高通量基因组测序中 测序深度,覆盖度
二代测序质控各参数标准

二代测序质控各参数标准一、引言二代测序(Next-GenerationSequencing,NGS)是一种高通量的基因组测序技术,广泛应用于生物医学研究、农业育种、疾病诊断等领域。
在二代测序过程中,质量控制(QualityControl,QC)是至关重要的一步,其中质控参数的设定和标准是关键。
本文将介绍二代测序质控各参数的标准。
二、样本质量评估1.完整性:样本应保持完整,无断裂或降解。
可通过测定样本的分子量、片段长度分布等指标进行评估。
2.浓度:样本浓度应在合理范围内,过高或过低的浓度都可能导致测序质量下降。
3.特异性:样本应具有特异性,不应包含其他杂质序列。
可通过序列特异性指数(Sequence-SpecificityIndex)进行评估。
三、测序数据质量评估1.序列深度:测序深度是指测得的有效序列数量。
理想情况下,测序深度应覆盖目标区域的每个碱基。
2.覆盖度:覆盖度是指测序序列对目标区域的整体覆盖程度。
理想情况下,应具有广泛的覆盖度,以保证准确性和可信度。
3.质量值分布:测序质量值应在合理范围内,过低或过高的质量值都可能导致错误率升高。
4.碱基错配率:碱基错配率是指非特异性碱基的比例。
应尽可能降低错配率,以保证结果的准确性。
四、质量控制标准1.严格控制样本质量和浓度,确保样本具有特异性。
2.确保测序深度和覆盖度达到预期要求,同时关注质量值和错配率。
3.对数据进行多维度分析,包括序列长度、GC含量、突变位点等,以确保结果的全面性和准确性。
4.根据实验需求和样本特性,制定合适的质控参数标准,并定期评估和调整。
5.建立完善的质控流程和标准,确保实验数据的可靠性和可信度。
五、结论二代测序质控各参数标准的设定和评估是质量控制的关键环节。
通过严格控制样本质量和浓度、确保测序深度和覆盖度、关注质量值和错配率、多维度分析数据等措施,可以提高二代测序的准确性和可信度。
同时,建立完善的质控流程和标准,定期评估和调整质控参数,可以确保实验数据的可靠性和可信度,为后续研究提供有力支持。
高通量基因组测序中测序深度覆盖度

1G=1024M测序深度是指测序得到的总碱基数与待测基因组大小的比值.假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M.测序深度=总数据量20M/基因组大小2M=10X覆盖度是指测序获得的序列占整个基因组的比例.由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap.例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的.核苷酸多态性位点SNP,插入缺失位点InDel,Insertion/Deletion、结构变异位点SV,StructureVariation位点.SBC可以协助客户,通过手段,分析不同间的结构差异,同时完成注释.技术路线提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段0.2~5Kb,加上接头,进行cluster制备Solexa或E-PCRSOLiD,最后利用Paired-EndSolexa或者Mate-PairSOLiD的方法对插入片段进行重测序.图1-1,以SOLiD为例,说明整个实验方案.2、外显子测序也称目标组捕获,是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法.是一种选择基因组的的高效策略,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势.外显子expressedregion是真核生物基因的一部分,它在剪接Splicing后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质.外显子是最后出现在成熟RNA中的基因序列,又称表达序列.既存在于最初的产物中,也存在于成熟的RNA分子中的核苷酸序列.在人类基因中大约有180,000,占人类基因组的1%,约30MB.。
DNA的质量监测通常有两个方法

2)DNA的质量监测通常有两个方法:首先OD260/OD280比值应该在1.8左右(1.7-1.9),否则意味着DNA样品中存在大量的蛋白质或RNA污染。
