欧洲药典附录3.1.3.-推荐下载
欧洲药典identification鉴别-概述说明以及解释

欧洲药典identification鉴别-概述说明以及解释1.引言1.1 概述欧洲药典identification鉴别是药品质量控制中的一项重要工作。
药品的identification鉴别可以理解为确定药品的真实性和纯度的过程,通过分析样品的特征和性质,与标准或已知样品进行比对,从而确保药品的质量和安全。
在欧洲药典中,identification鉴别是确定药品真实性的一种重要方法。
欧洲药典作为一个权威的药典体系,旨在保障欧洲地区药品的质量、安全和疗效。
其中,identification鉴别是其质量控制的核心内容之一。
通过进行identification鉴别,可以判断药品中的成分和杂质,并与标准进行比对,从而确保药品的质量符合规定标准。
此外,identification鉴别还能够帮助识别药品中可能存在的伪劣药品和假药,起到保护公众健康的重要作用。
在市场上,药品的质量状况千差万别,有些药品可能存在掺假、掺杂或不合格等情况。
通过进行identification鉴别,可以及时发现并排除这些潜在的风险,确保消费者使用的药品安全可靠。
此外,identification鉴别也有助于验证药品的纯度和稳定性。
药品的纯度和稳定性对于药物的疗效和安全性至关重要。
通过进行identification 鉴别,可以确定药品中各成分的含量和比例,以及检测药物是否受到环境因素的影响而发生变化。
这对于药品的生产和使用过程中的质量控制非常重要,可以保证药品的治疗效果和安全性。
综上所述,欧洲药典的identification鉴别是一项非常重要的工作。
它不仅可以确保药品的质量和安全,还可以帮助识别伪劣药品,保护公众健康。
随着科学技术的不断进步,我们对identification鉴别的研究和应用也将得到进一步的发展,为药品监管提供更加可靠和有效的手段。
1.2 文章结构文章结构部分的内容可以是对整篇文章的框架和组织方式进行说明。
这一部分旨在向读者介绍本文的结构及各部分的主要内容,以便读者能够更好地理解和阅读文章。
欧洲药典附录

欧洲药典附录 Prepared on 22 November 2020第二部分、附录附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取硫酸肼溶于水,加水稀释至,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以水溶解乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容。
欧洲药典适用性证书的变更更新的管理程序 中英对照 2013.07.13

Procedures for management of revisions/renewals of certificates of suitability to the European Pharmacopoeia monographs Certification of suitability to Monographs of the European Pharmacopoeia欧洲药典适用性证书PROCEDURES FOR MANAGEMENT OF REVISIONS/RENEWALS OF CERTIFICATES OF SUITABILITY TO THE EUROPEAN PHARMACOPOEIAMONOGRAPHS欧洲药典适用性证书的变更/更新的管理程序Introduction:介绍This document should be read in conjunction with the EDQM “Guideline on Requirements on Revision/Renewal of Certificates of Suitability to the European Pharmacopoeia monographs”(PA/PH/CEP (04) 2, as amended), which describes the conditions to be fulfilled as well as the documentation to be submitted for each request for revision.此文件应该与EDQM的“欧洲药典适用性证书修订与更新规定指南” (PA/PH/CEP (04) 2)联合起来阅读,后者描述了每个变更所要求满足的条件,以及要提供的文件资料。
The procedures for the management of revisions of certificates of suitability (CEPs) are described below and have been revised according to the revised European Regulation for Variations to Marketing Authorisation Applications.对于CEP证书变更管理的程序,在下面进行了描述,并且按照新修订的欧洲市场授权申请的有关法规进行了修订。
eu无菌附录 解读

eu无菌附录解读1.引言概述部分的内容可以简要介绍无菌附录的背景和重要性。
以下是一个示例:1.1 概述无菌附录是指在欧洲联盟(EU)法规体系下对医疗器械生产领域中的无菌产品制造过程提出的一系列要求和规范。
无菌产品制造过程的合规性对于确保产品质量和安全至关重要。
因此,EU无菌附录对于制药和医疗器械行业的从业人员来说具有重要的解读和意义。
在医疗器械生产过程中,无菌产品扮演着至关重要的角色,尤其是在手术等关键操作中。
一旦无菌产品受到污染或制造过程中存在缺陷,就会导致严重的医疗事故和患者感染。
因此,制定一套科学合理的无菌产品制造要求和标准非常重要。
EU无菌附录作为欧盟法规的一部分,旨在确保医疗器械制造企业在生产无菌产品时遵守规范,从而保证产品的质量和安全。
该附录包含了一系列关于无菌产品生产中必须遵循的要求和指导原则,涵盖了生产环境、工作人员培训、设备验证和监测等方面。
理解和解读EU无菌附录对于从事医疗器械生产的企业和从业人员至关重要。
它可以帮助制造企业建立清晰的目标和标准,确保生产过程的合规性,并最终提供高品质的无菌产品给医疗领域使用。
从长远来看,对EU 无菌附录的解读和应用也可以促进医疗器械行业的发展,推动技术创新和质量管理体系的不断完善。
在本文接下来的部分,我们将深入研究EU无菌附录的定义和背景,明确其要求和标准,并探讨其对医疗器械生产行业的解读和意义。
我们还将探讨未来发展趋势并提出建议,以促进无菌附录的进一步完善和实施。
让我们开始吧。
1.2文章结构1.2 文章结构本文将按照以下结构进行论述和解读EU无菌附录的相关内容:1. 引言:在本节中,将对文章的背景和引言进行概述,介绍EU无菌附录的重要性和应用领域。
2. 正文:本节主要分为两个部分,分别是无菌附录的定义和背景以及EU无菌附录的具体要求和标准。
2.1 无菌附录的定义和背景:首先,将对无菌附录的定义进行详细解释,包括无菌状态的概念和其在医疗和制药行业中的重要作用。
欧洲药典对无水物及干燥品的定义

欧洲药典对无水物及干燥品的定义1. 引言1.1 欧洲药典的重要性欧洲药典是欧洲医药行业的核心指导性文献,为确保药品的质量、安全和有效性提供了重要的参考依据。
欧洲药典作为药品质量管理的标准,对无水物及干燥品的定义和要求起着至关重要的作用。
欧洲药典不仅规定了无水物和干燥品的标准化定义,还明确定义了制备、储存和贮运过程中对其质量的要求。
无水物在欧洲药典中被定义为完全不含水分的固体药品或物质,其中水分含量不得超过特定的限制。
而干燥品则被描述为含有适量水分的物质,需符合特定的含水量标准。
欧洲药典对无水物及干燥品的规定严格,要求药品的含水量、纯度、稳定性等方面达到一定标准。
1.2 无水物及干燥品的定义引言欧洲药典作为制定和维护欧洲药品质量标准的权威性组织,对药品的无水物及干燥品提出了严格的要求。
在实际生产过程中,无水物及干燥品是药品制备过程中不可或缺的一环。
无水物是指不含水的固体或溶液,通常是通过干燥技术从水解物中去除水分而得到的。
而干燥品则是指含有水分的固体或溶液,需要通过干燥过程来去除其中的水分,使其达到一定的干燥程度。
在药品生产中,无水物及干燥品的质量直接影响到药品的稳定性、有效性和安全性。
欧洲药典对无水物及干燥品的要求十分严格,包括其化学成分、物理性质、纯度和稳定性等方面都有详细的规定。
欧洲药典还规定了对无水物及干燥品的检测方法,以确保药品质量符合标准。
欧洲药典对无水物及干燥品的定义和要求,为药品生产提供了明确的指导,保障了药品质量和安全性。
在日常生活中,我们可以通过选择合格的药品来保障自身健康和安全。
2. 正文2.1 无水物的定义无水物是指在物质中不存在任何水分的固体或液体化合物。
在药学领域,无水物通常是指没有结晶水或水合物的纯净化合物。
无水物的制备通常需要严格控制环境中的水分含量,以保证最终产品的纯度和稳定性。
无水物的特点包括在室温下不会吸收空气中的水蒸气,不会因水的存在而导致化学反应的发生。
在药品生产过程中,选择纯净的无水物作为原料可以提高产品的质量和稳定性,避免因水分含量导致药品的失效或变质。
欧洲药典在线查询全文
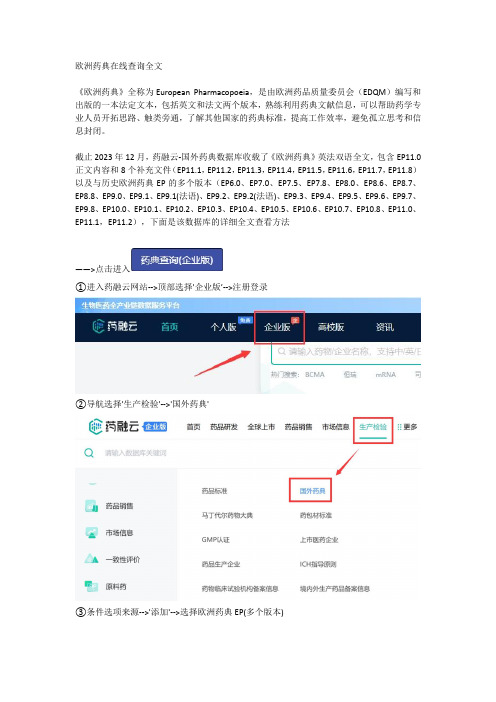
欧洲药典在线查询全文《欧洲药典》全称为European Pharmacopoeia,是由欧洲药品质量委员会(EDQM)编写和出版的一本法定文本,包括英文和法文两个版本,熟练利用药典文献信息,可以帮助药学专业人员开拓思路、触类旁通,了解其他国家的药典标准,提高工作效率,避免孤立思考和信息封闭。
截止2023年12月,药融云-国外药典数据库收载了《欧洲药典》英法双语全文,包含EP11.0正文内容和8个补充文件(EP11.1,EP11.2,EP11.3,EP11.4,EP11.5,EP11.6,EP11.7,EP11.8)以及与历史欧洲药典EP的多个版本(EP6.0、EP7.0、EP7.5、EP7.8、EP8.0、EP8.6、EP8.7、EP8.8、EP9.0、EP9.1、EP9.1(法语)、EP9.2、EP9.2(法语)、EP9.3、EP9.4、EP9.5、EP9.6、EP9.7、EP9.8、EP10.0、EP10.1、EP10.2、EP10.3、EP10.4、EP10.5、EP10.6、EP10.7、EP10.8、EP11.0、EP11.1,EP11.2),下面是该数据库的详细全文查看方法——>点击进入①进入药融云网站-->顶部选择'企业版’-->注册登录②导航选择'生产检验'-->'国外药典'③条件选项来源-->'添加'-->选择欧洲药典EP(多个版本)-----最后说下关于欧洲药典新版本EP11---《欧洲药典》EP11.0于2023年1月1日开始实施,是欧洲药品行业的权威参考书,EP11.0版本对之前版本的内容进行了全面更新和重新组织,新增和修改了600多个单体和试剂。
因此欧洲药典2024版将涵盖超过4000个全新的认证试剂,并提供7000多个化学物质的简要概述。
EP11.0版本的欧洲药典引入了一些重要的改进。
欧洲药典附录3.1.3.大全
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3% –1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3% –二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
欧洲药品GMP检查指南及附件(中英文)
GUIDE TO GOOD MANUFACTURINGPRACTICE FOR MEDICINAL PRODUCTS药品GMP检查指南.PIC/S July 2004Reproduction prohibited for commercial purposes.Reproduction for internal use is authorised,provided that the source is acknowledged.Editor: PIC/S SecretariatP.O. Box 5695CH-1211 Geneva 11e-mail: daniel.brunner@web site: :// 1 July 2004 PE 009-2TABLE OF CONTENT目录INTRODUCTION介绍 (1)CHAPTER 1 QUALITY MANAGEMENT 质量管理 (4)PRINCIPLE 原则 (4)QUALITY ASSURANCE 质量保证 (4)GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS (GMP) 药品GMP (6)QUALITY CONTROL 质量控制 (7)CHAPTER 2 PERSONNEL 人员 (10)PRINCIPLE 原则 (10)GENERAL 通则 (10)KEY PERSONNEL 关键人员 (10)TRAINING 培训 (13)PERSONAL HYGIENE 个人卫生 (14)CHAPTER 3 PREMISES AND EQUIPMENT 厂房和设备 (16)PRINCIPLE 原则 (16)PREMISES General总则 (16)Production Area 生产区域 (17)Storage Areas 储存区域 (19)Quality Control Areas 质量控制区域 (20)Ancillary Areas 辅助区域 (20)EQUIPMENT 设备 (21)CHAPTER 4 DOCUMENTATION 文件 (23)PRINCIPLE 原则 (23)GENERAL 总则 (23)DOCUMENTS REQUIRED 必需的文件 (25)MANUFACTURING FORMULA AND PROCESSING INSTRUCTIONS 生产方法和加工指示 (27)PACKAGING INSTRUCTIONS 包装指示 (28)BA TCH PROCESSING RECORDS 批加工记录 (29)BA TCH PACKAGING RECORDS 批包装记录 (30)PROCEDURES AND RECORDS 程序和记录 (32)CHAPTER 5 PRODUCTION 生产 (36)PRINCIPLE 原则 (36)GENERAL 通则 (36)PREVENTION OF CROSS-CONTAMINATION IN PRODUCTION 生产过程中防止交叉污染 (38)V ALIDATION 验证 (39)STARTING MA TERIALS 起始物料 (40)PROCESSING OPERA TIONS - INTERMEDIATE AND BULK PRODUCTS 加工操作:中间体和散装产品 (42)PACKAGING MATERIALS 包装材料 (42)PACKAGING OPERATIONS 包装操作 (43)FINISHED PRODUCTS 最终成品 (45)REJECTED, RECOVERED AND RETURNED MATERIALS 拒绝的,回收的和退回的物料46CHAPTER 6 QUALITY CONTROL 质量控制 (48)PRINCIPLE 原则 (48)GENERAL 通则 (48)GOOD QUALITY CONTROL LABORATORY PRACTICE 优良质量控制实验室实践 (49)DOCUMENTATION 文件 (49)SAMPLING 取样 (50)TESTING 检测 (52)CHAPTER 7 CONTRACT MANUFACTURE AND ANAL