03-BLAST(生物信息学国外教程2010版)
生物信息学-BLAST

南方医科大学实验报告姓名学号专业年级基础学院生物信息学教研室题目BLAST 日期实验者实验者一、实验目的一、实验目的1,了解BLAST算法原理算法原理2,掌握BLAST参数设定的意义参数设定的意义3,利用BLAST解决生物学问题,如寻找给定序列(DNA或者蛋白质)的同源序列。
或者蛋白质)的同源序列。
二、实验器材二、实验器材电脑电脑三、方法与步骤三、方法与步骤)或者用自己的序列。
给定人蛋白RBP4(NP_006735)或者用自己的序列。
1 限定物种为人(Homo sapiens ),在参考序列数据库中搜索,列出结果(具体比对不列)。
1)进入BLAST主页主页/Blast.cgi2)限定物种为人Homo sapiens 3)在参考序列数据库中搜索在参考序列数据库中搜索4)CLICK BLAST 2 分别限定物种为Mus musculus ,Rattus norvegicus ,Drosophila melanogaster ,Bos taurus ,Danio rerio,各列出E值最小的两条序列。
值最小的两条序列。
1)选择物种选择物种3)输入序列,选择数据库和BALST程序,点击BLAST 4)E值最小的两个序列值最小的两个序列5)其余物种依次重复其余物种依次重复四、结果与讨论四、结果与讨论限定物种为人(Homo sapiens ),在参考序列数据库中搜索,列出结果(具体比对不列)。
分别限定物种为Mus musculus ,Rattus norvegicus ,Drosophila melanogaster ,Bos taurus ,Danio rerio,各列出E值最小的两条序列。
值最小的两条序列。
Mus musculus Rattus norvegicus Drosophila melanogaster Bos taurus Danio rerio 。
NCBI网站BLAST使用方法介绍完整版

息学方法
BLAST
宿主菌
细胞转化
几周的时间 蛋白质分离纯化及性质测定
Gene family Or
Protein Family
几分钟的时间
Function annotation
BLAST
Web Access
Text
Wang LS, Gao PJ, cellulase,et al.
? RPS BLAST
– searches a database of PSSMs – tool for conserved domain searches
Basic Local Alignment Search Tool
? Widely used similarity search tool
? Heuristic approach based on
ACATGGACCCT ...
Protein Words
Query : GTQITVEDLFYNIATRRKALKN
WGoTrdQsize = 3 (default)
TQI
Word size can only be 2 or 3
Make a lookup table of words
QIT ITV
Basic Local Alignment Search Tool
?Why use sequence similarity? ?BLAST algorithm ?BLAST statistics ?BLAST output ?Examples
Why Do We Need Sequence Similarity Searching?
11-mer
GTACTGGACAT
WORD SIZE
BLAST使用方法

BLAST使用方法一、BLAST的安装和准备工作2.获取待比对的序列文件,可以是FASTA格式的DNA或蛋白质序列。
二、BLAST的常用参数和选项1. Program:指定使用哪种BLAST程序(如BLASTn、BLASTp等)。
2. Database:指定使用哪个数据库进行比对。
3. Query:指定待比对的序列文件。
4. E-value:期望值。
一种描述比对结果误差率的指标,值越小表示结果越可信。
通常情况下,E-value小于0.01被认为是显著结果。
5. Word size:BLAST在比对时使用的核心词的长度。
长度越大表示查全率(sensitivity)越高,但速度会减慢。
6. Gap open:允许在比对过程中插入空位(如插入一个碱基)。
Gap open参数定义了开放一个空位的惩罚分数。
7. Gap extension:允许空位的延伸。
Gap extension参数定义了延伸一个空位的惩罚分数。
三、使用BLAST进行比对1.命令行方式:-打开命令行界面,并定位到BLAST软件的安装目录。
