D-A 反应
第七章固相反应

第七章固相反应第七章固相反应固相反应在固体材料的⾼温过程中是⼀个普遍的物理化学现象,⼴义地讲,凡是有固相参与的化学反应都可称为固相反应。
例如固体的热分解、氧化以及固体与固体、固体与液体之间的化学反应等都属于固相反应范畴之内。
但从狭义上,固相反应常指固体与固体间发⽣化学反应⽣成新的固体产物的过程。
Tammann 等很早就研究了CaO 、MgO 、PbO 、CuO 和WO 3的反应,他们分别让两种氧化物的晶⾯彼此接触并加热,发现在接触⾯上⽣成着⾊的钨酸盐化合物,其厚度x 与反应时间t 的关系为C t K x +=ln ,确认了固态物质间可以直接进⾏反应。
因此Tammann 等提出:(1) 固态物质间的反应是直接进⾏的,⽓相或液相没有或不起重要作⽤;(2)固相反应开始温度远低于反应物的熔融温度或系统的低共熔温度,通常相当于⼀种反应物开始呈现显著扩散作⽤的温度,这个温度称为泰曼温度或烧结温度。
对于不同物质的泰曼温度与其熔点(m T )间存在⼀定的关系。
例如,对于⾦属为0.3~0.4m T ;盐类和硅酸盐则分别为0.57m T 和0.8~0.9m T 。
(3)当反应物之⼀存在有多晶转变时,则此转变温度也往往是反应开始变得显著的温度,这⼀规律称为海德华定律。
Tammann 等⼈的观点长期为化学界所接受,但随着⽣产和科学实验的发展,发现许多固相反应的实际速度⽐Tammann 理论计算的结果快得多,⽽且有些反应(例如MoO 3和CaCO 3的反应)即使反应物不直接接触也仍能较强烈地进⾏。
因此,⾦斯特林格等⼈提出,在固相反应中,反应物可转为⽓相或液相,然后通过颗粒外部扩散到另⼀固相的⾮接触表⾯上进⾏反应,表明⽓相或液相也可能对固相反应过程起重要作⽤。
显然这种作⽤取决于反应物的挥发性和体系的低共熔温度。
图7-1描述了物质A 和B 进⾏化学反应⽣成C 的⼀种反应历程:反应⼀开始是反应物颗粒之间的混合接触,并在表⾯发⽣化学反应形成细薄且含⼤量结构缺陷的新相,随后发⽣产物新相的结构调整和晶体⽣长。
化学反应工程 第二版课后习题

第一章习题1化学反应式与化学计量方程有何异同?化学反应式中计量系数与化学计量方程中的计量系数有何关系? ?水溶液流量为10m 3hr -1。
9 反应O 2H N 2NO 2H 222+→+,在恒容下用等摩尔H 2,NO 进行实验,测得以下数据总压/MPa0.0272 0.0326 0.0381 0.0435 0.0543 半衰期/s 265 186 135 104 67求此反应的级数。
10 考虑反应3P A →,其动力学方程为Vn k t n V r A A A d d 1=⋅-=-试推导在恒容下以总压表示的动力学方程。
11 A 和B 在水溶液中进行反应,在25℃下测得下列数据,试确定该反应反2A →R+SA 组分分压与时间关系见下表:t /sec 0 20 40 60 80 100 120 140 160 p A /MPa 0.1 0.096 0.080 0.056 0.032 0.018 0.008 0.004 0.002 试求在100℃,0.1MPa 下,进口物流中包含20%惰性物,A 组份流量为100mol·hr -1,达到95%转化率所需的平推流反应器的体积。
17间歇操作的液相反应A→R,反应速率测定结果列于下表。
欲使反应物浓度由c A0=1.3kmol·m-3降到0.3 kmol·m-3需多少时间?c A/kmol·m-30.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 1.0 1.3 2.0(−r A)/kmol·m-3min-0.1 0.3 0.5 0.6 0.5 0.25 0.10 0.06 0.05 0.045 0.0421c A0=c B0=5mol·m3,动力学方程式为−r A=kc A c B,其中k=100m3kmol-1min-1。