YSIS 合同加工和分析 (55)PRINCIPLE 原则 (55)GENERAL 通则 (55)THE CONTRACT GIVER 合同提供人 (55)THE CONTRACT ACCEPTOR 合同接受人 (56)THE CONTRACT 合同 (57)CHAPTER 8 COMPLAINTS AND PRODUCT RECALL 抱怨和产品召回 (59)PRINCIPLE 原则 (59)COMPLAINTS 抱怨 (59)RECALLS 召回 (60)CHAPTER 9 SELF INSPECTION 自检 (61)PRINCIPLE 原则 (61)ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS无菌药品的生产 (63)PRINCIPLE (63)GENERAL (63)BLOW/FILL/SEAL TECHNOLOGY (67)TERMINALL Y STERILISED PRODUCTS (67)ASEPTIC PREPARA TION (68)PERSONNEL (68)PREMISES (70)EQUIPMENT (71)SANITATION (71)PROCESSING (71)STERILISATION (73)STERILISATION BY HEA T (74)MOIST HEAT (75)DRY HEAT (75)STERILISATION BY RADIATION (75)STERILISATION WITH ETHYLENE OXIDE (76)FILTRATION OF MEDICINAL PRODUCTS WHICH CANNOT BE STERILISED IN THEIR FINAL CONTAINER (77)FINISHING OF STERILE PRODUCTS (77)QUALITY CONTROL (78)ANNEX 2 MANUFACTURE OF BIOLOGICAL MEDICINAL PRODUCTS FOR HUMAN USE人用生物药品的生产 (79)SCOPE (79)PRINCIPLE (79)PERSONNEL (80)PREMISES AND EQUIPMENT (81)ANIMAL QUARTERS AND CARE (82)DOCUMENTATION (82)PRODUCTION (83)QUALITY CONTROL (84)ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS 放射性药品的生产 (85)PRINCIPLE (85)PERSONNEL (85)PREMISES AND EQUIPMENT (85)PRODUCTION (86)QUALITY CONTROL (86)DISTRIBUTION AND RECALLS (86)ANNEX 4 MANUFACTURE OF VETERINARY MEDICINAL PRODUCTS OTHER THAN IMMUNOLOGICALS MANUFACTURE OF PREMIXES FOR MEDICATED FEEDING STUFFS 除为预混合加药饲料原料生产的免疫产品以外的,兽药产品的生产 (87)THE MANUFACTURE OF ECTOPARASITICIDES (88)THE MANUFACTURE OF VETERINARY MEDICINAL PRODUCTS CONTAINING PENICILLINS (88)RETENTION OF SAMPLES (point 1.4. viii and point 6.14.) (88)STERILE VETERINARY MEDICINAL PRODUCTS (88)ANNEX 5 MANUFACTURE OF IMMUNOLOGICAL VETERINARY MEDICAL PRODUCTS免疫兽药产品的生产 (89)PRINCIPLE (89)PERSONNEL (89)PREMISES (90)EQUIPMENT (93)ANIMALS AND ANIMAL HOUSES (94)DISINFECTION - WASTE DISPOSAL (94)PRODUCTION (95)STARTING MA TERIALS (95)QUALITY CONTROL (98)ANNEX 6 MANUFACTURE OF MEDICINAL GASES药用气体的生产 (99)1. PRINCIPLE (99)2. PERSONNEL (99)3. PREMISES AND EQUIPMENT (99)4. DOCUMENTA TION (100)5. PRODUCTION (101)6. QUALITY CONTROL (104)7. STORAGE AND RELEASE (105)ANNEX 7 MANUFACTURE OF HERBAL MEDICINAL PRODUCTS草药产品的生产 (108)PRINCIPLE (108)PREMISES (108)DOCUMENTATION (108)SAMPLING (109)QUALITY CONTROL (110)ANNEX 8 SAMPLING OF STARTING AND PACKAGING MA TERIALS起始物料和包装材料的取样 (111)PRINCIPLE (111)PERSONNEL (111)STARTING MA TERIALS (111)PACKAGING MATERIAL (112)ANNEX 9 MANUFACTURE OF LIQUIDS, CREAMS AND OINTMENTS流体,霜体和膏体药品的生产 (113)PRINCIPLE (113)PRODUCTION (113)ANNEX 10 MANUFACTURE OF PRESSURISED METERED DOSE AEROSOL PREPARATIONS FOR INHALATION吸入式剂量仪的气雾剂的生产 (115)PRINCIPLE (115)GENERAL (115)PREMISES AND EQUIPMENT (115)PRODUCTION AND QUALITY CONTROL (116)ANNEX 11 COMPUTERISED SYSTEMS 计算机化系统 (117)PRINCIPLE (117)PERSONNEL (117)V ALIDATION (117)ANNEX 12 USE OF IONISING RADIATION IN THE MANUFACTURE OF MEDICINAL PRODUCTS使用离子放射生产药品 (120)INTRODUCTION (120)RESPONSIBILITIES (120)DOSIMETRY (121)V ALIDATION OF THE PROCESS (121)COMMISSIONING OF THE PLANT (122)PREMISES (124)PROCESSING (124)DOCUMENTATION (126)MICROBIOLOGICAL MONITORING (126)ANNEX 13 MANUFACTURE OF INVESTIGA TIONAL MEDICINAL PRODUCTS观察期药品的生产 (127)PRINCIPLE (127)GLOSSARY (128)QUALITY MANAGEMENT (130)PERSONNEL (130)PREMISES AND EQUIPMENT (130)DOCUMENT A TION (131)PRODUCTION (132)QUALITY CONTROL (136)RELEASE OF BATCHES (137)SHIPPING (139)COMPLAINTS (139)RECALLS AND RETURNS (139)DESTRUCTION (140)ANNEX 14 MANUFACTURE OF PRODUCTS DERIVED FROM HUMAN BLOOD OR HUMAN PLASMA生产自人类血液或人体组织分离的产品 (143)PRINCIPLE (143)GLOSSARY (144)QUALITY MANAGEMENT (144)PREMISES AND EQUIPMENT (145)BLOOD AND PLASMA COLLECTION (145)TRACEABILITY AND POST COLLECTION MEASURES (146)PRODUCTION AND QUALITY CONTROL (147)RETENTION OF SAMPLES (148)DISPOSAL OF REJECTED BLOOD, PLASMA OR INTERMEDIATES (148)ANNEX 15 QUALIFICATION AND V ALIDATION 确认和验证 (149)PRINCIPLE (149)PLANNING FOR V ALIDATION (149)DOCUMENTATION (150)QUALIFICATION (150)PROCESS V ALIDATION (151)CLEANING VALIDATION (153)CHANGE CONTROL (154)REV ALIDATION (154)GLOSSARY (154)[ANNEX 16] [QUALIFIED PERSON AND BA TCH RELEASE]*经授权的人员和批放行 (157)ANNEX 17 PARAMETRIC RELEASE参数放行 (158)1. PRINCIPLE (158)2. PARAMETRIC RELEASE (158)3. PARAMETRIC RELEASE FOR STERILE PRODUCTS (158)4. GLOSSARY (160)[ANNEX 18] [GMP GUIDE FOR ACTIVE PHARMACEUTICAL INGREDIENTS] 17原料药GMP 指南 (161)GLOSSARY术语表 (162)GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS药品GMP指南INTRODUCTION介绍为进一步消除药品贸易壁垒,促进许可证的一致性,以及确保整个欧洲在研发,生产和控制药品中保持高标准的质量保证,根据药品检查协会(PIC)同意,药品检查使用一致的GMP原则,和药品检查合作计划表中的欧洲药品GMP及其附录。
欧洲药典3.1.4-无添加剂的PET包装材料的检测
EUROPEAN PHARMACOPOEIA 8.0 3.1.4.Polyethylene without additives forcontainersReference solution (n).Dissolve 60mg of plastic additive 16CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Reference solution (o).Dissolve 60mg of plastic additive 17CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Reference solution (p).Dissolve 60mg of plastic additive 16CRS and 60mg of plastic additive 17CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Plate :TLC silica gel GF 254plate R .Mobile phase A :hexane R .Mobile phase B :methylene chloride R .Application :20μL of the test solution S24,the reference solution (p)and the reference solutions corresponding to all the phenolic and non-phenolic antioxidants mentioned in the type composition of the material to be examined.Development A :over a path of 18cm with mobile phase A.Drying A :in air.Development B :over a path of 17cm with mobile phase B.Drying B :in air.Detection :examine in ultraviolet light at 254nm;spray with alcoholic iodine solution R and examine in ultraviolet light at 254nm after 10-15min.System suitability :reference solution (p):–the chromatogram shows 2clearly separated spots.Limit :any spots in the chromatogram obtained with test solution S24are not more intense than the spots in thecorresponding positions in the chromatograms obtained with the reference solutions.Plastic additive 22.Liquid chromatography (2.2.29).Test solution .Evaporate 25mL of solution S2to dryness in vacuo at 45°C.Dissolve the residue in 10mL of toluene R and 10mL of a 10g/L solution of tetrabutylammonium hydroxide R in a mixture of 35volumes of toluene R and 65volumes of anhydrous ethanol R .Boil under a reflux condenser for 3h.Allow to cool and filter if necessary.Reference solution .Dissolve 30mg of plastic additive 22CRS in 50mL of toluene R .Add 1mL of this solution to 25mL of blank solution S2and evaporate to dryness in vacuo at 45°C.Dissolve the residue in 10mL of toluene R and 10mL of a 10g/L solution of tetrabutylammonium hydroxide R in a mixture of 35volumes of toluene R and 65volumes of anhydrous ethanol R .Boil under a reflux condenser for 3h.Allow to cool and filter if necessary.Column :–size :l =0.25m,Ø=4.6mm;–stationary phase :aminopropylsilyl silica gel for chromatography R (5μm).Mobile phase :anhydrous ethanol R ,hexane R (11:89V/V ).Flow rate :2mL/min.Detection :spectrophotometer at 227nm.Injection :20μL.Run time :10min.System suitability :–resolution :minimum 7between the peaks due to the “diol”component and to the diluent of the reference solution.Limit :the area of the peak due to the “diol”component from plastic additive 22in the chromatogram obtained with the test solution is less than the corresponding peak in the chromatogram obtained with the reference solution.Amides and stearates .Thin-layer chromatography (2.2.27).Test solution .Use test solution S24described in the test for non-phenolic antioxidants.Reference solution (q).Dissolve 20mg of stearic acid (plastic additive 19CRS )in 10mL of methylene chloride R .Reference solution (r).Dissolve 40mg of oleamide (plastic additive 20CRS )in 20mL of methylene chloride R .Reference solution (s).Dissolve 40mg of erucamide (plastic additive 21CRS )in 20mL of methylene chloride R .Plate :TLC silica gel GF 254plate R (2plates).A.Mobile phase :anhydrous ethanol R ,trimethylpentane R (25:75V/V ).Application :10μL of the test solution S24and reference solution (q).Development :over a path of 10cm.Drying :in air.Detection :spray with a 2g/L solution ofdichlorophenolindophenol,sodium salt R in anhydrous ethanol R and heat in an oven at 120°C for a few minutes to intensify the spots.Limit :any spot corresponding to plastic additive 19in the chromatogram obtained with test solution S24is identical in position to (R F =about 0.5)but not more intense than the spot in the chromatogram obtained with reference solution (q).B.Mobile phase A :hexane R .Mobile phase B :methanol R ,methylene chloride R (5:95V/V ).Application :10μL of the test solution S24and the reference solutions (r)and (s).Development A :over a path of 13cm with mobile phase A.Drying A :in air.Development B :over a path of 10cm with mobile phase B.Drying B :in air.Detection :spray with a 40g/L solution of phosphomolybdic acid R in anhydrous ethanol R .Heat in an oven at 120°C until spots appear.Limit :any spots corresponding to plastic additive 20or plastic additive 21in the chromatogram obtained with test solution S24are identical in position to (R F =about 0.2)but not more intense than the corresponding spots in the chromatograms obtained with reference solutions (r)and (s).01/2008:30104corrected 6.03.1.4.POLYETHYLENE WITHOUT ADDITIVES FOR CONTAINERS FOR PARENTERAL PREPARATIONS AND FOR OPHTHALMIC PREPARATIONSDEFINITIONPolyethylene without additives is obtained by thepolymerisation of ethylene under high pressure in the presence of oxygen or free-radical-forming initiators as catalyst.CHARACTERSAppearance :beads,granules,powder or,after transformation,translucent sheets of varying thickness or containers.Solubility :practically insoluble in water,soluble in hot aromatic hydrocarbons,practically insoluble in anhydrous ethanol,in hexane and in methanol.It softens at temperatures beginning at 65°C.Relative density:0.910to 0.937.IDENTIFICATIONIf necessary,cut the samples of the material to be examined into pieces of maximum dimension on a side of not greater than 1cm .A.Infrared absorption spectrophotometry (2.2.24).General Notices (1)apply to all monographs and other texts3833.1.5.Polyethylene with additives for containers EUROPEAN PHARMACOPOEIA8.0Preparation:to0.25g add10mL of toluene R and boilunder a reflux condenser for about15min.Place a fewdrops of the solution on a sodium chloride disc andevaporate the solvent in an oven at80°C.Absorption maxima:at2920cm−1,2850cm−1,1465cm−1, 730cm−1and720cm−1.The spectrum obtained is identical to that obtained with the material selected for the type sample.If the material to be examined is in the form of sheets,the identification may be performed directly on a cut piece of suitable size.B.Additives(see Tests).TESTSIf necessary,cut the samples of the material to be examined into pieces of maximum dimension on a side of not greater than1cm.Solution S1.Place25g in a borosilicate-glassflask with a ground-glass neck.Add500mL of water for injections R and heat under a reflux condenser for5h.Allow to cool and decant.Keep part of the solution for the test for appearance of solution.Filter the rest through a sintered glassfilter(16) (2.1.2).Use solution S1within4h of preparation.Solution S2.Place2.0g in a conical borosilicate-glassflask with a ground-glass neck.Add80mL of toluene R and boil under a reflux condenser with constant stirring for1h30min. Allow to cool to60°C and add with continued stirring120mL of methanol R.Filter the solution through a sintered-glassfilter(16)(2.1.2).Rinse theflask and thefilter with25mL of a mixture of40mL of toluene R and60mL of methanol R,add the rinsings to thefiltrate and dilute to250mL with the same mixture of solvents.Prepare a blank solution.Solution S3.Place100g in a conical borosilicate-glassflask with a ground-glass neck.Add250mL of0.1M hydrochloric acid and boil under a reflux condenser with constant stirring for1h.Allow to cool and decant the solution. Appearance of solution.Solution S1is clear(2.2.1)and colourless(2.2.2,Method II).Acidity or alkalinity.To100mL of solution S1add0.15mL of BRP indicator solution R.Not more than1.5mL of0.01M sodium hydroxide is required to change the colour of the indicator to blue.To100mL of solution S1add0.2mL of methyl orange solution R.Not more than1.0mL of0.01M hydrochloric acid is required to reach the beginning of the colour change of the indicator from yellow to orange. Absorbance(2.2.25):maximum0.2,determined between wavelengths of220nm and340nm on solution S1. Reducing substances.To20mL of solution S1add1mLof dilute sulfuric acid R and20mL of0.002M potassium permanganate.Boil under a reflux condenser for3min and cool immediately.Add l g of potassium iodide R and titrate immediately with0.01M sodium thiosulfate,using0.25mLof starch solution R as indicator.Carry out a blank titration. The difference between the titration volumes is not more than 0.5mL.Substances soluble in hexane.Place10g in a250mL conical borosilicate-glassflask with a ground-glass neck.Add100mL of hexane R and boil under a reflux condenser for4h,stirring constantly.Cool in iced water andfilter rapidly through a sintered-glassfilter(16)(2.1.2)maintaining the solution at 0°C(thefiltration time must be less than5min;if necessary thefiltration may be accelerated by applying pressure to the solution).Evaporate20mL of thefiltrate in a tared glass dish on a water-bath.Dry the residue in an oven at100-105°C for1h.The mass of the residue obtained is within10per cent of the residue obtained with the type sample and does not exceed5per cent.Additives.Thin-layer chromatography(2.2.27).Test solution.Evaporate50mL of solution S2to dryness in vacuo