- 输入命令,指定BLAST程序、数据库、查询文件和其他参数。
例如:blastn -db nt -query query.fasta -out output.txt -evalue 0.01-运行命令,BLAST将开始进行比对并生成结果文件。
2.网页方式(以NCBIBLAST为例):- 打开NCBI网站的BLAST页面()。
-选择需要使用的BLAST程序(如BLASTn、BLASTp等)。
-上传待比对的序列文件,或者粘贴序列文本到输入框中。
-选择适当的数据库和其他参数。
-点击“BLAST”按钮,等待比对完成。
四、解读BLAST结果1. E-value:表示在随机比对中获得与查询序列相似度更高的结果的期望概率。
E-value越小表示比对结果越显著。
2. Bitscore:用于表示比对结果的质量。
Bitscore越高表示比对结果越可信。
生物信息学 实验三 数据库搜索-BLAST
实验三数据库搜索—BLAST1. Nucleotide BLAST在Nucleotide中输入登录号搜索人类MAPK9(NM_139069.2)基因,send to 为coding sequences,作为Query 序列,或者下载complete sequences,在Blastn 中限制序列搜索范围为272-1420(编码区)。
分别用megablast, discontiguous megablast 和 blastn 进行搜索。
这三个搜索的参数不同之处,主要体现在单词单位,megablast的单词单位默认为28,可选范围从16-256, discontiguous megablast的单词单位默认为11,可选为11和12, blastn单词单位默认为11,可选范围为7,11和15。
Megablast 可以快速搜索到与query 高度相似的序列;discontiguous megablast用于寻找与 query 高度相似的序列; blastn则用于寻找与 query 有一定相似度的序列。
单词单位越小,敏感度越高,也就是说,Megablast敏感度最差,discontiguous megablast 居中,blastn 最高。
Megablast的搜索速度最快,discontiguous megablast居中,blastn最差。
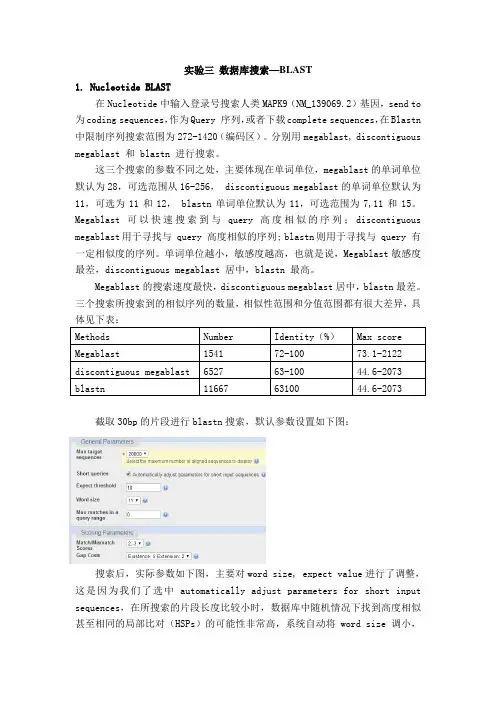
三个搜索所搜索到的相似序列的数量,相似性范围和分值范围都有很大差异,具Methods Number Identity(%)Max score Megablast154172-10073.1-2122 discontiguous megablast652763-10044.6-2073 blastn116676310044.6-2073截取30bp的片段进行blastn搜索,默认参数设置如下图:搜索后,实际参数如下图,主要对word size, expect value进行了调整,这是因为我们了选中automatically adjust parameters for short input sequences,在所搜索的片段长度比较小时,数据库中随机情况下找到高度相似甚至相同的局部比对(HSPs)的可能性非常高,系统自动将 word size 调小,提高敏感度,而将 E-value 调大,确保有搜索结果出现。
04-Multiple sequence alignment(生物信息学国外教程2010版)

Page 179
Multiple sequence alignment: outline
[1] Introduction to MSA Exact methods Progressive (ClustalW) Iterative (MUSCLE) Consistency (ProbCons) Structure-based (Expresso) Conclusions: benchmarking studies [2] Hidden Markov models (HMMs), Pfam and CDD
Multiple sequence alignment: outline
[1] Introduction to MSA Exact methods Progressive (ClustalW) Iterative (MUSCLE) Consistency (ProbCons) Structure-based (Expresso) Conclusions: benchmarking studies [2] Hidden Markov models (HMMs), Pfam and CDD
Page 185
Use ClustalW to do a progressive MSA
http://www.ebi. /clustalw/