其次,琼脂糖电泳分析时应主要以超螺旋条带为主。
最多不超过三条带(分别为超螺旋DNA,线性化DNA和环状DNA)。
否则意味质粒DNA的质量不高,应该重新制备。
2.限制性内切酶的活性1)限制性内切酶一般需要低温保存,而且反复的升降温过程对酶活性的损害很明显。
因而为了确保在有效期内的限制性内切酶不会失活,限制性内切酶的日常保存和使用应当很小。
2)建议购买具有保温功能的冻存盒保存限制性内切酶(-20度),而且取用限制性内切酶时,也应该使用具有保温功能的冻存盒,尽量防止酶的温度反复出现大的波动。
3.限制性内切酶的用量1)限制性内切酶的单位定义通常为:在合适的温度下,完全消化1ugDNA底物所需的酶量定义为一个单位。
2)在这个单位定义中,有几个不确定因素:首先是底物,不同的酶单位定义是选择的底物可能不同(常用的几个底物DNA包括:Lambda DNA ,AD2 DNA 和一些质粒DNA);第二个不确定因素是限制性内切酶在底物DNA上的酶切位点的个数。
由于单位定义中要求完全消化,因而底物上某个酶的酶切位点的个数的多少,就直接影响了该酶的单位定义。
3)因而,在进行酶切时,用1ul酶(一般10IU/ul)消化1ugDNA的通常做法是很不科学的,这也导致在实际工作中,大家要进行多次预实验才能确定最合适酶切条件。
4)以前,我推荐了一个在线的双酶切设计软件,double digestion designer, 可以精确地计算酶切时的限制性内切酶的用量。
使用中,能够注意到,用来进行双酶切的两个酶的用量有时竟然相差近20倍(EcoRI + NheI),而且发现,小片段PCR产物(100-500bp)进行酶切时,需要的酶量比质粒DNA酶切时用量多10倍以上。
5)该软件目前可以免费使用,用户名和密码都是test。
高通量名词解释

高通量测序常用名词汇总一代测序技术: 即传统的Sanger 测序法,Sanger 法是根据核苷酸在待定序列模板上 的引物点开始,随机在某一个特定的碱基处终 并且在每个碱基后面进行荧光标 止, 记,产生以A 、T 、C 、G 结束的四组不同长度的一系列核苷酸,每一次序列测定由一套四个单独的反应构 成,每个反应含有所有四种脱氧核苷酸三磷 (dNTP ),并混入限量的一种不同的双脱氧酸核苷三磷酸(ddNTP )。
由于ddNTP 缺乏延伸所需要的 3-0H 基团,使延长的寡聚核苷酸选择性地在 G 、A 、T 或C 处终止,使反应得到一组长几百至几千碱基的链终止产物。
它们具有相比,二代测序技术可以一次对几十万到几百万条核酸分子同时进行序列测定, 从而使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序Deepsequencing )。
NGS 主要的平台有 Roche ( 454 & 454 +) , Illumina ( HiSeq2000/2500、GA IIx 、MiSeq ), ABI SOLiD 等。
是DNA 或RNA 分子上具有遗传信息的特定核苷酸序列 基因通过复制把遗传信息传递给下一代,使后代出现与亲代相似的性状。
DNA : Deoxyribonucleic acid,脱氧核糖核酸,一个脱氧核苷酸分子由三部分组成:含氮碱基、脱氧核糖、磷酸。
脱氧核糖核酸通 3',5'-磷酸二酯键按一定的顺序彼此相连构成长过链,即DNA 链,DNA 链上特定的核苷酸序列包含有生物的遗传信息, 是绝大部分生物遗传信息的载体。
RNA : Ribonucleic ,,核糖核酸,一个核糖核苷酸分子由碱基,核糖和磷酸构成。
Acid核糖核苷酸经磷酯键缩合而成长链状分子称之 为RNA 链。
RNA 是存在于生物细胞以及部分病 毒、类病毒中的遗传信息载体。
不同种类的 RNA 链长不同,行使各式各样的生物功能,如参与蛋白质生物合成的RNA 有信使RNA 、转移RNA 和核糖体 RNA 等。
《宏基因组高通量测序技术应用于感染性疾病病原检测中国专家共识》(2021)要点汇总

《宏基因组高通量测序技术应用于感染性疾病病原检测中国专家共识》(2021)要点感染性疾病一直备受临床关注。
新型冠状病毒肺炎全球大流行,给世界经济和人类健康带来严峻挑战,更让病原的精准快速检测成为关注的热点。
近年来,因物价、政策等方面的灵活性,独立实验室已广泛开展基于高通量测序(也称新一代测序技术,NGS)的病原学检测实验。