求:(1)反应在平推流反应器中进行时出口转化率为多少?(2)欲用全混流反应器得到相同的出口转化率,反应器体积应多大?(3)若全混流反应器体积V R=0.001m3,可达到的转化率为多少?已知k=1m3kmol-1hr-1,c B0=3kmol·m-3,c A饱和=0.02kmol·m-3,水溶液流量为10m3hr-1。
无机化学常用公式小全

无机化学常用公式小全1:理想气体状态方程pv=nrt(该公式的各类变体参见第四章)2:实际气体状态方程。
⎢p实+a⎢⎢(v实-nb)=nrt⎢v⎢⎢⎢⎢⎢3:混合气体的分压定律p总=∑pipi=p总gxi=p总4:graham气体蔓延定律=u(b)b(b)=n(b)n(b)或m(b)=m(a)m(a)n(b)m(b)w(b)=vm,x(剂)=n(剂)=n液n质+n剂n(剂)5:质量摩尔浓度,物质的量浓度,质量浓度和摩尔分数则表示方法。
c(b)=n液n质+n剂n(质)6:raoult定律n质+n剂n(质)n剂n(质)p=p*g(x剂)∆p=p*g(x质)=p*k’b=kb(k=p*k’)7:沸点升高公式∆tb=k’∆p=kbb∆tf=k’∆p=kfb8:溶液渗透压公式∏=crt∏v=nrt常用物理量一览表(无论x是什么量)1:∆x=x(终)-x始2:w=-p外g∆v3:(热力学第一定律)∆u=q+w4:恒容反应热(1)∆u=qv(2)qv=-cg∆t(3)qv1∆t1⇒qv1=qv21(推论1)∆t2∆t2m1q1mq=22(推论2)m2∆t1m2∆t25:恒压反应热(1)h=u+pvqp=∆u-w=∆u+p外∆v=(u2-u1)+(p2v2-p1v1)=(u2+p2v2)-(u1+p1v1)=∆h6:qp与qv的关系(1)qp=qv+∆nrt(2)∆rhm=(3)∆rhm=∆rum+∆νrt(备注:左右两边的单位为j·mol-1)7:标准分解成冷的应用领域∆rhm=∑vi∆fh(生成物)-v∆h(∑mifm反应物)θθθ∆rhm=∑vi∆ch(m反应物)-∑vi∆ch(m生成物)(注意反应物与生成物的前后顺序。
)s=klnω(k=1.38⨯10-23jk-1)(注:该公式只在恒温可逆过程中成立)tθθθ∆rsm=∑vis(m生成物)-∑vis(m反应物)10:吉布斯自由能∆ru=q+w体+w非⇒q=∆ru+w体+w非=∆ru+(-p∆v)+w非=∆rh-w非qr=t∆s≥∆rh-w非-(∆h-t∆s)≥-w非-⎢⎢(h2-h1)-(t2s2-t1s1)⎢⎢≥-w非-⎢⎢(h2-t2s2)-(h1-t1s1)⎢⎢≥-w非-[g2-g1]=-∆g≥-w非(g=h-ts)11:标准生成吉布斯自由能∆rgm=∑vi∆fg(生成物)-v∆g(∑ifm反应物)m∆rgm=∆rhm-t∆rsm1:反应平均速率v(a)=2:反应瞬时速率c(a)2-c(a)1t2-t1∆c(a)⎢∆c(a)⎢dc(a)v(a)=lim-⎢=∆t→0∆tdt⎢⎢aa+bb=cc+ddv=k⎢⎢c(a)⎢⎢⎢⎢c(b)⎢⎢(注:k是反应速率常数,m,n称为a,b浓度的幂指数,不一定等于反应系数,三者皆可以通过实验测得。
D-A反应
H COOMe
H C
COOMe
+ C
MeOOC H
H H COOMe + COOMe
COOMe HH COOMe
COOMe H
COOMe
H
A
10
2.内向加成原理 优先生成内向加成产物
