DETERMINATION OF 240PU239PU RATIO IN ENVIRONMENTAL SAMPLES BASED ON THE MEASUREMENT OF Lxα-RAY ACTI
GC-MS测定聚氨酯产品中的N,N-二甲基甲酰胺

doi:10.19677/j.issn.1004-7964.2021.01.009GC-MS 测定聚氨酯产品中的N,N-二甲基甲酰胺胡苹,李大刚,詹迎旭(黎明职业大学材料与化学工程学院,福建泉州362000)摘要:以乙酸乙酯为提取溶剂超声提取聚氨酯产品中的N,N-二甲基甲酰胺,提取液经滤膜过滤后,选择HP-INNOWAX 色谱柱为固定相,在离子监测模式(SIM)下扫描,进行气相色谱—质谱法测定,并以保留时间和特征碎片的离子丰度比定性,外标法定量。
结果表明,N,N-二甲基甲酰胺在0.2~10mg/L 浓度范围内呈良好的线性,方法检出限为0.46mg/kg,在3个加标水平下的平均回收率为92.9%~98.7%,相对标准偏差(RSD,n=6)为6.5%~7.7%。
该方法准确可靠、方便快捷,适用于实际聚氨酯产品中N,N-二甲基甲酰胺含量的测定。
关键词:GC-MS;聚氨酯;N,N-二甲基甲酰胺中图分类号:TS 57文献标志码:ADetermination of N,N-Dimethylformamide in Polyurethane Products byGas Chromatography-Mass Spectrometry(Department of Materials and Chemical Engineering,Liming Vocational University,Quanzhou 362000,China)Abstract:A method based on gas chromatography-mass spectrometry (GC-MS)was developed to determine the content of N,N-dimethylformamide (DMF)in polyurethane (PU)products.DMF in PU sample was ultrasonically extracted using ethyl acetate as solvent,and the extracting solution was then purified with membrane filter.Under the ion monitoring (SIM)mode,DMF was determined using GC-MS with HP-INNOWAX chromatographic column as the stationary phase.Retention time and the abundance ratio of the characteristic fragment ions were adopted for the qualitative analysis,whilequantification analysis was performed using an external standard method.Results indicate that a linear calibration curve ofDMF can be obtained in the range of 0.2-10mg/L,and the detection limit is 0.46mg/kg.The spiked average recoveries varied from 92.9%to 98.7%at three spiked levels with relative standard deviation (RSD,=6)of 6.5%-7.7%.The present method is accurate,reliable and convenient,and could be applied in the determination of DMF in PU products.Key words:GC-MS;polyurethane products;N,N-dimethylformamide收稿日期:2020-03-16基金项目:2019黎明职业大学科研团队项目(LMTD201901)第一作者简介:胡苹(1985-),男,理学硕士,工程师,研究方向:消费品中有害物质的检测,Tel:150****3102,E-mail:***************。
Determination of total and unbound warfarin

Journal of Chromatography A,1314 (2013) 54–62Contents lists available at ScienceDirectJournal of ChromatographyAj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c h r o maDetermination of total and unbound warfarin and warfarin alcohols in human plasma by high performance liquid chromatography with fluorescence detectionTommaso Lomonaco a ,Silvia Ghimenti a ,Isabella Piga a ,Massimo Onor b ,Bernardo Melai a ,Roger Fuoco a ,Fabio Di Francesco a ,c ,∗aDepartment of Chemistry and Industrial Chemistry,University of Pisa,Pisa,Italy bInstitute of Chemistry of Organometallic Compounds,CNR,Pisa,Italy cInstitute of Clinical Physiology,CNR,Pisa,Italya r t i c l ei n f oArticle history:Received 23April 2013Received in revised form 23August 2013Accepted 25August 2013Available online 31 August 2013Keywords:WarfarinWarfarin alcoholsHigh performance liquid chromatography Sample preparation techniquesa b s t r a c tTwo analytical procedures are presented for the determination of the total content and unbound fraction of both warfarin and warfarin alcohols in human plasma.Chromatographic separation was carried out in isocratic conditions at 25◦C on a C-18reversed-phase column with a mobile phase consisting of a 70%buffer phosphate 25mM at pH =7,25%methanol and 5%acetonitrile at a flow rate of 1.2mL/min.Fluores-cence detection was performed at 390nm (excitation wavelength 310nm).Neither method showed any detectable interference or matrix effect.Inter-day recovery of the total warfarin and warfarin alcohols at a concentration level of 1000ng/mL was 89±3%and 73±3%,respectively,whereas for their unbound fraction (at a concentration level of 10ng/mL)was 66±8%and 90±7%,respectively.The intra-and inter-day precision (assessed as relative standard deviation)was <10%for both methods.The limits of detection were 0.4and 0.2ng/mL for warfarin and warfarin alcohols,respectively.The methods were successfully applied to a pooled plasma sample obtained from 69patients undergoing warfarin therapy.© 2013 Elsevier B.V. All rights reserved.1.IntroductionWarfarin [3-(␣-acetonylbenzyl)-4-hydroxycoumarin,WAR],the most common anticoagulant drug,is prescribed for the treat-ment of many diseases and the prevention of thromboembolic events [1].WAR is a weakly acidic drug (p K a of 5.19at T =25◦C)with an enolic group (Fig.1a)[2].It has an asymmetric carbon center and is commercially available as a racemic mixture.The two enantiomers (R-WAR and S-WAR)have a different anticoag-ulant potency (S-WAR is 3–4times more potent than R-WAR),metabolism and interaction with other drugs [3,4].WAR is com-pletely absorbed after oral administration (F =1),is highly bound to site I of albumin (≈99%)with a high affinity (K d =3.4±0.7M),has a small volume of distribution (0.11–0.18L/kg),and low clearance (0.0005–0.0079L/h/kg)[5–8].Only the unbound fraction can carry out a therapeutic action and cross the biological membranes (e.g.salivary glands [9]).The drug is metabolized in the liver by cytochrome (CYP)P450to inactive hydroxylated metabolites (OH-WAR)(major pathway)and by ketone reductases to warfarin∗Corresponding author at:Department of Chemistry and Industrial Chemistry,University of Pisa,Pisa,Italy.Tel.:+390502219308;fax:+390502219260.E-mail address:fdifra@dcci.unipi.it (F.Di Francesco).alcohols (WAROHs,Fig.1b)[10].This