at45°C.Dissolve the evaporation residue with5mL of methylene chloride R.Prepare a blank solution from the blank solution corresponding to solution S2.Reference solution.Dissolve20mg of plastic additive15CRS and20mg of plastic additive08CRS in methylene chloride R and dilute to10mL with the same solvent.Plate:TLC silica gel G plate R.Mobile phase A:hexane R.Mobile phase B:methanol R,methylene chloride R(5:95V/V). Application:10μL.Development A:over a path of13cm using mobile phase A. Drying A:in air.Development B:over a path of10cm using mobile phase B. Drying B:in air.Detection:spray with a40g/L solution of phosphomolybdic acid R in ethanol(96per cent)R and heat at120°C until the spots appear in the chromatogram obtained with the reference solution.System suitability:reference solution:–the chromatogram shows2separated spots.Limit:no spot appears in the chromatogram obtained with the test solution,except for a spot which may be at the solvent front from thefirst development and which correspondsto oligomers.Disregard any spots corresponding to those obtained in the chromatogram with the blank solution. Extractable heavy metals(2.4.8):maximum2.5ppm. Evaporate50mL of solution S3to about5mL on a water-bath and dilute to20mL with water R.12mL of solution complies with test A.Prepare the reference solution using2.5mL of lead standard solution(10ppm Pb)R.Sulfated ash(2.4.14):maximum0.02per cent,determined on5.0g.01/2008:30105corrected7.5 3.1.5.POLYETHYLENE WITH ADDITIVES FOR CONTAINERS FOR PARENTERAL PREPARATIONS AND FOR OPHTHALMIC PREPARATIONS DEFINITIONPolyethylene with additives is obtained by the polymerisation of ethylene under pressure in the presence of a catalyst or by copolymerisation of ethylene with not more than25per cent of higher alkene homologues(C3to C10).PRODUCTIONA certain number of additives are added to the polymer in order to optimise their chemical,physical and mechanical properties in order to adapt them for the intended use.All these additives are chosen from the appended list which specifies for each product the maximum allowable content. They may contain at most3antioxidants,1or several lubricants or antiblocking agents as well as titanium dioxide as an opacifying agent when the material must provide protection from light.–butylhydroxytoluene(plastic additive07):maximum0.125per cent;–pentaerythrityl tetrakis[3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate](plastic additive09):maximum0.3per cent;–1,3,5-tris(3,5-di-tert-butyl-4-hydroxybenzyl)-S-triazine-2,4,6(1H,3H,5H)-trione(plastic additive13):maximum0.3per cent;–octadecyl3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate, (plastic additive11):maximum0.3per cent;384See the information section on general monographs(cover pages)。
propoxycarbazone
EUROPEAN COMMISSIONHEALTH & CONSUMER PROTECTION DIRECTORATE-GENERALDirectorate E – Food Safety: plant health, animal health and welfare, international questionsE1 - Plant healthPropoxycarbozoneSANCO/4067/2001-Final30 September 2003COMMISSION WORKING DOCUMENT - DOES NOT NECESSARILY REPRESENTTHE VIEWS OF THE COMMISSION SERVICESReview report for the active substance propoxycarbazoneFinalised in the Standing Committee on the Food Chain and Animal Health at its meeting on3 October 2003 in view of the inclusion of propoxycarbazone in Annex I of Directive91/414/EEC.1. Procedure followed for the evaluation processThis review report has been established as a result of the evaluation of the new active substancepropoxycarbazone, made in the context of the work provided for in Articles 5 and 6 of Directive91/414/EEC concerning the placing of plant protection products on the market, with a view tothe possible inclusion of this substance in Annex I to the Directive.In accordance with the provisions of Article 6(2) of Directive 91/414/EEC, the Germanauthorities received on25 January 2000 an application from Bayer AG (now BayerCropScience), hereafter referred to as the applicant, for the inclusion of the active substancepropoxycarbazone in Annex I to the Directive. The German authorities indicated to theCommission on 10 March 2000 the results of a first examination of the completeness of thedossier, with regard to the data and information requirements provided for in Annex II and, for atleast one plant protection product containing the active substance concerned, in Annex III to theDirective. Subsequently, and in accordance with the requirements of Article 6(2), a dossier onpropoxycarbazone was distributed to the Member States and the Commission.The Commission referred the dossier to the Standing Committee on the Food Chain andAnimal Health in the meeting of the working group ‘legislation’ thereof on10 March 2000,during which the Member States confirmed the receipt of the dossier.In accordance with the provisions of Article 6(3), which requires the confirmation at Communitylevel that the dossier is to be considered as satisfying, in principle, the data and informationrequirements provided for in Annex II and, for at least one plant protection product containingthe active substance concerned, in Annex III to the Directive and in accordance with theprocedure laid down in Article 20 of the Directive, the Commission confirmed in its Decision 2000/463/EC 1 of 17 July 2000 that these requirements were satisfied.Within the framework of that decision and with a view to the further organisation of the works related to the detailed examination of the dossier provided for in Article 6(2) and (4) of Directive 91/414/EEC, it was agreed between the Member States and the Commission that Germany would, as rapporteur Member State, carry out the detailed examination of the dossier and report the conclusions of its examination accompanied by any recommendations on the inclusion or non-inclusion and any conditions relating thereto, to the Commission as soon as possible and at the latest within a period of one year.Germany submitted to the Commission on26 March 2001 the report of its detailed scientific examination, hereafter referred to as the draft assessment report, including, as required, a recommendation concerning the possible inclusion of propoxycarbazone in Annex I to the Directive.On receipt of the draft assessment report, the Commission forwarded it for consultation to all the Member States as well as to Bayer AG being the sole applicant on4 May 2001.The Commission organised further an intensive consultation of specialised scientific experts from a representative number of Member States, to review the draft assessment report and the comments received thereon (peer review), in particular on each of the following disciplines :- identity and physical /chemical properties ;- fate and behaviour in the environment ;;- ecotoxicology- mammalian toxicology ;- residues and analytical methods ;questions.