Page 186
Feng-Doolittle MSA occurs in 3 stages
[1] Do a set of global pairwise alignments (Needleman and Wunsch’s dynamic programming algorithm) [2] Create a guide tree
This insertion could be due to alternative splicing
Blast软件及常用数据库介绍

blastall:通用检索命令 -p(program name):为需要使用的程序名 blastn:为核酸序列对比搜索程序 -d(database name):指定所使用的数据库 的名称 -i (input file):待搜索的序列文件 -o(output file):指定保存结果的文件
2011-12-22
对比对结果分析比对结果登入ncbi主页点击进入对核酸进行blast点击进入直接输入fasta格式的未知核酸序或者本地上传一个fasta格式的核酸序列文件选择一个合适的数据库进行比对点击运行图形结果匹配序列列表输入的序列在库里比对到的序列genebank库包含了所有已知的核酸序列和蛋白质序列以及与它们相关的文献著作和生物学注释它是由美国国立生物技术信息中心ncbi建立和维护的
12
2011-12-22
blast软件及常用数据库介绍
如何在windows操作系统下安装使用本 操作系统下安装使用本 如何在 软件? 地BLAST软件? 软件
STEP3
执行Blast比对
2011-12-22
blast软件及常用数据库介绍
13
具体步骤 1.将所需比对的序列转化为fasta格式
2.执行比对命令
BLAST软件及常用数据库介绍 软件及常用数据库介绍
制作人:faneds
BLAST的概述:
Blast,全称Basic Local Alignment Search Tool, 即“基于局部比对算法的搜索工具” ,能够实现 比较两段核酸或者蛋白序列之间的同源性的功能, 具有较快的比对速度和较高的比对精度,适用于 多种序列比对的情况,在常规双序列比对分析中 应用最为广泛。
3.对比对结果分析
2011-12-22
blast软件及常用数据库介绍
Blast使用入门
引用次数:36501
引用次数:35799
引用次数:12894
引用次数:4179
移除Query序列中之低复杂度以及有串接重复现象的区域
Query word
将长序列转换成短序列
W=1
KNTMYVIIILTWNLTMTNDMKNHRCHSTTRTLMTNIRKTH
W=4
KNTM YVII ILTW NLTM TNDM KNHR CHST TRTL MTNI RKTH
全局比对 Smith-Waterman算法 局部比对
Fasta算法
Blast算法
建立评分矩阵
Pam250
blosum62
执行比对 (动态规划算法) Needleman-Wunsch Smith-Waterman
确定最佳途径
当面向数据之海的时候,该怎么办?
生物信息学:努力在数据的海洋里畅游
BLAST (Basic Local Alignment Search Tool) is a set of similarity search programs that explore all of the available sequence databases for protein or DNA. BLAST (基本局部相似性比对搜索工具 ) 是一套用来探索可供使用的序列 数据库中所有DNA或者蛋白质的 相似性搜索程序 Local:局部 研究对象:DNA或者蛋白质 搜多对象:数据库
Blast
Query sequence
Database
Subject sequence Subject sequence Subject sequence Subject sequence ……
1. 兼顾搜寻的速度以及搜寻结果的精确度 2. BLAST使用启发式搜索代替动态规划算法来找出相关的序列, 在速度上比完全只使用动态规划大约快上50倍左右
生物信息学-blast
筛选结果
点击开始搜索
其他一些显示格式参数
18
提交任务
返回查询号(request id)
修改完显示格式后点 击进入结果界面
可以修改显示结果格式
19
结果页面(一)
图形示意结果
20
结果页面(二)
目标序列描述部分
带有genbank的链接,点击可以进入 相应的genbank序列
匹配情况,分值,e值
21
结果页面(三)
匹配序列列表
31
分析过程(八)
具体匹配情况
32
单机版的Blast使用(一)
为什么使用单机版的Blast? 1.特殊的数据库要求。 2.涉及序列的隐私与价值。 3.批量处理 4.其他原因??