reduction is more effec-tively catalyzed by nicotinamide adenine dinucleotide phosphate (NADPH)than by nicotinamide adenine dinucleotide (NADH [11]).This latter reduction generates a second chiral center (C-11)and makes two pairs of diastereoisomers possible,namely RS/SR-Warfarin alcohols and RR/SS-Warfarin alcohols [12,13].After the administration of a single dose of WAR,S-7-hydroxywarfarin and RS-Warfarin alcohol are the two major metabolites observed in human plasma [14].Gebauer reported that 7-OH-WAR is an inactive metabolite with a half maximal inhibitor concentration (IC 50)of 250M,whereas WAROHs show a low anticoagulant activity with IC 50of 12.5M (IC 50of WAR is 2.2M)[15].Around 40million patients are currently undergoing WAR ther-apy and this number has been increasing steadily [16].Positive clinical outcomes during WAR therapy depend on maintaining the level of WAR within a narrow therapeutic range.This is challeng-ing due to the large inter-and intra-individual variability in patient responses,as many factors (e.g.diet,liver disease,comorbidities,other drugs,and genetic variability)may interact with the ther-apy [17].Furthermore,after about 60years of clinical use,the anticoagulation mechanism of WAR has never been completely clarified and the possible role of WAROHs has received little atten-tion in the literature.A few studies indicate that the anticoagulant activity of WAROHs may be an additional variable involved in0021-9673/$–see front matter © 2013 Elsevier B.V. All rights reserved./10.1016/j.chroma.2013.08.091T.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–6255Fig.1.Structures of WAR(a)and WAROHs(b)(*asymmetric center).Carbon4 ,10,6,7,8are the hydroxylation sites of WAR by cytochrome P450.the anticoagulant mechanism of WAR[13].However,pharmaco-logical data concerning these metabolites are scarce.Lewis et al. reported that the plasmatic concentrations of WAROHs after a sin-gle dose(100mg)were steady for20days,and the long half-life may strengthen its potency and contribution to the overall antico-agulant activity.Unlike WAR,WAROHs are directly eliminated by renal excretion without any further transformation[13].The international normalized ratio(INR)is the primary assay used in monitoring WAR therapy[18].When a patient is started on WAR therapy,INR monitoring should be performed on a daily basis until the INR is within the therapeutic range and should be performed two or three times a week for one to two weeks[19]. Therapeutic monitoring is essential in some situations to assess anticoagulant affecting factors,especially when the INR is diffi-cult to target.Knowledge of the plasma concentration of WAR and WAROHs is valuable for clinical decisions and enables severe intox-ication due to interference with other drugs and food to be treated effectively[18].Sensitive methods to measure WAR concentrations and its hydroxylated metabolites in biological samples have been pro-posed[19–21],but to our knowledge no study has been published on analytical methods for the determination of the unbound frac-tion of WAROHs in human plasma and other biologicalfluids.Most of the recently reported methods for measuring WAR in biological samples use high performance liquid chromatography(HPLC)with mass spectrometric detection(MS)[22,23].These methods have a high sensitivity,but are too expensive for routine analyses in a clinical laboratory.WAR has the advantage of a nativefluorescence if excited at 310nm,which can be successfully exploited for detection at low concentrations[24].This work illustrates an HPLC method withfluorescence detec-tion(FLD)to determine the total content and unbound fraction of WAR and WAROHs in human plasma.These methods are not expen-sive,show a good recovery and sensitivity,and are thus suitable for use in pharmacokinetic studies and for therapy monitoring.The proposed methods are currently being used in a clinical trial,whose results will be published in a future paper.2.Materials and methods2.1.Chemicals and reagentsRacemic WAR,i.e.3-(␣-acetonylbenzyl)-4-hydroxycoumarin sodium salt(purity≥98.0%),sodium borohydride(purity≥98.0%), sulfuric acid(purity≥99.5%),ethanol(purity≥99.5%)and deuter-ated methanol were purchased from Sigma Aldrich(Milan,Italy). Sodium phosphate monobasic(purity≥99.0%),potassium phos-phate dibasic(purity≥99.0%),dichloromethane(purity≥99.5%), hexane(purity≥95.0%),formic acid(purity≥99.5%),acetoni-trile and methanol were purchased from Sigma Aldrich at HPLC grade.HPLC grade water was produced by a Milli-Q Reagent Water System(Millipore,USA).The thromboplastin reagent (HemosIL RecombiPlasTin2G),thromboplastin diluent(HemosIL RecombiPlasTin2G Diluent),calibration plasma samples,normal control assay,low and high abnormal control assays used both for INR measurements and quality control were supplied by the Instrumentation Laboratory(Milan,Italy).2.2.Synthesis of warfarin alcoholsWAROHs were obtained in our laboratory by reduction of a racemic WAR standard solution with sodium borohydride[25]. WAR(a):1H NMR(CD3OD,ı/ppm):7.98–7.94(1H,m),7.67–7.61 (1H,m),7.41–7.18(7H,m),4.12(1H,dd,J=0.7Hz,CH at C-9),2.22 (2H,ddd,J=30.0,14.0and6.9Hz,CH2at C-10),1.74(3H,s).13C NMR(CD3OD,ı/ppm):207.1(1C,carbonyl at C-11),163.0,159.9, 152.7,143.7,131.7,128.1,127.8,127.1,126.8,125.9,125.7,124.0, 