O H
O
O
H
O H
O
H O
H H
O OO
内向
O O
HO H
外向
A
11
键迁移重排
邻近共轭体系的一个原子或基团的键迁移至新 的位置,同时共轭体系发生转移,这种分子内非 催化的异构化协同反应称为键迁移重排。
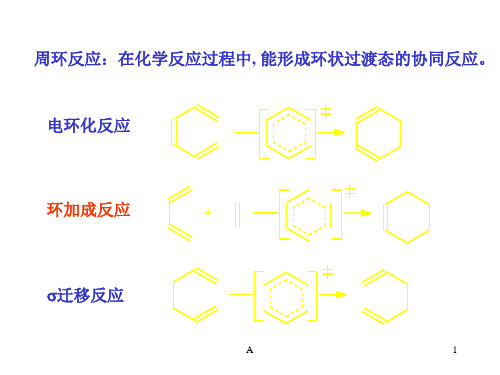
周环反应:在化学反应过程中, 能形成环状过渡态的协同反应。 电环化反应
环加成反应
+
迁移反应
A
1
环加成反应(Cycloaddition Reactions)
在光或热作用下,两个电子共轭体系的两端同时生成键而形成环状化 合物的反应。
hv +
[ 2+2 ] 环加成
+
[ 4+2 ] 环加成
括号中的数字表示两个体系中参与反应的电子数。
i C 1 C 2 C 3 C 4 C 5 [3 ,3]迁 移C 1 C 2 C 3 C 4 C 5
j
CCCCC
1 ' 2 ' 3 ' 4 ' 5 '
CCCCC
1 ' 2 ' 3 ' 4 ' 5 '
2
1
3
1'
3'
2'
2
1
3 [3,3]迁 移
化学反应速率4个公式

化学反应速率4个公式1. 平均反应速率(Average Reaction Rate)平均反应速率是指反应物质浓度在一段时间内的平均变化率。
对于一般的反应A+B→C+D,平均反应速率可以表示为:v=Δ[A]/Δt=-Δ[B]/Δt=Δ[C]/Δt=Δ[D]/Δt其中v表示平均反应速率,Δ[A]、Δ[B]、Δ[C]、Δ[D]分别表示反应物A、B和生成物C、D的浓度变化量,Δt表示时间间隔。
该公式表示反应物物质浓度的变化量与时间的比值。
2. 瞬时反应速率(Instantaneous Reaction Rate)瞬时反应速率是指在其中一特定时刻的反应速率。
由于反应速率在反应过程中可能会发生变化,因此瞬时反应速率需要通过微分来进行计算。
对于一般的反应A+B→C+D,瞬时反应速率可以表示为:v = -d[A]/dt = -d[B]/dt = d[C]/dt = d[D]/dt其中v表示瞬时反应速率,d[A]/dt、d[B]/dt、d[C]/dt、d[D]/dt 分别表示反应物A、B和生成物C、D的浓度随时间变化的微分。
该公式表示反应物物质浓度的变化率。
3. 反应速率定律(Rate Law)反应速率定律是描述反应速率与反应物浓度的关系的数学公式。
对于一般的反应A+B→C+D,反应速率定律可以表示为:v=k[A]^m[B]^n其中v表示反应速率,k为反应速率常数,[A]、[B]分别表示反应物A和B的浓度,m和n为反应物浓度的阶数,可以根据实验结果来确定。
4. Arrhenius公式(Arrhenius Equation)Arrhenius公式是描述反应速率与温度的关系的数学公式,可用于计算反应速率常数。
Arrhenius公式可以表示为:k=Ae^(-Ea/RT)其中k为反应速率常数,A为预指数因子,Ea为活化能,R为气体常数,T为反应温度。
该公式表示反应速率常数与温度的关系。
通过测定不同温度下的反应速率常数,可以确定活化能。
平衡常数计算公式

平衡常数计算公式平衡常数(Ka)是指在给定条件下,化学反应达到平衡时,反应物和生成物之间浓度的相对关系。
平衡常数的计算公式可以使用两种方法:浓度法和活度法。
一、浓度法1.对于一般的平衡反应:aA+bB⇌cC+dD平衡常数Ka的计算公式为:Ka=[C]c[D]d/[A]a[B]b其中,[A]、[B]、[C]和[D]分别表示反应物A、B和生成物C、D的摩尔浓度。