NGS技术应用的成功案例,对部分感染性疾病的病原学诊断起到决定性作用。
然而目前,临床对NGS应用的适应证认识不足,送检存在较大的盲目性和随意性;操作过程复杂,缺乏规范;提供检验的主体多元而检测质量良莠不齐;结果解释缺乏可信的标准,误导临床的案例屡见不鲜,限制了该技术的临床应用和普及。
一、共识制定过程二、分类和术语(一)分类宏基因组高通量测序技术(mNGS)通过对临床样本的DNA或RNA 进行鸟枪法测序,可以无偏倚地检测多种病原微生物(包括病毒、细菌、真菌和寄生虫)。
二代和三代测序平台均可用于该项技术。
按从临床样本中提取的核酸类型可以分为宏基因组测序和宏转录组测序。
按照测序模式可以分为单端测序和双端测序。
(二)术语和定义NGS:也称高通量测序,是一种可以同时对数十万到数百万条DNA分子序列进行读取的测序技术。
mNGS:m指宏基因组,也称元基因组,是标本中全部生物(人、微生物)基因组的总称。
诊断宏基因组学:指用于临床诊断目的的宏基因组学。
临床宏基因组学:含义与诊断宏基因组学类似,但应用场景更为广泛。
微生物组:指某一个系统、生态环境或特定区域中全部微生物的总和。
试剂盒基因组:游离脱氧核糖核酸(cfDNA):读长:文库:比对:深度:覆盖率:相对丰度:Q值:标定对照:无模板对照:检出限(LOD):正常无菌部位:三、mNGS技术应用于感染性疾病的适应证(一)临床适应证共识1对常规微生物学检查容易明确病原体的感染,如尿路感染通过尿培养手段,不建议mNGS。
共识2患者表现为发热或发热症候群,病因未明确(符合不明原因发热定义),考虑感染或不除外感染,但规范性经验抗感染治疗无效,考虑应用常规技术检测的同时,或在其基础上,开展mNGS。
测序常用名词解释整理
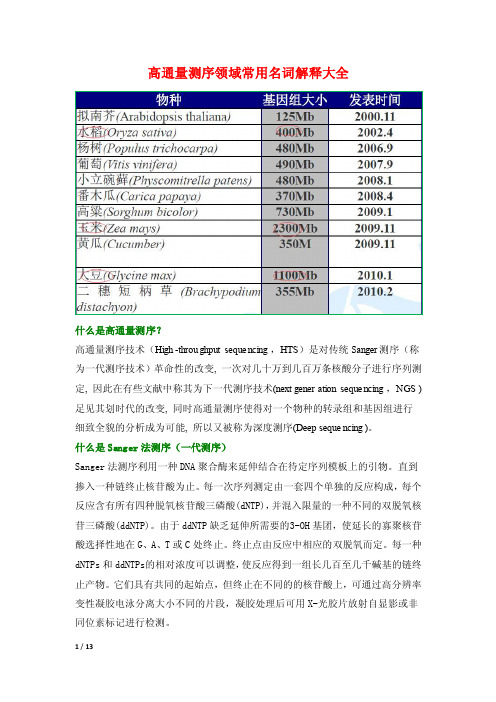
高通量测序领域常用名词解释大全什么是高通量测序?高通量测序技术(High-throug hputsequen cing,HTS)是对传统Sa nger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next genera tionsequen cing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能,所以又被称为深度测序(Deep sequen cing)。
什么是San ger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNT P缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNT Ps和dd NTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequen cing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
高通量测序基础知识
高通量测序基础知识简介陆桂什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变,一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
高通量测序常用名词解释
什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
高通量测序基础知识
高通量测序基础知识简介陆桂什么是高通量测序?高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变,一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。
直到掺入一种链终止核苷酸为止。
每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。
由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。
终止点由反应中相应的双脱氧而定。
每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。
它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。