123.9,122.7,99.5,42.8,26.4,24.6.ESI-QTOF-MS:m/z309.1[M+H]+.WAROHs(b):1H NMR(CD3OD,ı/ppm):7.99–7.95(1H,m), 7.60–7.50(1H,m),7.50–7.14(7H,m),4.70–4.63(1H,m,CH at C-11), 3.71–3.68(1H,m,CH at C-9),2.37(2H,m,CH2at C-10),1.27–1.19 (3H,m).13C NMR(CD3OD,ı/ppm):161.2,159.9,152.7,142.9,131.6, 127.9,127.6,127.5,125.8,125.6,125.2,123.7,122.9,122.6,116.0, 65.8(1C,hydroxyl at C-11),40.2,22.8,22.4.ESI-QTOF-MS:m/z 311.1[M+H]+.Carbonyl reduction at C-11generates a second chiral carbon, which makes four stereoisomers(RR,RS,SR,and SS)possible.These can be seen as two pairs of diastereoisomers(RR-SS and RS-SR), with a different chromatographic behavior.Fig.1shows the struc-tures of WAR(1a)and WAROHs(1b).The HPLC separation of the reduced solution revealed that the reaction with sodium borohydride was completed and WAROHs were generated.The product of sodium borohydride reduction was confirmed by comparing the1H NMR and13C NMR spectra of WAR and WAROHs.2.3.Plasma samplesPlasma samples were obtained from the local oral anticoagu-lant center with the permission of the Ethics Committee of the local hospital(AOUP,Pisa,Italy).Human whole blood samples were col-lected into vacuum tubes containing109mM(3.2%)sodium citrate (Vacutest Kima,Italy)and immediately centrifuged at3000rpm for10min to obtain platelet-poor plasma.The plasma sample was divided into two aliquots:thefirst was immediately used for the determination of INR,whereas the second was stored at−80◦C together with the nominally healthy volunteer samples until the WAR and WAROHs were measured.2.4.Preparation of calibration standards and quality control samplesA phosphate buffer solution(PBS)(1M,pH=7.0)was prepared by dissolving18.30g of sodium phosphate monobasic and49.20g56T.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–62of potassium phosphate dibasic in water.This solution was diluted to25mM to obtain the PBS used for the HPLC analyses and for the preparation of standard solutions.Stock solutions of WAR(950g/mL)and WAROHs (1060g/mL)were prepared by dissolving weighed amounts of the pure compounds in water and methanol respectively.The solutions were used to prepare another stock solution containing WAR(96g/mL)and WAROHs(105g/mL)in PBS25mM,which was further diluted with PBS to obtain standard working solutions of WAR and WAROHs at1,2,5,10,15,20,30,100,200,500,1000, 1500,2000and3000ng/mL levels.All the solutions were protected from light and stored in a refrig-erator at4◦C.The stability of the working solutions over2months was investigated.Pooled patient plasma samples(PPPSs)were obtained by pool-ing samples from69patients undergoing WAR therapy,whereas control plasma samples(CPSs)were obtained from10volunteers not taking WAR.Aliquots of these latter samples were spiked with known amounts of WAR and WAROHs to obtain standard plasma samples(SPSs)at different concentration levels.SPSs were stored at−80◦C,and some were used to check the stability over2months and after thaw–freeze cycles.2.5.Sample preparationSeveral preparation procedures were tested to determine the concentrations in plasma of total WAR and WAROHs as well as the unbound fraction.2.5.1.Procedure A:precipitation of plasma proteins(totalWAR/WAROHs)A solution(1mL)containing equal volumes of plasma and ace-tonitrile was vortex-mixed(30s)and centrifuged at5000rpm for 5min at room temperature.The supernatant(10L)was injected into the HPLC system.2.5.2.Procedure B:protein precipitation and ultrafiltration(total WAR/WAROHs)A solution(1mL)containing equal volumes of plasma and ace-tonitrile was vortex-mixed for30sec and centrifuged at5000rpm for5min at room temperature.An aliquot of200L of super-natant was diluted to1mL with PBS25mM at pH=7.This solution was transferred into an Amicon tube(Amicon®Ultra-4,Millipore) with a molecular weight cut-off of3kDa and then centrifuged at 5000rpm for60min at20◦C.25L of the ultrafiltrate sample were injected into the HPLC system without any further treatment.2.5.3.Procedure C:liquid–liquid extraction(total WAR/WAROHs)A modified version of Naidong’s procedure was used[26].In brief,2mL of H2SO40.5M,500L of ethanol,and4mL of a mixture of dichloromethane/hexane(1:5,v/v)were added to a plasma sam-ple(500L).The resulting mixture was vortex-mixed for30s and then centrifuged at5000rpm for5min at room temperature.The upper organic phase was transferred to a screw top V-Vial(Sigma Aldrich,Italy)and evaporated to dryness under a gentle stream of nitrogen at room temperature.The vials were weighed in all steps of this extraction procedure.The residue was dissolved in1mL of PBS25mM at pH=7and then injected(15L)into the HPLC system.2.5.4.Procedure D:ultrafiltration(unbound fractionWAR/WAROHs)An aliquot of the plasma sample(1mL)was transferred into an Amicon tube(Amicon®Ultra-4,Millipore)with a molecular weight cut-off of3kDa and centrifuged at5000rpm for60min at20◦C.25L of the ultrafiltrate sample were injected into the HPLC system without any further treatment.2.6.EquipmentThe HPLC system(Jasco,Japan)was equipped with an autosam-pler(AS2055),a quaternary low-pressure gradient pump(PU 2089),afluorescence detector(FP2020),and an ultraviolet detector (UV2070).The column temperature was controlled by a HT3000 thermostat(ClinLab,USA).The HPLC system was controlled using ChromNAV TM software(Jasco,Japan).Chromatographic separation