33
单机版的Blast使用(二)
单机版Blast的基本操作过程 1.下载单机版的Blast程序 ftp:///blast/executables/ 目录下,下载对应的操作系统版本。 2.解压程序包(blast.tar.gz) 命令是: $ tar zxvf blast.tar.gz
5
序列相似性比较和序列同源性分析
序列相似性比较: 就是将待研究序列与DNA或蛋白质序列库进行比较, 用于确定该序列的生物属性,也就是找出与此序列相似 的已知序列是什么。完成这一工作只需要使用两两序列 比较算法。常用的程序包有BLAST、FASTA等;
序列同源性分析: 是将待研究序列加入到一组与之同源,但来自不同物种 的序列中进行多序列同时比较,以确定该序列与其它序 列间的同源性大小。这是理论分析方法中最关键的一步。 完成这一工作必须使用多序列比较算法。常用的程序包 有CLUSTAL等;
6
Blast简介(一)
BLAST种类及使用方法
BLAST种类及使用方法BLAST(Basic Local Alignment Search Tool)是一种广泛使用的序列比对算法,可用于比较DNA,RNA或蛋白质序列的相似性。
它是生物信息学领域中最常用的工具之一,可以帮助研究人员识别新的序列,注释基因功能,鉴定物种间的进化关系等。
1.BLASTN:BLASTN用于比对DNA序列。
它可以将一个查询DNA序列与已知的DNA序列数据库进行比较,找到相似的序列。
BLASTN通常用于物种鉴定、基因组注释和寻找同源基因等方面的研究。
2.BLASTP:BLASTP用于比对蛋白质序列。
它可以将一个查询蛋白质序列与已知的蛋白质数据库进行比较,找到相似的蛋白质序列。
BLASTP 通常用于寻找同源蛋白质,预测蛋白质功能和结构,以及识别蛋白质家族等方面的研究。
3.BLASTX:BLASTX用于比对DNA序列与蛋白质数据库的比对。
它通过将DNA序列翻译成蛋白质序列,然后与已知的蛋白质数据库进行比对,找到相似的蛋白质序列。
BLASTX通常用于从未知的DNA序列中预测蛋白质编码区域,注释基因功能等方面的研究。
4. TBlastN:TBlastN用于比对蛋白质序列与DNA数据库的比对。
与BLASTX相反,TBlastN将已知的蛋白质序列与DNA数据库进行比对,找到相似的DNA序列。
TBlastN通常用于寻找蛋白质在基因组中的编码区域,确定启动子和转录因子结合位点等方面的研究。
5. TBlastX:TBlastX用于比对转录本与转录本数据库的比对。
它可以将一个查询转录本序列与已知的转录本数据库进行比对,找到相似的转录本。
TBlastX通常用于寻找新的转录本和预测基因表达模式等方面的研究。
使用BLAST有以下几个步骤:1.准备查询序列:将待比对的DNA、RNA或蛋白质序列准备成文本文件,确保序列格式正确,并确保序列长度适合比对任务。
2. 选择数据库:根据研究需求,选择适当的数据库。
常用生物信息学软件BLAST
Blast的主程序是blastall。程序的输入文件是query序列(-i 参数)和库文件(-d 参数),比对类型的 选择(-p 参数)和输出文件(-o 参数)由用户指定。其中“-p”参数有 5 种取值: -p blastp:蛋白序列与蛋白库做比对。 -p blastx:核酸序列对蛋白库的比对。 -p blastn:核酸序列对核酸库的比对。 -p tblastn:蛋白序列对核酸库的比对。 -p tblastx:核酸序列对核酸库在蛋白级别的比对。 这些元素就构成了blast的基本运行命令(以blastn为例): blastall -i query.fasta -d database_prefix -o blast.out -p blastn 其中如果"-o"参数缺省,则结果输出方式为屏幕输出。下面以一个blastn比对为例,来说明比对全过程: Query序列(query.fasta): >gi|45593933|gb|AY551259.1| Oryza sativa precursor microRNA 319c gene AGGAAGAGGAGCTCCTTTCGATCCAATTCAGGAGAGGAAGTGGTAGGATGCAGCTGCCGATTCATGGATA CCTCTGGAGTGCATGGCAGCAATGCTGTAGGCCTGCACTTGCATGGGTTTGCATGACCCGGGAGATGAAC CCACCATTGTCTTCCTCTATTGATTGGATTGAAGGGAGCTCCACATCTCT >gi|45593932|gb|AY551258.1| Oryza sativa precursor microRNA 319b gene CATATTCTTTTAATTTGATGGAAGAAGCGATCGATGGATGGAAGAGAGCGTCCTTCAGTCCACTCATGGG CGGTGCTAGGGTCGAATTAGCTGCCGACTCATTCACCCACATGCCAAGCAAGAAACGCTTGAGATAGCGA AGCTTAGCAGATGAGTGAATGAAGCGGGAGGTAACGTTCCGATCTCGCGCCGTCTTTGCTTGGACTGAAG GGTGCTCCCTCCTCCTCGATCTCTTCGATCTAATTAAGCTACCTTGACAT 库文件Database(db.seq,已经运行formatdb -i db.seq -p F -o T建库): >fake_seq AGGAAGAGGAGCTCCTTTCGTTCCAATTCAGGAGAGGAAGTGGTAGGATGCAGCTGCCGATTCATGGATA CCTCTGGAGTGCATGCAGCAATGCTGTAGGCCTGCACTTGCATGGGTTTGCATGACCCGGCGAGATGAAC CCACCATTGTCTTCCTCTATTGATTGGATTGAAGGGAGCTCCACATCTCT 运行命令: blastall -i query.fasta -d db.seq -o blast.out -p blastn 运行结果: BLASTN 2.2.8 [Jan-05-2004] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. Query= gi|45593933|gb|AY551259.1| Oryza sativa precursor microRNA 319c gene, complete sequence
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BLAST search output: top portion
database query
program
taxonomy
page 112
BLAST search output: taxonomy report summarizes species with matches
BLAST search output: graphical output
page 112
BLAST search output: tabular output
High scores low E values
Cut-off: .05? 10-10?