随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。
通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。
NGS技术在肿瘤个体化治疗中的优势及局限性
NGS技术在肿瘤个体化治疗中的优势及局限性肿瘤个体化治疗是一种基于个体基因组信息的治疗策略,旨在根据不同患者的基因组特征,制定个体化的治疗方案,从而提高治疗效果。
随着高通量测序技术的不断进步,尤其是下一代测序(Next Generation Sequencing,NGS)技术的问世,肿瘤个体化治疗进入了一个新的时代。
本文将重点探讨NGS技术在肿瘤个体化治疗中的优势及局限性。
首先,NGS技术具有高通量的特点,能够快速、准确地测序大量的DNA或RNA序列。
相比传统的测序方法,NGS技术在样本处理、实验时间和经济成本上都有明显的优势。
通过大规模测序,可以同时检测多个基因的突变、融合、复制数变异等信息,为肿瘤个体化治疗提供了更多的生物学信息基础。
其次,NGS技术可以检测多种类型的变异。
除了已知的突变类型,如基因点突变、插入缺失等,NGS技术还可以检测到新的突变类型,如复杂结构变异、基因重排等。
这使得NGS技术在个体化治疗中能够更全面地了解肿瘤的基因组特征,从而更准确地制定治疗方案。
另外,NGS技术还可以通过分析肿瘤中的循环肿瘤DNA (circulating tumor DNA,ctDNA),实现无创的肿瘤监测。
ctDNA是肿瘤细胞释放到血液中的DNA片段,可以通过血液样本进行测序分析。
通过监测ctDNA的变化,可以实时了解肿瘤的进展和治疗效果,并根据变化调整治疗方案,实现个体化的治疗。
然而,NGS技术在肿瘤个体化治疗中也存在一些局限性。
首先,NGS技术虽然能够高通量地测序基因组,但仍然受到测序深度和覆盖度的限制。
特别是对于样本中低频突变的检测,仍然存在一定的假阴性或假阳性的可能性。
因此,在解读NGS结果时需要结合临床病史和其他检测方法,进行综合分析。
其次,NGS技术在分析和解读大规模测序数据时需要复杂的生物信息学分析流程。
研究人员需要具备专业的生物信息学知识和数据处理能力,才能更好地解读和分析NGS结果。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
高通量基因组测序中,什么是测序深度和覆盖度?
1G=1024M
测序深度是指测序得到的总碱基数与待测基因组大小的比值。
假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。
(测序深度=总数据量20M/基因组大小2M=10X)
覆盖度是指测序获得的序列占整个基因组的比例。
由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。
例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。
序的个体,通过序列比对,可以找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel,Insertion/Deletion)、结构变异位点(SV,
技术路线
提取基因组DNA,利用Covaris进行随机打断,电泳回收所需长度的DNA片段(0.2~5Kb),加上接头, 进行cluster制备(Solexa)或E-PCR (SOLiD),最后利用Paired-End(Solexa)或者Mate-Pair(SOLiD)的方法对插入片段进行重测序。
图1-1,以SOLiD为例,说明整个实验方案。
高效策略,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel 等具有较大的优势。
外显子(expressed region)是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质。
外显子是最后出现在成熟RNA中的基因序列,又称表达序列。
既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。
在人类基因中大约有180,000外显子,占人类基因组的1%,约30MB。