of WAR and WAROHs was car-ried out with a TC–C18150mm×4.6mm,5m reversed-phase column(Agilent Technologies,USA)connected to a TC–C18 12.5mm×4.6mm,5m guard column(Agilent Technologies, USA).A few selected samples were also analyzed by an HPLC-ESI-Q-TOF to validate the analytical methods.The LC–MS analyses were carried out using a1200Infinity HPLC coupled to a6530Accurate-Mass Quadrupole Time-Of-Flight(Q-TOF)mass spectrometer with a Jet Stream ESI interface(Agilent Technologies,USA).The HPLC system was controlled by MassHunter TM software(Agilent Tech-nologies,USA).Chromatographic separation of WAR and WAROHs was carried out with a Zorbax Extend–C1850mm×2.1mm, 1.8m reversed-phase column(Agilent Technologies,USA)con-nected to an Eclipse Plus12.5mm×4.6mm,5m guard column (Agilent Technologies,USA).NMR spectra were recorded by a Bruker Avence400MHz (Bruker Corporation,USA).Absorption andfluorescence spectra were recorded by a Lambda 25spectrophotometer(PerkinElmer,USA)and a LS45spectrofluo-rimeter(PerkinElmer,USA),respectively.A centrifuge Centrifugette4206(ALC,Italy)and a Centrifuge 5804R equipped with an A-4-44swinging bucket rotor(Eppendorf, Italy)were used for sample centrifugation.The plasma samples were centrifuged using an Amicon®Ultra-4 device(Millipore,Italy),equipped with a cap,afilter device,and a centrifuge tube.The INR measurements were carried out at the clinical chemical laboratory of the local hospital(AOUP,Pisa,Italy)by an automatic ACL TOP700system(Instrumentation Laboratory,USA)equipped with an autosampler.Spectrometric INR measurements were per-formed at a wavelength of671nm to minimize optical interference (e.g.hemoglobin and bilirubin).All data were analyzed using GraphPad Prism v.5(GraphPad Software Inc.,USA).2.7.Chromatographic and detection conditionsChromatographic separation was carried out by the Jasco HPLC in isocratic mode with a mobile phase consisting of25%methanol, 5%acetonitrile,and70%phosphate buffer25mM at pH=7at aflow rate of1.2mL/min.The total run time was20min.The column tem-perature was set at25◦C.Fluorescence detection was performed at excitation and emission wavelengths of310and390nm,respec-tively.A few selected samples were analyzed by HPLC-ESI-Q-TOF. Chromatographic separation was carried out by HPLC in isocratic mode with a mobile phase of35%acetonitrile and65%water con-taining1%formic acid at aflow rate of0.25mL/min at25◦C.The injection volume was4L.The ESI operating conditions were:dry-ing gas(N2,purity>98%):350◦C and10L/min;capillary potential 4.5kV;nebulizer gas35psig;sheath gas(N2,purity>98%):375◦C and11L/min.The fragmentor was kept at100V and the collision energy(CID)for the MS/MS experiments was20V.The collision gas was nitrogen(purity99.999%).High-resolution MS and MS/MST.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–6257Fig.2.Full scan and product ion mass spectra of[M+H]+of WAR(a),RR/SS-Warfarin alcohols(b)and RS/SR-Warfarin alcohols(c).spectra were achieved in positive mode in the range100–350m/z. The protonated molecule[M+H]+with m/z311.1and309.1were monitored for WAROHs and WAR identification,respectively.The total HPLC-ESI-Q-TOF run time was10min.3.Results and discussionValidation of the proposed methods was performed in accor-dance with the IUPAC guidelines[27]and included an evaluation of interferences,matrix effect,calibration curves,limits of detection (LOD)and quantification(LOQ),intra-day and inter-day precision, stability,and recovery.3.1.Sample preparationThree preparation procedures were compared to determine total WAR and WAROHs in plasma samples.The aim was tofind the most simple and reliable sample preparation method to remove any endogenous components(e.g.proteins,salts,lipids)that might interfere with the analyte determination.Three standard plasma samples with a WAR and WAROHs concentration of1000ng/mL were prepared.Each of them was split into three aliquots and processed according to A,B and C procedures.Mean values(n=9) for each aliquots refer to three repeated injections.Procedure A(protein precipitation with1:1addition of ace-tonitrile)showed very good levels of recovery and precision for both WAR(95±2%)and WAROHs(100±2%).However,this treat-ment did not remove all the proteins,thus increasing the pressure in the column and shifting the retention time(data not shown). These drawbacks were avoided in procedure B by ultrafiltrating the samples at3kDa.Lower but still good levels of recovery and a better precision than the previous procedure were observed(WAR: 85±1%and WAROHs:86±1%).A liquid–liquid extraction(proce-dure C)with an acidification step to release WAR and WAROHs from plasma proteins was optimized by comparing single and mul-tiple extractions.The second extraction did not increase recovery58T.