page 113
BLAST search output: alignment output
Outline of today’s lecture
Step 4: optional parameters
You can... • choose the organism to search • turn filtering on/off • change the substitution matrix • change the expect (e) value • change the word size • change the output format
page 109
(c) Query: human insulin NP_000198 Program: blastp Database: C. elegans RefSeq Option: conditional compositional score matrix adjustment
Note that the bit score, Expect value, and percent identity all change with the compositional score matrix adjustment
BLAST
BLAST (Basic Local Alignment Search Tool) allows rapid sequence comparison of a query sequence against a database. The BLAST algorithm is fast, accurate, and web-accessible.
Expect Word size
Scoring matrix
Filter, mask
page 108
Step 4a: optional blastn search parameters
Expect Word size
Match/mismatch scores Filter, mask
page 108
page 109
(e) Query: human insulin NP_000198 Program: blastp Database: C. elegans RefSeq Option: Mask for lookup table only
Filtering (the filtered sequence is the query in lowercase and grayed out)
Then click “BLAST”
page 102
page 103
Step 1: Choose your sequence
Sequence can be input in FASTA format or as accession number
page 103
Example of the FASTA format for a BLAST query
BLAST Practical use Algorithm Strategies Finding distantly related proteins: PSI-BLAST Hidden Markov models BLAST-like tools for genomic DNA PatternHunter Megablast BLAT, BLASTZ
page 109
(d) Query: human insulin NP_000198 Program: blastp Database: C. elegans RefSeq Option: Filter low complexity regions
Note that the bit score, Expect value, and percent identity all change with the filter option
page 104
Choose the BLAST program Program Input
1 blastn blastp blastx tblastn DNA DNA
Database 1
protein 6 DNA
protein protein
6
protein 36 DNA
tblastx
DNA
DNA
Fig. 2.9 page 32
Step 2: Choose the BLAST program
page 104
Step 2: Choose the BLAST program
blastn (nucleotide BLAST) blastp (protein BLAST) blastx (translated BLAST) tblastn (translated BLAST) tblastx (translated BLAST)
Copyright notice
Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by Jonathan PevsnerCopyright © 2009 by John Wiley & Sons, Inc. These images and materials may not be used without permission from the publisher. We welcome instructors to use these powerpoints for educational purposes, but please acknowledge the source. The book has a homepage at including hyperlinks to the book chapters.
BLAST: background on sequence alignment
Outline of today’s lecture
BLAST Practical use Algorithm Strategies Finding distantly related proteins: PSI-BLAST Hidden Markov models BLAST-like tools for genomic DNA PatternHunter Megablast BLAT, BLASTZ
protein databases
nucleotide databases
page 106
Step 4a: Select optional search parameters
organism Entrez! algorithm
page 107
Step 4a: optional blastp search parameters
page 102
Four components to a BLAST search
(1) Choose the sequence (query) (2) Select the BLAST program (3) Choose the database to search
(4) Choose optional parameters
page 109
(e) Query: human insulin NP_000198 Program: blastp Database: C. elegans RefSeq Option: Mask for lookup table only
Note that the bit score, Expect value, and percent identity could change with the “mask for lookup table only” option
page 106
(a) Query: human insulin NP_000198 Program: blastp Database: C. elegans RefSeq Default settings: Unfiltered (“composition-based statistics”)
Our starting point: search human insulin against worm RefSeq proteins by blastp using default parameters
November 29, 2010
BLAST:
Basic local alignment search tool
BL A S T !
Jonathan Pevsner, Ph.D. Bioinformatics pevsner@ Johns Hopkins School of Medicine
5’ GTG GGT 5’ TGG GTA 5’ GGG TAG
page 105
Step 3: choose the database
nr = non-redundant (most general database) dbest = database of expressed sequence tags dbsts = database of sequence tag sites gss = genomic survey sequences