manual to assist manufacturers. 本手册覆盖了所有制造商及分销商当他们计划在制造并且在美国分销医疗器械包括其套件, 托盘及包装时必须考虑的质量体系及 GMP 基本要求。本手册也包括流程模型,表格样本。
The Quality System regulation outlines the minimum elements of a system for designing and producing a medical device. Manufacturers of medical devices commonly find that their quality needs are broader than these basic elements because of the additional need to meet company quality claims as required by paragraph 501(c) of the Federal Food, Drug, and Cosmetic (FD&C) Act and to meet customer needs and requirements. 质量体系法规描述设计和生产医疗器械最少体系要素。医疗器械制造商通常会发现为了满足 其公司质量声明(如 FD&C 法案 501(c)章所要求)的额外要求和客户需求和要求,他们的 质量需求比这些基本要素(范围)更宽。 The DSMA staff and the Office of Compliance (OC) in the Center for Devices and Radiological Health (CDRH) provided valuable assistance in preparing this manual. CDRH 的 DSMA 工作人员及顺应性办公室(OC)在本手册的准备中提供了富有价值的辅助。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
FCCA Quality System Audit Outline FCCA 质量体系审核纲要 (Hardline 杂货 (Quality System part质量体系部分 Factory Quality System工厂质量体系 1.0 Factory Facilities and Environment工厂设施和环境 1.0.1 There is sufficient lighting on: Production, revising, finishing, inspection, packing and loading areas?
在生产,修理,加工,检验,包装及装载的区域是否有足够的照明? 1.0.2 The facility maintains clean and organized production, finishing and packing areas. 工厂是否保持清洁 , 在生产,加工和包装区域是否有秩序 ?
1.0.3 Facility has separate inspection area with inspection table and proper ventilation. 工厂是否有单独的检验区与检验台并且通风良好 ?
1.0.4 Facility has documented pests/mildew and moisture control program, which includes frequent inspections. (In-house or 3rd party
工厂是否有害虫 /霉菌和湿度的控制程序文件 ? 是否有经常巡查 (公司内部或第三方检查 ? 1.0.5 No broken windows or leaking roofs that may result to product contamination was observed during audit.
在审核其间有没有发现窗户破损及房顶漏水可能导致产品污染。 1.0.6 (Critical Factory implements strict sharp tools control procedure to prevent scissors, knives, blades, broken glasses, and needles to be mixed with product. (严重 工厂是否实行严格的利器控制程序,以防止剪刀、小刀、刀片、碎玻璃及针等混入产 品中。
1.1 Machine Calibration and Maintenance机器校准和维护 1.1.1 Factory has documented system and procedure for scheduled equipment cleaning and repairs.
工厂是否有书面的文件系统和程序计划安排设备的清洁及维修。 1.1.2 Factory machines and equipments appear to be clean and in good running condition. 工厂的机器和设备是否清洁及运行良好。
1.1.3 Machines, equipments and tools are properly labeled with date of last maintenance/calibration and schedule. 机器、设备和工具是否有最近的维护 /校准日期及计划日期的标识。 1.1.4 Machines, equipments and tools that need to be repaired are properly labeled to avoid accidental use.
需要维修机器、设备和工具是否有维修标识以避免意外使用。 1.1.5 Factory has proper, clean and organized storage area of critical tooling (i.e. injection moulds with labeled shelves.
工厂是否有适当,整洁的存储区域储存关键模具 (比如 :注射模具 , 并且放在有标识的架子上。 1.1.6 Factory has proper documentation and updated inventory of machines, tools, spare parts, and equipments.
工厂有适当的机器、工具、零部件和设备的库存文件,并保持更新。 1.1.7 Factory has maintenance team with suitable skill level and equipments to perform necessary repair and calibration on machines.
工厂是否拥有一定技术水平的保养团队和设备可以执行必要的机器维修和校准的工作。
2.0 Quality Management System质量管理体系 2.0.1 Factory has established Quality Management System that is appropriate to their products and procedures.
工厂是否建立起符合他们产品和生产流程的质量管理体系。 2.0.2 Workers & Supervisors are familiar to these quality policies and objectives. 工人与主管是否熟悉这些品质政策和目标。 2.0.3 Factory has documented customer complaint system and documented recall program. 工厂是否建立了顾客投诉体系及产品召回程序。
2.0.4 (Critical Factory QC team is independent from Production division. (严重 工厂 QC 团队是否独立于生产部门。 2.0.5 Production management and QC team discuss and work together in solving Quality issues/ concerns. (Documented
是否有书面记录显示生产管理和 QC 团队共同讨论、解决质量问题及其他相关的问题 .
2.0.6 Factory has systems and procedures in place to control the risk of physical, chemical, and biological contamination that may damage the product and personnel as well. 工厂是否有系统和程序去控制那些可能会影响产品或对人造成伤害的物理、化学和微生物污染 风险。
2.0.7 Factory conducts risk assessments to identify hazards from chemicals, raw materials, process equipments, and tools.
工厂是否进行风险评估,以识别化学品、原材料、工艺设备和工具中带来的危害。
2.0.8 Is factory accredited with any international, national or customer quality standards association (e.g. ISO 9001, etc.?
工厂是否取得了国际的 , 国家的或客户的质量标准组织认证证书 (例如 : ISO 9001证书 , 等 .? 3.0 Incoming Materials Control来料控制
3.0.1 Has the factory taken adequate measures to assure raw materials conformance to required specifications before use?
工厂是否检测原物料以确认是否与要求的明细规格一致 ? 3.0.2 Proper first in-first out (FIFO system on materials are practiced. 工厂是否实施物料先进先出 (FIFO体系。 3.0.3 Factory has procedures (instructions, guidelines, and documented records for quality inspection on incoming raw materials, accessories, and components.
工厂是否有进仓原物料、配件和部件的质量检验程序 , 作业指导书 , 及记录文件。
3.0.4 Is needed testing equipment available, and maintained in good condition? 所需的来料测试仪器是否配备及保持在一个良好的状态 ? 3.0.5 Are raw materials properly labeled, stored, and traceable? 所有的原物料是否有合适的标识 , 储存及可溯性? 3.0.6 Factory has documented process and reference samples that ensure incoming raw materials conform to specifications.
工厂是否有文件程序和参考样品以确保来料符合规格。 3.0.7 (Critical Factory has proper system on material segregation to avoid accidental contamination from rejected items.
(严重 工厂是否建立起适当的物料控制体系 , 以隔离不合格的原材料及避免意外污染 ?
3.0.8 Factory properly separate good quality items from rejects and identifies non-conforming (rejects materials for replacement.