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–62Fig. 3.HPLC chromatograms of a standard solution of WAR and WAROHs (100ng/mL).The product ions traces are m/z251.1and175.0for WAROHs(a)and m/z 251.1and163.0for WAR(b).Elution order and retention times:2(RR/SS-Warfarin alcohols,r t:3.8min),3(WAR,r t:6.0min),4(RS/SR-Warfarin alcohols,r t:8.4min).significantly,thus a single extraction was applied for the quantita-tive analysis of WAR and WAROHs in plasma.In fact,procedure C showed the highest efficiency in separating the analytes from the matrix,as well as good levels of recovery and precision(see Section 3.4.3).Procedure C was thus selected for a more in-depth evaluation of the analytical performance.The unbound plasmatic fraction of WAR and WAROHs was determined by sample ultrafiltration with a cut-off membrane. Unlike other studies[20,22,28],the proposed procedure did not require any further treatment.3.2.HPLC–ESI-Q-TOF assay for the determination of warfarin and warfarin alcoholsThe ESI-Q-TOF gave the optimum sensitivity for WAR and WAROHs in positive ionization mode(data not shown).The prod-uct ion scan of the protonated[M+H]+ion for WAR(m/z309.1) displayed clear product ions at m/z251.1and163.0,whereas for WAROHs(m/z311.1),product ions at m/z251.1and175.0were recorded(Fig.2).The HPLC chromatogram of a standard solution of WAR and WAROHs shows three well separated peaks with reten-tion time of3.8,6.0and8.4min(Fig.3).The linear regression of the peak area values versus concentra-tion wasfitted in the concentration range of1–1500ng/mL for both WAR and WAROHs.The seven-point calibration curves(n=3at each concentration)were evaluated by Deming regression analysis, and the best-fit models for WAR and WAROHs were respectively: y=(1750±10)x,(R2=0.9999)and y=(820±10)x,(R2=0.9999).A blank plasma sample spiked at a low level of1ng/mL of WAR and WAROHs was prepared and analyzedfive times with the procedure C.The LOD and LOQ values were calculated,in accordance with IUPAC guidelines[29],as three and ten times the standard deviation(s b)of the“low level spiked blank”,and were 0.6and1.9ng/mL for WAR,and0.8and2.8ng/mL for WAROHs. 3.3.HPLC-fluorescence assay for the determination of warfarinand warfarin alcohols3.3.1.Selection offluorescence wavelengthsWAR shows goodfluorescence when excited at310nm[24]. WAR contain acidic moieties whosefluorescence quantum yields depend on pH,which need to be adjusted to give maximumflu-orescence.Thefluorescence yield of WAR decreases significantly at acidic pH[30],whereas no data were available concerning the dependence of WAROHsfluorescence on pH.Absorbance andfluo-rescence spectra at different pH values(4,5,6and7)were recorded to obtain the maximumfluorescence intensity for WAROHs.Two standard solutions containing10g/mL and0.2g/mL of WAROHs were prepared in PBS25mM at pH=7.Three aliquots of each solu-tion(1mL)were acidified at pH4,5and6by adding10,7and 5L of phosphoric acid1M respectively.The absorbance spectra of the solution at10g/mL(Fig.4a)were recorded in the range 200–400nm with a scan rate of0.2nm/s,whereas thefluorescence spectra of the solution at0.2g/mL(Fig.4b)were recorded by excit-ing at310nm and measuring the emission in the range310–500nm with a scan rate of0.5nm/s.The optimum excitation(310nm)and emission(390nm)wave-lengths were selected for HPLC-FLD measurements.3.3.2.Chromatographic separationFig.5shows the HPLC chromatograms of standard solution of WAR and WAROHs(a),and control plasma samples(CPSs), and pooled patient plasma samples(PPPSs)ultrafiltered(b)and extracted(c).The chromatogram of the standard solutions of WAR and WAROHs shows three well separated peaks with retention times of11.1,13.1and14.6min(Fig.5a).The WAROHs peaks were identified according to Hermans and Thijssen[31].These authors separated the two diastereoisomers of WAROHs by thin-layer chromatography using silica gel and a mobile phase consisting of diethylether and acetic acid(99:1.5,v/v). In these conditions,the RS/SR-Warfarin alcohols traveled faster than the RR/SS-Warfarin alcohols,whereas the opposite is true in reversed phase chromatography[30].Considering the results of these studies and comparing the chro-matograms of PPPSs and standard solutions of WAROHs,the peaks at11.1and14.6min(Fig.5)were assigned to RR/SS-Warfarin alco-hols and the RS/SR-Warfarin alcohols,respectively.3.4.Analyticalfigure of merits3.4.1.Limit of detection(LOD)and quantitation(LOQ)A blank plasma sample spiked at0.5ng/mL of WAR and WAROHs was prepared and analyzedfive times with procedure C.The LOD and LOQ values were calculated in accordance with IUPAC guide-lines[29],as three and ten times the standard deviation(s b)of the “low level spiked blank”,and were0.4and1.3ng/mL for WAR,and 0.2and0.7ng/mL for WAROHs.3.4.2.Calibration curvesA working range of100–3000ng/mL was chosen for the total content determination of both WAR and WAROHs.The WAR plas-matic concentration range reported in the literature is about 0.5–2.5g/mL[21,32],thus this extended range allowed for a safety margin to detect any potential overdose due to drug-drug interactions[16].The seven-point calibration curves(n=3at each concentration)were evaluated by the Deming regression analysis,T.