- regulatoryThe meetings for this consultation were organised on behalf of the Commission by the Pesticide Safety Directorate (PSD) in York, United Kingdom, from November 2001 to July 2002.The report of the peer review (i.e. full report) was circulated, for further consultation, to Member States and the sole applicant on 11 October 2002.The dossier, draft assessment report and the peer review report (i.e. full report) including in particular an outline resumé of the remaining technical questions, were referred to the Standing Committee on the Food Chain and Animal Health, and specialised working groups of this Committee, for final examination, with participation of experts from the 15 Member States. This final examination took place from March to October 2003, and was finalised in the meeting of the Standing Committee on 3 October 2003.The present review report contains the conclusions of this final examination; given the importance of the draft assessment report, the peer review report (i.e. full report) and the comments and clarifications submitted after the peer review as basic information for the final examination process, these documents are considered respectively as background documents A, B and C to this review report and are part of it.These review did not reveal any open question, which would have required the consultation of the Scientific Committee on Plants.2. Purposes of this review reportThis review report, including the background documents and appendices thereto, have been developed and finalised in support of the Directive 2003/119/EC2 concerning the inclusion of propoxycarbazone in Annex I to Directive 91/414/EEC, and to assist the Member States in decisions on individual plant protection products containing propoxycarbazone they have to take in accordance with the provisions of that Directive, and in particular the provisions of article 4(1) and the uniform principles laid down in Annex VI.This review report provides also for the evaluation required under Section A.2.(b) of the above mentioned uniform principles, as well as under several specific sections of part B of these principles. In these sections it is provided that Member States, in evaluating applications and granting authorisations, shall take into account the information concerning the active substance in Annex II of the directive, submitted for the purpose of inclusion of the active substance in Annex I, as well as the result of the evaluation of those data.In parallel with the provisions of Article 7(6) of Regulation 3600/92 for existing active substances, the Commission and the Member States will keep available or make available this review report for consultation by any interested parties or will make it available to them on their specific request. Moreover the Commission will send a copy of this review report (not including the background documents) to the applicant.The information in this review report is, at least partly, based on information which is confidential and/or protected under the provisions of Directive 91/414/EEC. It is therefore recommended that this review report would not be accepted to support any registration outside the context of Directive 91/414/EEC, e.g. in third countries, for which the applicant has not demonstrated possession of regulatory access to the information on which this review report is based.3. Overall conclusion in the context of Directive 91/414/EECThe overall conclusion from the evaluation is that it may be expected that plant protection products containing propoxycarbazone will fulfil the safety requirements laid down in Article 5(1)(a) and (b) of Directive 91/414/EEC. This conclusion is however subject to compliance with the particular requirements in sections 4, 5, 6 and 7 of this report, as well as to the implementation of the provisions of Article 4(1) and the uniform principles laid down in Annex VI of Directive 91/414/EEC, for each propoxycarbazone containing plant protection product for which Member States will grant or review the authorisation.Furthermore, these conclusions were reached within the framework of the uses which were proposed and supported by the main data submitter and mentioned in the list of uses supported by available data (attached as Appendix IV to this Review Report).Extension of the use pattern beyond those described above will require an evaluation at Member State level in order to establish whether the proposed extensions of use can satisfy the requirements of Article 4(1) and of the uniform principles laid down in Annex VI of Directive 91/414/EEC.4. Specific conclusions which are highlighted in this evaluation4.1 Residues of propoxycarbazone in foodstuffsThe review has established that the residues arising from the proposed uses, consequent on application consistent with good plant protection practice, have no harmful effects on human or animal health. The Theoretical Maximum Daily Intake (TMDI) for a 60 kg adult is0,26% of the Acceptable Daily Intake (ADI), based on the FAO/WHO European Diet (August 1994). This low intake value reflects the current limited use pattern for this active substance.4.2 Exposure of operators, workers and bystandersThe review has identified acceptable exposure scenarios for operators, workers and bystanders, which require, however, confirmation for each plant protection product in accordance with the relevant sections of the above mentioned uniform principles.Ecotoxicology4.3The review has also concluded that under the proposed and supported conditions of use there are no unacceptable effects on the environment, as provided for in Article 4 (1) (b) (iv) and (v) of Directive 91/414/EEC, provided that certain conditions are taken into account as detailed in section 7 of this report.5. Identity and Physical/chemical propertiesThe main identity and the physical/chemical properties of propoxycarbazone are given in Appendix I.The active substance shall have a minimum purity of974g/kg technical product (based on a pilot plant production).The review has established that for the active substance notified by the applicant (Bayer AG), none of the manufacturing impurities considered are, on the basis of information currently available, of toxicological or environmental concern.6. Endpoints and related informationIn order to facilitate Member States, in granting or reviewing authorisations, to apply adequately the provisions of Article 4(1) of Directive 91/414/EEC and the uniform principles laid down in Annex VI of that Directive, the most important endpoints as identified during the evaluation process are listed in Appendix II.7. Particular conditions to be taken into account on short term basis by Member States in relation to the granting of