2.对于涉及气体的平衡反应:aA+bB⇌cC+dD+eE平衡常数Ka的计算公式为:Ka=(PC)c(PD)d(PE)e/(PA)a(PB)b其中,PA、PB、PC、PD和PE分别表示气体反应物A、B和生成物C、D、E的分压。
3.对于涉及溶液的平衡反应:aA+bB⇌cC+dD平衡常数Ka的计算公式为:Ka=[C]c[D]d/[A]a[B]b[H2O]w其中,[H2O]表示反应体系中水的摩尔浓度或活度。
二、活度法活度是一种标量,表示溶液中溶质的有效浓度。
它可以用来描述溶液中分子之间的相互作用。
活度系数(γ)是活度与摩尔浓度之间的比值。
通常情况下,Ka的计算公式可以表示为:K a=γCγD/γAγB其中,γA、γB、γC和γD分别表示溶质A、B和溶质C、D的活度系数。
活度系数的计算涉及理想化和非理想化的溶液行为模型,如Debye-Hückel理论、van Laar方程或Flory-Huggins理论。
这些模型是根据溶质和溶剂之间相互作用的种类和强度来建立的。
总结:平衡常数的计算公式可以使用浓度法或活度法。
浓度法适用于任何类型的反应,包括涉及气体或溶液的反应。
活度法则更精确,适用于非理想溶液的情况。
具体计算中,需要确定参与反应的物质的浓度或活度,并根据反应方程式中的摩尔比例关系,计算各个物质的浓度或活度。
脱氮气的d-a反应

脱氮气的d-a反应
脱氮气的d-a反应可以应用于有机合成中,用于构建含氮杂环
化合物或者其他有机分子。
这种反应通常需要催化剂的存在,以促
进反应的进行。
在实验室条件下,通常会选用适当的试剂和溶剂来
促进这种反应的进行。
从机理上来看,脱氮气的d-a反应可以被描述为亲电加成反应,其中双键上的氮原子作为亲电子试剂与另一个分子中的双键发生反应。
这种反应通常会形成新的碳-碳或碳-氮键,并且会释放出氮气
分子。
总的来说,脱氮气的d-a反应在有机合成中具有重要的应用,
可以用于构建复杂的有机分子结构,同时也为化学研究提供了重要
的反应途径。
在实际应用中,需要考虑反应条件、催化剂选择、试
剂选择等因素,以实现高效、高选择性的反应过程。
生化知识点

1>.美国的著名大学(哈佛、麻省、斯坦佛、普林斯顿等)文理皆必修生化。
<2>.人体基因工程计划:上个世纪的三个计划:曼哈顿、阿波罗、人体基因工程:人类23对染色体(23对DNA分子)测序,几十万个基因,大肠杆菌8000个基因,基因改造(治病),WATSON和CRICK开玩笑,女儿赛过爱因斯坦和玛丽莲梦露,儿子聪明高飞低潜力大无穷的超人。
<3>.诺贝尔奖金(90多万美元,最高荣誉)的分布:化学,医学生理学领域不说独占鳌头也是多抢多占,如蛋白质的螺旋和折迭(化学)、G蛋白、第二信使学说的三代科学家三次获奖,光合作用机制,更不要说核酸领域了(复制、转录、逆转录、RNA复制等中心法则中的内容)不想获得诺贝尔奖的科学家不是好科学家。
今天的考研是为了明天的诺贝尔奖!对映体一个不对称碳原子的取代基在空间里的两种取向是物体与镜像的关系,并且两者不能重叠。
这两种旋光异构体称为对映体。
两个对映体具有程度相同但方向相反的旋光性(D+与L-;D-与L+)和不同的生物活性,其他物理和化学性质完全相同。
含n个C*的化合物,其旋光异构体的数目是2n, 组成2n /2对对映体。
任一旋光化合物都只有一个对映体,它的其他旋光异构体在理、化性质都与之不同,不是对映体的旋光异构体称非对映体。
仅一个手性碳构型不同的非对映体称差向异构体(有几种情况)。
异头物单糖由直链结构变成环状结构后,羰基碳成为新的手性碳(异头碳),导致C1差向异构化,产生两个非对映体,称之。
α、β异头物判断:有2种方式。
见P9-10。
多不饱和脂肪酸家族:分为ω-3和ω-6系列(指离羧基最远的双键到甲基末端3个碳和6个碳)。