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–6259Fig.4.Absorbance(a)andfluorescence(b)spectra of WAROHs at different pH values.Fluorescence spectra were obtained by exciting at310nm.Fig.5.HPLC chromatograms of a standard solution(10ng/mL)(a),and control plasma samples(CPSs,dashed line)and pooled patient plasma samples(PPPSs,solid line)ultrafiltered(b)and extracted(c).Elution order and retention times:1(7-OH-WAR,r t:6.8min),2(RR/SS-Warfarin alcohols,r t:11.1min),3(WAR,r t:13.1min),4 (RS/SR-Warfarin alcohols,r t:14.6min),and5(10-OH-WAR,r t:15.9min).and the best-fit models for WAR and WAROHs were respectively: y=(7750±10)x,(R2=0.9999)and y=(15,330±10)x,(R2=0.9999).The literature reports that the unbound plasmatic fraction of WAR is about1%of the total[33],thus a working range of 1–30ng/mL was chosen for both WAR and WAROHs.The seven-point calibration curves(n=3at each concentration)were evalu-ated by the Deming regression analysis,and the best-fit models for WAR and warfarin WAROHs were respectively:y=(13,100±10)x, (R2=0.9999)and y=(26,400±10)x,(R2=0.9999).3.4.3.Recovery and precisionProcedure C:liquid–liquid extraction:The extraction recovery of WAR and WAROHs from SPSs was calculated as the percentage of the analyte concentration measured in the extracted sample with respect to the initial spiked concentration.Three samples spiked at three different concentration levels(200,1000and 2000ng/mL)were prepared and each one was evaluated in tripli-cate within the same day and on three consecutive days.The WAR and WAROHs recovery and the corresponding intra-and inter-day relative standard deviation(RSD%)are reported in Table1.The dif-ference between WAR(89%)and WAROHs(73)recovery was likely due to different partition ratios.In fact,in procedure A,which is simply based on protein precipitation with acetonitrile without any partition equilibrium involved,the recovery was similar for both molecules(95for WAR,and100for WAROHs).These results confirmed that procedure C is suitable for a reliable determination of the total concentration of both analytes in human plasma.Procedure D:ultrafiltration:The recovery of WAR and WAROHs with procedure D was calculated in triplicate standard work-ing solutions at three different concentration levels(2,10and 20ng/mL).Standard solutions had to be used in this case because human plasma samples with a known amount of unbound WAR and WAROHs could not be produced and certified standards are not available.In fact,spiking an SPS with a warfarin solution results in a sample in which the unbound fraction is known to be about 1%.Small variations of this percentage,which might be possible due to differences between plasma samples,would result in large errors in the absolute values of the unbound fraction.The WAR and60T.Lomonaco et al./J.Chromatogr.A1314 (2013) 54–62Table1Extraction recovery,intra-and inter-day precision of the determination of WAR and WAROHs in plasma.Concentration(ng/mL)Recovery%Intra-day a RSD Recovery%Inter-day b RSDExpected MeasuredWAR191167862%882%928830894%893%19071670872%882%WAROHs208153705%743%1010740724%733%20801500713%722%a Calculated from three replicates at each concentration value.b Calculated from three replicates at each concentration in three days.Table2Recovery,intra-and inter-day precision of the determination of WAR and WAROHs in standard solutions.Concentration(ng/mL)Recovery%Intra-day a RSD Recovery%Inter-day b RSDExpected MeasuredWAR 1.9 1.3659%6812%9.6 6.3655%668%19.612.9662%664%WAROHs 2.1 1.9872%916%10.49.4859%907%21.419.1911%894%a Calculated from three replicates at each concentration value.b Calculated from three replicates at each concentrations in three days.WAROHs recovery and the corresponding intra-day and inter-day relative standard deviation(RSD%)are reported in Table2.The precision was also estimated in SPSs at the three different concentration levels(200,1000and2000ng/mL).Assuming that the unbound fraction was1%,the estimated WAR and WAROHs concentrations in the ultrafiltrate samples were2,10and20ng/mL. The inter-and intra-day relative standard deviations are reported in Table3.The estimated recoveries of the two methods were used to cal-culate the real concentrations of WAR and WAROHs in plasma samples.3.4.4.InterferenceThe possible effect of endogenous interfering substances on the measured concentrations of WAR and WAROHs(total and unbound fraction)was investigated by comparing results obtained by HPLC-fluorescence and HPLC–ESI-Q-TOF in triplicate on SPSs with a concentration of1000ng/mL and on PPPSs with unknown concentrations.The concentration values obtained by the HPLC–ESI-Q-TOF,assumed as reference values,and the HPLC-fluorescence were in good agreement in all the samples(with differences always lower than10%).Table3Intra-and inter-day precision of the determination of unbound WAR and WAROHs in SPS samples.Estimatedconcentration(ng/mL)Intra-day a RSD Inter-day b RSDWAR 1.94%9%9.33%6%18.74%9%WAROHs 2.14%6%10.13%7%20.42%8%a Calculated from three replicates at each concentration value.b Calculated from three replicates at each concentrations in three days.3.4.5.Matrix effectThe shape and position of the WAR and WAROHs peaks were not affected by the presence of the plasma matrix(Fig.5),which suggests the absence of any detectable matrix effect.This hypothe-sis was confirmed by comparing the slopes of the calibration curves obtained with working solutions,ultrafiltered and extracted PPPSs (Table4).For both methods,these slopes were not significantly different at a confidence level of95%.3.4.6.StabilityThe stability of both standard working solutions and human plasma samples was evaluated following the experimental plan reported in Table5.The concentrations of each analyte at t=0h were used as the reference value.The sample’s stability was eval-uated by analysis of variance(ANOVA)at a confidence level of95%. Standard working solutions were stable throughout the duration of a typical sequence of