authorisations of plant protection products containing propoxycarbazoneOn the basis of the proposed and supported uses, the following particular issues have been identified as requiring particular and short term (within 12 months at the latest) attention from the Member States, in the framework of any authorisations to be granted, varied or withdrawn, as appropriate:In this overall assessment Member States- should pay particular attention to the potential of propoxycarbazone and its metabolites for groundwater contamination, when the active substance is applied in regions with vulnerable soil and/or climate conditions;- should pay particular attention to the protection of aquatic ecosystems, especially of aquatic plants.Risk mitigation measures should be applied where appropriate.8. List of studies to be generatedNo further studies were identified which were considered at this stage, and under the current inclusion conditions necessary in relation to the inclusion of propoxycarbazone in Annex I.9. Updating of this review reportThe technical information in this report may require periodic updating to take account of technical and scientific developments as well as of the results of the examination of any information referred to the Commission in the framework of Articles 7, 10 or 11 of Directive 91/414/EEC. Such adaptations will be examined and finalised in the Standing Committee on the Food Chain and Animal Health, in connection with any amendment of the inclusion conditions for propoxycarbazone in Annex I of the Directive.APPENDIX IIdentity, physical and chemical propertiesPROPOXYCARBAZONECommon name (ISO) Propoxycarbazone (The given data belong to thesodium salt Propoxycarbazone-sodium if not specifiedotherwise)MKH 65 61Development Code (for new activesonly)Chemical name (IUPAC) parent2-(4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carboxamidosulfonylbenzoicacid-methylestersodium saltsodium (4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-ylcarbonyl)(2-methoxycarbonylphenylsulfonyl)azanideChemical name (CA) parentmethyl 2-[[[(4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carbonyl]amino]sulfonyl]benzoatesodium saltbenzoic acid, 2-[[[4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carbonyl]amino] sulfonyl]-, methyl ester,sodium saltCIPAC No 655 (parent); 655.011 (sodium salt)CAS No 145026-81-9 (parent); 181274-15-7 (sodium salt)EEC No not assignedFAO SPECIFICATION not availableMinimum purity 974 g/kgMolecular formula C15H18N4O7S (parent); C15H17N4NaO7S (sodium salt) Molecular mass 398.4 g/mol (parent); 420.4 g/mol (sodium salt)Structural formulaMelting point 230 – 240 °C,the melting occurs under decompositionBoiling point Not measurable, decomposition at 230 – 240 °C Appearance odourless and colourless crystalline powder (PAS, TAS) Relative density 1.42 g/cm³ at 20°CVapour pressure Cannot be determined directly due to its extreme low value;upper limit: < 9 ⋅ 10-8 Pa at 70 °C< 1 ⋅ 10-8 Pa at 20 °C (extrapolated)Henry's law constant < 1 ⋅ 10-10 Pa m3 mol-1 at 20 °C (calculated)Solubility in water (g/L) pH 8.8: 42 (unbuffered)pH 9.0: 42pH 7.2: 42pH 4.5: 2.9Solubility in organic solvents (g/L) n-heptane < 0.1xylene < 0.10.11-octanol <2-propanol <0.1ethyl acetate < 0.1polyethylen glycol 5.2acetonitrile 0.90acetone 0.50dichloromethane 1.5dimethylsulfoxide 190Partition co-efficient (log P ow) pH 9: - 1.59pH 7: - 1.55pH 4: - 0.30Hydrolytic stability (DT50) Stable at 25 ° C at pH 4 –9Dissociation constant The free acid of the sodium salt is produced by protonationof the active substance under acidic conditions.It has a pKa-value of 2.1 in aqueous solutions.Quantum yield of direct photo-Φ = 0.75transformation in water at λ >290nmFlammability Not highly flammable in the sense of EU guideline A.10.Does not undergo spontaneous combustion in the sense ofEU guideline A.16 and in 1L BCC-Test.Explosive properties Not explosive in the sense of EU guideline A.14UV/VIS absorption (max.) 230 nmPhotostability in water (DT50) DT50 18.1 days (phenyl-labelled)DT50 40.9 days (triazolinone-labelled)23 June 2003APPENDIX IIEND POINTS AND RELATED INFORMATIONPROPOXYCARBAZONE1 Toxicology and metabolismAbsorption, distribution, excretion and metabolism in mammalsRate and extent of absorption: Rapid but incomplete absorption (25 – 30 %) within48 hoursDistribution: Widely distributed, highest residues in the liverPotential for accumulation: No evidence for accumulationRate and extent of excretion: Rapid and nearly complete excretion (>88 % of theadministered dose within 48 hours) primarily via faeces(>66 %)Toxicologically significant compounds: Parent compound, plant metabolite 2-hydroxypropoxyMKH 6561 (M01) was not of greater toxicologicalsignificance than parentMetabolism in animals: Limited metabolism: unchanged parent compound in urine(>90 %) and faeces (>80 %); metabolism via cleavage ofthe amide bond, sulfonamide methylester (M05),sulfonamide acid (M06) and saccharin (M07)Acute toxicityRat LD50 oral: > 5000 mg/kg bwRat LD50 dermal: > 5000 mg/kg bwRat LC50 inhalation: > 5.03 mg/l airSkin irritation: Non-irritantEye irritation: Non-irritantSkin sensitization (test method used andNot sensitising (M & K)result):Short term toxicityTarget / critical effect: Decreased body weight gain, increased water intake;irritation of forestomach epithelium (rats); decreased foodconsumption and relative heart weights (dog)Lowest relevant oral NOAEL / NOEL: 1yr dog: 2000 ppm (56 mg/kg bw/d)Lowest relevant dermal NOAEL / NOEL: >1000 mg/kg bw/d (rat)Lowest relevant inhalation NOAEL / NOEL:No data – not requiredGenotoxicity No genotoxic potential23 June 2003Long term toxicity and carcinogenicityTarget / critical effect: Decreased body weight gain; increased urine pH value andrenal pelvic mineralization in rats onlyLowest relevant NOAEL: 2yr rat: 1000 ppm (43 mg/kg bw/d)Carcinogenicity: No carcinogenic potentialReproductive toxicityTarget / critical effect - Reproduction: No reproductive toxicity at limit dose level (rat)Lowest relevant reproductive NOAEL /NOEL:>16000 ppm (>1000 mg/kg bw/d; rat)Target / critical effect - Developmental toxicity: Embryotxic effects (reduced fetus number, reduced fetal and placental weights, increased post-implantation loss, retarded skeletal ossification) at a maternally toxic dose (1000 mg/kg bw/d) in rabbitsLowest relevant developmental NOAEL / NOEL: Rabbit:500 mg/kg bw/d (developmental toxicity) 100 mg/kg bw/d (maternal toxicity)Delayed neurotoxicity No acute or subchronic (90-day) neurotoxicity in rats(NOAEL acute: >2000 mg/kg bw; NOAEL subchronic:>1321 mg/kg bw/d)Other toxicological studies Plant metabolite M01 has a very low acute oral toxicity(LD50: >5000 mg/kg bw), was not genotoxic (Ames-Test, invitro chromosome aberration test Chinese