chromatographic analyses(storage in the autosampler for about24h at room temperature)and for at least2 months at4◦C.In fact,no significant variations in concentrations were observed.The results also showed that the total concentration of WAR and WAROHs in human plasma samples was stable after two cycles of thaw–freeze,whereas the unbound plasmatic fraction showed a decrease of about20%after the second thaw–freeze cycle. No significant degradation occurred in the extracted or ultrafil-tered samples throughout the typical sequence of chromatographic analyses(storage in the autosampler for about24h at room tem-perature)and for at least2months of storage at4◦C.Similar results were obtained for both total and unbound concentrations of plasma samples processed after24h storage at room temperature.The results are consistent with data reported in the literature for WAR [19–34].3.4.7.Robustness of the methodA+10%and−10%variation in the volume of H2SO4(0.5M), ethanol,and the mixture of dichloromethane/hexane(1:5,v/v) were tested in the analysis of a plasma sample spiked with 1000ng/mL of WAR and WAROHs to verify the robustness of the。
西北太平洋表层海水中^(239+240)Pu浓度及^(240)Pu^(239)Pu同位素比

Vol. 55,No. 4Apr 2021第55卷第4期2021年4月原子能科学技术AtomicEnergyScienceandTechnology西北太平洋表层海水中239+240 Pu 浓度及240P u /239P u 同位素比李思璇1黄雯娜2许宏宀,郭秋菊1"1.北京大学物理学院核物理与核技术国家重点实验室,北京100871&2.浙江省辐射环境监测站,浙江杭州310012)摘要:钚是与核工业密切相关的敏感元素,是来源于人类核活动、以痕量或超痕量水平存在于环境中 的重要锕系元素%与陆地土壤中钚的环境行为不同,输入到海洋环境中的钚会随洋流路径进行远距 离迁移扩散%因此,对于包括我国近海在内的西北太平洋区域海水,除受全球沉降影响外,还长期受 到美国太平洋核试验场(PPG)所造成区域污染的显著影响%本文利用从相关报道中收集的数据,对西北太平洋表层海水中钚浓度及同位素比的分布特征进行了分析%结果表明,000年至今,西北太平洋表层海水中239D240 Pu 浓度和240 Pu/239 Pu 同位素比分别在1. 15〜22. 3 mBq/m 3和0. 184〜0. 31间 变化,其中,39D240 Pu 浓度分布与西北太平洋各区域的环境条件等密切相关,而240 Pu/239 Pu 分布则相对 均匀,后者在除中国南海以外的西北太平洋地区均值为0.247土 0.025("),据此估算得美国太平洋核试验场区域污染输入的钚对该海域表层海水中钚的贡献约占其总活度的45% %此外,本文还对福岛核电站附近海域中核事故前后钚的相关数据进行了分析对比,未观察到该事故对西北太平洋海域中 钚分布的影响%关键词:西北太平洋;表层海水;钚;PPG 中图分类号:TL75. 1文献标志码:A 文章编号"000-6931(2021)04-0751-10doi 10 7538/yzk2020 youxian 0082239+240Pu Concentration and 240Pu/239Pu Isotopic Ratio in Surface Seawater of Northwest PacificLI Sixuan 1 , HUANG Wenna 2 , XU Hon g 2'* , GUO Qiuju *(1. State Key Labrratrry of Nuclear Physics and Technology , School of Physics ,Peking University , Beijing 100871 , China &2. Radiation Monitoring Station of Zhejiang Province , Hangzhou 310012 , China )Abstract: Plutonium is a sensitive element closely related to nuclear industry. It is an.mportantact.n.deelementor.g.natedfrom human nuclearact.v.t.esandex.sts.ntheenv.ronmentattraceorultratracelevels.D.f erentfromthebehav.orofpluton.um.n theterrestr.alenv.ronment !pluton.um entered.ntothe mar.neenv.ronmentcould be收稿日期:2020-02-10;修回日期:2020-05-31基金项目:国家自然科学基金资助项目(11775009)作者简介:李思璇(1994-),女,浙江宁波人,博士研究生,辐射防护与环境放射性专业% 通信作者:许 宏,E-mail : 705793140@752原子能科学技术第55卷driven by ocean currents a nd then transported for a long distance.In addition to the global subsidence,the sea water in the Northwest Pacific region,including China,s coastal waters,is significantly affected by the regional pollution caused by the US Pacific Proving Ground(PPG)for a long time.With data assembled from published articles, the distributions of Pu concentration and isotopic ratios in surface seawater of Northwest Pacific were analyzed in this work.After2000,Z39+Z40Pu concentration and Z40Pu/Z39Pu isotopic ratio in surface seawater of this region vary in 1.15-22.3mBq/m<and0.1840.31,respectively.The spatial distribution of239+240Pu concentration is closely relatedto local environmental conditions,while that of Z40Pu/Z39Pu isotopic ratio is rather more homogeneous.Despite the China South Sea,the mean value of Z40Pu/Z39Pu isotopic ratio for Northwest Pacific Ocean is0.247+0.025(1#).According to that,the contribution of Pu from PPG is estimated to be around45%in surface seawater.In addition,Pu data in marine environment off Japan Fukushima Dai-chi Nuclear Power Plant were also collected and compared,among which no indication of Pu released from the accident could beobserved.Key words:Northwest Pacific;surface seawater;plutonium;PPG钚(Pu)的原子序数为94,是与核工业等人类核活动密切相关的敏感锕系元素,其存在于环境中的重要同位素包括Z38Pu(T1/2=87.7a)+ Z39Pu(T1/z=24110a)J40P u(T1/2=6563a)和241Pu(3i/(=14.1a),其中以239Pu和240Pu的环境浓度最大由于钚兼具放射性和化学毒性,且除(41Pu外均为长寿命的%放射性核素,释放到环境中后可长期存在,因此,在核设施安全运行、乏燃料及放射性废物后处理和处置等领域,钚在各类环境介质中的分布及行为特性等都备受关注%目前环境中以痕量或超痕量水平存在的钚的主要来源为1945—1980年间有核国家开展的543次大气层核试验(总当量约440Mt)⑵%此外核工业、核事故以及核废料处置等对环境中钚的含量也有较重要贡献。
塑料测试方法中英文对比表
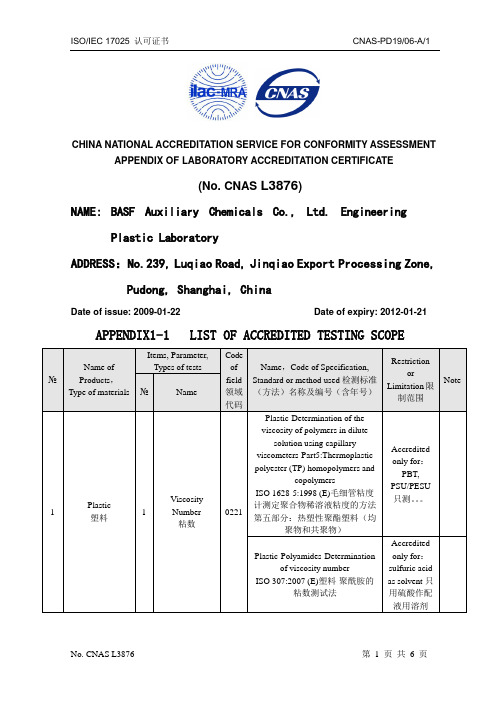
CHINA NATIONAL ACCREDITATION SERVICE FOR CONFORMITY ASSESSMENT APPENDIX OF LABORATORY ACCREDITATION CERTIFICATE(No. CNAS L3876)NAME:BASF Auxiliary Chemicals Co., Ltd. EngineeringPlastic LaboratoryADDRESS:No.239, Luqiao Road, Jinqiao Export Processing Zone,Pudong, Shanghai, ChinaDate of issue: 2009-01-22 Date of expiry: 2012-01-21 APPENDIX1-1 LIST OF ACCREDITED TESTING SCOPE№Name ofProducts,Type of materialsItems, Parameter,Types of testsCodeoffield领域代码Name,Code of Specification,Standard or method used检测标准(方法)名称及编号(含年号)RestrictionorLimitation限制范围Note №Name1 Plastic塑料1ViscosityNumber粘数0221Plastic-Determination of theviscosity of polymers in dilutesolution using capillaryviscometers-Part5:Thermoplasticpolyester (TP) homopolymers andcopolymersISO 1628-5:1998 (E)毛细管粘度计测定聚合物稀溶液粘度的方法第五部分:热塑性聚酯塑料(均聚物和共聚物)Accreditedonly for:PBT,PSU/PESU只测。
【放射化学系列】铀、镎、钚的放射化学

~.
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .... . . . . . . . . . . . . . . . . . . ...~i~ .
~."
1.
11.
Iii.
.... ... .. .. ... ... .... . ... .. . ... .. .. ..". . .. .. . . .. . . . . . . ..iv
Ratiochemical Determination of Plutonium in Marine Samples by Extraction.Chrmatography:.............”.44 The Determination of Plutonim in EnVir~ntal Samples by Extraction with Tridodecylamine ................46 .
8.
Determination Ikanlum in NaturalMaters of
Afte=Aaion-Exchange Sepa.ration. ..........................26 Uranium kalyais by Liquid Scfnttl18tlon Counting .........28
VI.
~
Introduction.................................. ....” . .‘h ...........28 “’. \
Discussionoft heProcedures. ..................................38 Procedures:
液体闪烁计数法测定天然铀样品中的铀

液体闪烁计数法测定天然铀样品中的铀王朝阳;陈忠恭;李绪平【摘要】建立了液体闪烁计数法测定天然铀样品中铀含量的分析方法.对影响液闪测量的主要因素,如α/β甄别、淬灭效率等进行了研究,确定了最佳脉冲甄别参数值和淬灭校正曲线.与铀的容量法进行了方法比对,结果吻合较好.方法的精密度良好,对浓度为0.985 7 mg/g的铀标准溶液平行测定7次,相对标准偏差为1.9%.该法易于操作、分析速度快,可用于核燃料循环前段天然铀样品中铀的分析测定.【期刊名称】《核化学与放射化学》【年(卷),期】2013(035)002【总页数】4页(P112-115)【关键词】液体闪烁计数法;天然铀;测定【作者】王朝阳;陈忠恭;李绪平【作者单位】中核四○四有限公司第二分公司,甘肃兰州 732850;中核四○四有限公司第二分公司,甘肃兰州 732850;中核四○四有限公司第二分公司,甘肃兰州732850【正文语种】中文【中图分类】TL271液体闪烁测量(简称液闪)技术是20世纪50年代初期发展起来的一种测量低能β射线的行之有效的方法,在α核素测定方面, 由于β核素对被测α核素的干扰,使其应用受到了限制。
随着20世纪80年代后期带有脉冲形状甄别功能的液闪谱议的问世,α核素的液闪测定技术得到了快速发展[1]。
在α核素的液闪测量过程中,该法避免了α能谱法中最棘手的样品几何条件与标准源不一致和测量源自吸收的问题,并可实现4π立体角测量,大幅提高了探测效率和测量准确度,对α粒子的探测效率接近100%。
目前,关于用液闪技术测定α核素的报道大多针对超铀核素238Pu、239Pu、240Pu、241Am、237Np等[1-6],对于铀的液闪分析技术研究报道相对较少[7-13],而且这些关于铀的报道各有侧重,有的侧重于对234U/238U、235U/238U 原子比值测定[8-9],有的侧重于对铀的绝对测量[11],还有的侧重于样品的预处理[7,10,12-13]。
湖北省政府采购合同预付款比例规定法律依据

湖北省政府采购合同预付款比例规定法律依据The legal basis for the determination of prepayment ratio in government procurement contracts in Hubei Province.In Hubei Province, the legal basis for the determination of the prepayment ratio in government procurement contracts can be found in various laws, regulations, and policies. These include the Government Procurement Law of thePeople's Republic of China, the Hubei Province Government Procurement Regulations, and other relevant administrative regulations and notices.Under these legal instruments, the prepayment ratio for government procurement contracts is usually determined based on factors such as contract value, project nature, and supplier creditworthiness. The purpose behind establishing a prepayment ratio is to strike a balance between protecting both parties' interests and ensuring efficient completion of government projects.Although specific provisions might vary depending on the type and value of the procurement contract, common principles apply. For instance, for small-scale projects with lower contract values, a higher prepayment ratio may be allowed to facilitate smooth project execution and alleviate financial burdens on suppliers. Conversely,larger projects with higher contract values might have lower prepayment ratios to minimize potential risks associated with large upfront payments.Furthermore, guidelines issued by relevant departments or authorities at both the national and provincial levels can also provide additional clarification or specific requirements regarding prepayment ratios in government procurement contracts. These guidelines aim to enhance transparency and fairness in government procurement processes while promoting responsible financial management.It should be noted that the determination of prepayment ratios is subject to periodic revisions based on changing circumstances or adjustments to relevant policies. Therefore, it is essential for both suppliers and procuringentities to stay informed about any updates or amendments related to this matter.Overall, when it comes to determining the prepayment ratioin government procurement contracts in Hubei Province, legislative acts such as laws, regulations, and policies serve as vital reference points. These instruments outline key principles that help maintain a fair and balanced approach while supporting efficient project implementation across various sectors.我的问题是:湖北省政府采购合同预付款比例规定法律依据在湖北省,关于政府采购合同预付款比例的法律依据可以在各种法律、法规和政策中找到。
pu239截面数据

ENDF/B-VII.1 PU-239 UR fission cross section
10
1
Inf. Dil. 100 b 1b
Cross section (barns)
10
0
10
-2
Energy (MeV)
ENDF/B-VII.1 PU-239 UR capture cross section
ENDF/B-VII.1 PU-239 resonance total cross section
10
3
total
Cross section (barns)
10
2
101
10
-4
10
-3
Energy (MeV)
ENDF/B-VII.1 PU-239 resonance total cross section
(n,n*26) (n,n*27) (n,n*28) (n,n*29) (n,n*30)
16
18
20
Energy (MeV)
ENDF/B-VII.1 PU-239 Inelastic levels
16 *10-3 14
Cross section (barns)
12 10 8 6 4 2 0 2 4 6 8 10 12 14
(n,n*31) (n,n*32) (n,n*33) (n,n*34) (n,n*35)
16
18
20
Energy (MeV)
ENDF/B-VII.1 PU-239 Inelastic levels
4.0 *10-3 3.5
Cross section (barns)
3.0 2.5 2.0 1.5 1.0 0.5 0.0 2 4 6 8 10 12 14