hamster V79cells), and caused no effects in the rat after subacutefeeding of 10000 ppmMedical data Limited data (new compound); no human health problemsobservedSummaryValueStudySafetyfactor ADI: 0.4 mg/kg bw Rat, 2yr study 100AOEL systemic: 0.3 mg/kg bw/day Rabbit,developmentalstudy (approx. oralabsorption rate25%)100ARfD (acute reference dose): Not allocated Not necessaryDermal absorption10 % default value (due to physical and chemicalproperties)14 April 2003 2 Fate and behaviour in the environment2.1 Fate and behaviour in soilRoute of degradationAerobic:Mineralization after 100 days: phenyl-label:9.1 to 41.9 % (88-98d); 21.7 to 49.0% (180-361d)(n=4)triazolinone-label:1.3 to 8.9 % (93-117d);2.6 to 12.6 % (182 –365d)(n=4)Non-extractable residues after 100 days: phenyl-label:6.5 to 29.5 % (88-98d); 8.2 to 28.3 % (180-361d)(n=4)triazolinone-label:8.9 to 64.9 %(93-117d); 17.9 to 65.7 % (182-365d)(n=4)Major metabolites above 10 % of applied active substance: name and/or code% of applied rate (range and maximum) sulfonamide methyl ester (M05):Range: <10 % at day 100, max. 20.9 % (day 6) saccharin (M07):Range: 17.3 % at day 100 in 1 soil, max. 26.7 % (day 14)4-Hydroxy saccharin (M08):Range: 9.3 to 18.1 % at day 100, max. 19.5 % (day 36)N-methyl propoxy triazolinone amide (M09): Range: <10 % at day 100, max. 13.2 % (day 253)N-methyl propoxy triazolinone (M10):Range: 12.6 to 34.3 % at day 100, max. 55.2 % (day 182)Supplemental studiesAnaerobic:not required, application in springSoil photolysis:stable to photolysisRemarks:noneRate of degradationLaboratory studiesDT50lab (20 °C, aerobic): DT50lab (20°C, aerobic): propoxycarbazone-sodium:14 April 2003range: 22.8 – 98.8 (220.4*) d (n = 8), 1st order,median: 60.6 d, r²: 0.818 – 0.997,DT50calc (20°C, aerobic): sulfonamide methyl ester(M05):3.0 - 22.3 d(n = 3), 1st order, median: 3.1, r²: 0.898 - 1DT50calc (20°C, aerobic): saccharin (M07):5 - 57 d(n = 3), 1st order, median: 27.3, r²: 0.909 - 1DT50lab (20°C, aerobic): 4-Hydroxy saccharin (M08):178.4 d (r²:0.899), 185.5 d (r²:0.838), 953.8* d(r²:0.227) 1st order (n = 3),DT50lab (20°C, aerobic): N-methyl propoxy triazolinoneamide (M09):83.8 d (r²:0.777), 90.1 d (r²:0.855), >>1 year*(r²:0.025), 1st order (n = 3)DT50lab+calc (20°C, aerobic): N-methyl propoxytriazolinone (M10):38.7 - 75.6 d, 1st order (n = 3)* soil with low microbial activity (not representative) DT90lab (20 °C, aerobic): DT90lab (20°C, aerobic): propoxycarbazone-sodium:range: 76 – 328 (733*) d [n = 8], r²: 0.818 -.0.997,1st order, median: 202 d* soil with low microbial activity (not representative) DT50lab (10 °C, aerobic): DT50lab (10°C, aerobic): calculated from lab (20 °C):50.2 – 217.4 d (485* d), median 133 d (n = 8), 1storder* soil with low microbial activity (not representative)3 field trials with average temperatures ~10 °C in the firweeks after application showed DT50 between 4.9 and20 daysDT50lab (20 °C, anaerobic): DT50lab (20°C, anaerobic): not requiredField studies (country or region)DT50f from soil dissipation studies: DT50f: Southern Europe (France): 2.7 / 9.1d, √1st order[n = 2], recalc. 1st order: 12 and 37 dNorthern Europe (Germany, France, UK): 6.6 - 21.0 d,1st order [n = 4] and 4.9 d √1st order [n = 1], recalc. 1storder: 9.1 dDT50 (total residues):DT50f: Southern Europe (France): 15–29 dDT50f: Northern Europe (Germany, France, UK):12–56 dDT90f from soil dissipation studies: DT90f: Southern Europe (France): 30 / 101 d, √1storder [n = 2], recalc. 1st order: 40 and 123 dNorthern Europe (Germany, France, UK): 22 - 71 d, 1st4] and 54 d √1st order [n = 1], recalc. 1st order: 49 d14 April 2003Soil accumulation studies: not requiredSoil residue studies: Not requiredRemarks:e.g. effect of soil pH on degradation ratenoneAdsorption/desorptionK f / K oc:K d:pH dependence: K oc: propoxycarbazone-sodium:12.9 –106.2 L/kg [n=5], median: 28.8 L/kg sulfonamide methyl ester (M05):K oc: 35 L/kg (column leaching study), 71.1 L/kg (calculated, PCKOCWIN)saccharin (M07):K oc: 4.6 to 15.5 L/kg [n = 5], median: 5.2 L/kg4-Hydroxy saccharin (M08):K oc: 456.9 to 2872.7 L/kg [n = 5], median: 2033.8 L/kg N-methyl propoxy triazolinone amide (M09):K oc: 10.4 to 551.5 L/kg [n = 5], median: 99.9 L/kgN-methyl propoxy triazolinone (M10):K oc: 8.9 to 75.5 L/kg [n = 5], median: 20.6 L/kgpropoxycarbazone-sodium:K d: 0.22 – 1.7. L/kg [n = 5]sulfonamide methyl ester (M05):0.161 L/kg (column leaching study)saccharin (M07):K d: 0.02 to 0.25 L/kg [n = 5]4-Hydroxy saccharin (M08):K d: 7.5 to 46.3 L/kg [n = 5]N-methyl propoxy triazolinone amide (M09):K d: 0.26 to 3.90 L/kg [n = 5]N-methyl propoxy triazolinone (M10):K d: 0.22 to 1.22 L/kg [n = 5]propoxycarbazone-sodium, M05, M07, M08, M09,M10: noMobilityLaboratory studies:Column leaching: not requiredAged residue leaching: Loamy sand (80 % sand, 4 % clay, 0.5 % C org), ageingtime 28 d (phenyl-label) and 29 d (triazolinone-label),irrigation 508 mm, incubation at 20 °C and FC in the14 April 2003darkleachates contained 85.8 % (phenyl-label) and 89.0 %(triazolinone-label) of the applied radioactivity, containin76.5 % propoxycarbazone-sodium, 3.1 - 3.6 % carboxyacid (M04), 0.8 % sulfonamide acid (M06), 4.3 % sacch(M07) and 7.9 % N-methyl propoxy triazolinone (M10).Sulfonamide methyl ester (M05) and N-methyl propoxytriazolinone amide (M09) were not detectedField studies:Lysimeter/Field leaching studies: 70 g as/ha, spring application, 2 lysimeters over 3years, single application in year 1 and 2annual average concentration in leachate (µg/l):propoxycarbazone-sodium: 0.009, 0.004, 0.002N-methyl propoxy triazolinone(M10):0.002,0.018,0007total radioactive residue: 0.061, 0.057, 0.049maximum concentration in leachate (µg/l):propoxycarbazone-sodium: 0.02N-methyl propoxy triazolinone(M10): 0.04total radioactive residue: 0.099Remarks: under vulnerable conditions,propoxycarbazone-sodiumand N-methyl propoxy triazolinone (M 10) might leachinto ground water14 April 2003 2.2 Fate and behaviour in waterAbiotic degradationHydrolytic degradation: pH 4: stable (25°C)pH 7: stable (25°C)pH 9: stable (25°C)Major metabolites: nonePhotolytic degradation: phenyl label: propoxycarbazone-sodium: DT50: 18.1 dtriazolinone label: propoxycarbazone-sodium: DT50: 40. Major metabolites: metabolite saccharin (M07): 22 % of TAR (day 19, endof study)metabolite N-methyl propoxy triazolinone (M10): 13.6 %TAR (day 19, end of study)Biological degradationReadily biodegradable: not required (see water sediment study)Water/sediment study:DT50 water:DT90 water:DT50 whole system:DT90 whole system:Distribution in water / sediment systems (active substance)Distribution in water / sediment systems (metabolites) 10.6 and 90.8 d35.4 and 302.0 d12.0 and 189.0 d39.9 and 627.0 dwater: 97 - 97.9 % AR (0 d)0.7 - 47.2 % AR after 100 dsediment: max. 18.2 % AR (3 d) - 21.4 % AR (100 d) 0.2 - 21.4 % AR after 100 dcarboxylic acid (M04)water: max. 0.1 % AR (30 d) - 50.2 % AR (30 d)0.1 - 34.6 % AR after 100 dsediment: max. 0.0 % AR (0-100 d) - 19.3 % AR (62d)0.0 - 13.2 % AR after 100 dsulfonamide acid (M06)water: max. 1.6 % AR (100d) - 16.2 % AR (100d) sediment: max. 0 (0-100 d)- 3.2 % AR (100d)N-methyl propoxy triazolinone (M10)water: max. 3.1 % AR (100 d) - 21.2 % AR (100 d) 3.1 - 21.2 % AR after 100 dsediment: max. 3.8 % AR (100 d) - 13.2 % AR (100 d) 3.8 - 13.2 % AR after 100 d14 April 2003 Degradation in the saturated zone degradation in the saturated zone: not required Remarks: none14 April 2003 2.3 Fate and behaviour in airVolatilityVapour pressure: Cannot be determined directly due to its extreme lowvalue; upper limit: < 9 ⋅ 10-8 Pa at 70 °C< 1 ⋅ 10-8 Pa at 20 °C (extrapolated)Henry's law constant: < 1 ⋅ 10-10 Pa m3 mol-1 at 20 °C (calculated)Photolytic degradationDirect photolysis in air: no dataPhotochemical oxidative degradation in air DT50: Calculations according to Atkinson (AOPWin 1.75): t½: 4.5 h (c OH = 0.5 · 106 cm-3, 24 h day)Volatilisation: from plant surfaces: < 10 % within 24 hoursfrom soil: < 10 % within 24 hours Remarks: no short or long range transport expected14 April 20033 EcotoxicologyTerrestrial VertebratesAcute toxicity to mammals: LD 50 >5000 mg/kg bw (rat)Acute toxicity to birds: LD 50 >2000 mg/kg bw (bobwhite quail) Dietary toxicity to birds: LC 50 >10000 ppm (bobwhite quail)Reproductive toxicity to birds: NOEL 1250 ppm (bobwhite quail and mallard duck)Aquatic OrganismsGroup Test sub-stanceTime-scaleEnd-pointToxicity (mg/L)Laboratory testsAcute toxicity fish (LC50):O. mykiss a.s. acute (static) mortality>77.6 Long term toxicity fish (NOEC):P.promelasa.s. long-term (flow-through)Mortalit y,hatch, growth, beha-viour 105Bioaccumulation fish:Not relevant (Log P OW : 0.03) D. magna a.s. acute(stat.) mortalit y >107 D. magnaMKH 7017 (M10) acute (stat.) mortalit y >100D. magna MKH 7018 (M04) acute (stat.) mortalit y >100Acute toxicity invertebrate (EC50)D. magna STJ 4934 (M05)acute (stat.)mortalit y>100Chronic toxicity invertebrate (NOEC): D. magnaa.s. chronic (flow-through) Mortalit y,growth,reprodu ction110Acute toxicity algae (EC50):Selenastr umcapricorn utuma.s. chronic biomass 1.57。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
– 二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3%–1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基)- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%– 二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3%– 二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;– didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;– dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;– 三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;– 增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
–铝碳酸镁:限量:0.5%;–烷基酰胺:限量:0.5%;–烯烃酰胺:限量:0.5%;–硅铝酸钠:限量:0.5%;– 二氧化硅: 限量:0.5%;–苯甲酸钠: 限量:0.5%;– 脂肪酸酯及其盐类: 限量:0.5%;–磷酸三钠: 限量:0.5%;–液态石蜡: 限量:0.5%;– 氧化锌: 限量:0.5%;– 滑石: 限量:0.5%;–氧化镁: 限量:0.5%;– 硬脂酸钠或硬脂酸锌或两者的混合物:限量:0.5%;–二氧化钛: 限量:4%;材料提供者须能证明样品组成的数量和质量都符合要求。
特性外观:粉末,珠子,颗粒或在转换后,不同厚度的片材或容器。
溶解性:几乎不溶于水,溶于热的芳香烃,几乎不溶于乙醇,乙烷和甲醇。
它们的软化温度在 65 °C和 165 °C之间,它们燃烧时都会有蓝色火焰。
鉴定:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
A.红外吸收分光光度法(2.2.24)准备:在0.25g样品中加入10ml的甲苯R并在回流冷凝器下加热约15分钟;放置几滴从氯化钠滑落获得的溶剂,然后在80°C的烘箱上将溶剂烘干。
最大吸收值:2920 cm− 1, 2850 cm− 1, 1475 cm− 1,1465 cm− 1, 1380 cm− 1, 1170 cm−1, 735 cm− 1 和 720 cm− 1.所得出的光谱和材料选出的标准样品的光谱是一样的。
如果材料以表格的形式进行检查,鉴定应该直接决定裁剪成合适大小的片状。
B.它符合相应的现场添加剂的补充测试。
C.在一个铂钳锅内,用1g硫酸氢钾R混合20mg,然后加热直到完全熔化。
允许对其进行冷却并加入20ml的稀硫酸。
轻轻地进行加热。
过滤掉产生的溶剂。
在滤液中添加1ml的磷酸和1ml的强过氧化氢溶液R。
如果该物质是不透明的钛白色,那么将会变成橙黄色。
试验:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
S1试液.使用S1试液需要4小时的准备。
称取25g材料,置于带磨砂口的硼硅玻璃烧瓶中,加入500ml的注射用水R,然后加热回流约5个小时。
冷却并转移到烧杯中。
保留部分提取液用于测试S1试液的外观,然后用烧结玻璃过滤器过滤剩余溶液。
S2试液.精密称取2.0g材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入80ml的甲苯R然后加热搅拌回流约90分钟。
冷却至60°C然后加入120ml的甲醇R并持续搅拌。
用烧结玻璃过滤器将溶液进行过滤。
取25ml由40ml的甲苯R和60ml的甲醇R混合的溶液来冲洗烧瓶和过滤器,合并洗液和滤液,并用相同的混合溶剂稀释至250ml。
同法制备空白溶液。
S3试液.称取100g的材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入250ml的0.1M盐酸,加热搅拌回流1个小时。
冷却并收集溶液。
S1试液的外观。
清晰无色。
酸度或碱度。
量取100ml的S1试液,加入0.15ml的BRP指示剂R。
加入约1.5ml的0.01M氢氧化钠溶液,直到溶液颜色蓝色。
量取100ml的S1试液,加入0.2ml的甲基橙溶液R,加入约1ml的0.01M盐酸,直到溶液变为黄色再变为橙色。
吸光率:S1试液在波长范围220nm-340nm之间的吸光率最大值不超过0.2。
还原性物质。
量取20ml的S2试液,加入1ml的稀盐酸R和20ml的0.002M高锰酸钾。
加热回流3分钟然后直接冷却。
加入1g碘化钾R然后直接用0.01M硫代硫酸钠进行滴定测量法测量,用0.25ml的淀粉溶液R作为指示液。
同法空白滴定。
滴定体积的区别小于3.0ml。
可溶于己烷的物质。
称取10g材料,置于250ml带磨砂口的的圆锥形硼硅酸盐玻璃烧瓶中。
加入100ml的己烷然后加热搅拌回流4个小时。
在冰水中冷却然后用烧结玻璃过滤器迅速过滤(过滤时间必须少于5分钟;如有必要可将溶液加压来加速过滤)并让溶液保持在0°C左右。
量取20ml的滤液,放在恒重过的硼硅酸盐玻璃皿上,在水浴中蒸发。
将滤渣放在100-105°C 的烘箱上进行干燥一小时。
所得滤渣不得大于标准样品的10%,而且不超过5%。
可提取出的铝:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用铝标准溶液(200ppm Al)R加0.1M的盐酸稀释成标准溶液。
波长:铝发射的波长为396.15nm,光谱采用396.25nm。
确定所用的盐酸中没有铝。
可提取出的钛:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用钛标准溶液(100ppm Ti)R加0.1M的盐酸稀释成标准溶液。
波长:钛发射的波长为336.12nm,光谱采用336.12nm。
确定所用的盐酸中没有钛。
可提取出的锌:限量:1ppm。
原子吸收光谱测定法。
测试溶液:S3试液。
标准溶液。
用锌标准溶液(10ppm Zn)R加0.1M的盐酸稀释成标准溶液。
来源:锌空心阴极电子管。
波长:213.9nm雾化装置:空气乙炔火焰确定所用的盐酸中没有锌。
可提取出的重金属:限量:2.5ppm将50ml的溶液S3在水浴中蒸发至5ml然后加水R稀释成20.0ml。
取12ml溶液按照测试A进行。
用2.5ml的铅标准溶液(10ppm Pb)准备标准溶液。
硫酸灰分:限量:1.0%,称取5.0g试验。
此限制不适用于被二氧化钛处理过的材料。
补充测试只有在材料定期检查时,这些测试可全部或部分进行实施。
酚类抗氧化剂。
液相色谱法。
溶剂混合:氰化甲烷R,四氢呋喃R(50:50 V/V)S21试液。
精密量取50ml的S2试液,在45°C真空蒸干后,使残渣溶于5.0ml的混合溶剂中。
用S2试液的空白溶液同法制备空白样品。
S22试液。
精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的二氯甲烷中。
用S2试液的空白溶液同法制备空白样品。
S23试液.精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的相同量的氰化甲烷R和10g/l的四氢呋喃R里的叔丁基化过氧氢溶液的混合剂。
封上烧瓶静置1小时。
准备一个空白溶液。
仅在有必要对规定的所检查的物质组成进行酚类抗氧化剂分析时准备以下标准溶液。
标准溶液(a)。
溶解25.0mg的二叔丁基对甲酚CRS(增塑剂07)和60.0mg的增塑剂08CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的该溶液稀释成50.0ml。
标准溶液(b)。
溶解60.0mg的增塑剂09CRS和60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释成50.0ml。
标准溶液(c)。
溶解60.0mg的增塑剂11CRS和60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(d)。
溶解25.0mg的增塑剂07CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(e)。
溶解60.0mg的增塑剂08CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(f)。
溶解60.0mg的增塑剂13CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(g)。
溶解60.0mg的增塑剂09CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(h)。
溶解60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶剂稀释至50.0ml。
标准溶液(i)。
溶解60.0mg的增塑剂11CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶剂(j)。
溶解60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(k)。
溶解20.0mg的增塑剂18CRS于10.0ml的同等量的氰化甲烷R和10g/l的四氢呋喃R里的叔丁基化过氧氢溶液的混合剂。
允许放在密封容器内静置一个小时。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
A.如果该物质检查出含有增塑剂07和/或增塑剂08,请实施以下测试:-尺寸:l = 0.25 m, O = 4.6mm;-固定相:十八烷基甲硅烷基硅胶的气相色谱R (5 μm).移动相:水R,氰化甲烷R流量:2 mL/min检测方法:分光光度计,280nm注入:20μL的 S21试液,相应的空白溶液,标准溶液(a),和要么标准溶液(d)要么标准溶液(e)要么标准溶液(d)和(e)。