如亚油酸和γ-亚麻酸为ω-6系列,而α-亚麻酸为ω-3系列。
人体内二者不能互转!且二者对血脂的影响不同皂化值:皂化1g油脂所需的KOHmg数;碘值:100g油脂卤化时所吸收的碘的克数;乙酰值:中和1g乙酰化物所释放的乙酸所需要的KOHmg数;酸值:中和1g油脂中的游离脂肪酸所需的KOHmg数氨基酸洗脱顺序的判定:氨基酸与树脂的亲合力主要决定于它们之间的静电吸引,其次是氨基酸侧链与树脂聚苯乙烯之间的疏水相互作用;亲和力愈大愈难洗脱!肽的理化性质:①肽键的酰氨氢不解离,肽的酸碱性质主要决定于肽键中的游离末端α-NH2、α-COOH及侧链R基上的可解离基团;②肽中末端α-羧基的pKa值比游离氨基酸的大,末端α-氨基的pKa值比游离氨基酸的小;③游离的α-氨基、α-羧基和R基可发生与氨基酸中相应的类似反应,如茚三酮反应等;④蛋白质部分水解后所得的肽若不发生消旋,则有旋光性,短肽的旋光度约等于组成氨基酸的旋光度之和,较长的肽的旋光度则不是简单加和α角蛋白经充分伸展后可转变成β角蛋白,即β折叠片结构。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
狄尔斯–阿尔德反应[编辑]
维基百科,自由的百科全书
(重定向自狄尔斯-阿德尔反应)
跳转至:导航、搜索
RXNO:0000006
(英语:)又叫Diels–Alder反应、双烯加成反应,其中狄尔斯又译作第尔斯–阿尔德又译作阿德尔、阿德耳。
狄尔斯–阿尔德反应是一种有机反应(具体而言是一种环加成反应),共轭双烯与取代烯烃(一般称为亲双烯体)反应生成取代环己烯[1][2][3]。
即使新形成的环之中的一些原子不是碳原子,这个反应也可以继续进行。
一些狄尔斯–阿尔德反应是可逆的,这样的环分解反应叫做逆狄尔斯–阿尔德反应或逆Diels–Alder反应(retro-Diels–Alder)。
1928年德国化学家奥托·迪尔斯和他的学生库尔特·阿尔德首次发现和记载这种新型反应,他们也因此获得1950年的诺贝尔化学奖。
狄尔斯–阿尔德反应用很少能量就可以合成六元环,是有机化学合成反應中非常重要的碳碳键形成的手段之一,也是现代有机合成里常用的反应之一。
反应有丰富的立体化学呈现,兼有立体选择性、立体专一性和区域选择性等。
目录
[隐藏]
∙ 1 发现
∙ 2 机理
∙ 3 立体化学
∙ 4 合成中的价值
∙ 5 参见
∙ 6 参考资料
发现[编辑]
本条目没有列出任何参考或来源。
(2012年2月1日)
維基百科所有的內容都應該可供查證。
请协助添加来自可靠来源的引用以改善这篇条目。
无法查证的内容可能被提出
异议而移除。
狄尔斯-阿尔德反应是1928年由德国化学家奥托·迪尔斯(Otto Paul Hermann Diels)和他的学生库尔特·阿尔德(Kurt Alder)发现的,他们因此获得1950年的诺贝尔化学奖。
最早的关于狄尔斯–阿尔德反应的研究可以上溯到1892年。
齐克(Zinke)发现并提出了狄尔斯-阿尔德反应产物四氯环戊二烯酮二聚体的结构;稍后列别捷夫(Lebedev)指出了乙烯基环己烯是丁二烯二聚体的转化关系。
但这两人都没有认识到这些事实背后更深层次的东西。
1906年德国慕尼黑大学研究生阿尔布莱希特(Albrecht)按导师惕勒(Thiele)的要求做环戊二烯与酮类在碱催化下缩合,合成一种染料的实验。
当时他们试图用苯醌替代其他酮做实验,但是苯醌在碱性条件下很容易分解。
实验没有成功。
阿尔布莱希特发现不加碱反应也能进行,但是得到了一个没有颜色的化合物。
阿尔布莱希特提了一个错误的结构解释实验结果。
1920年德国人冯·欧拉(von Euler)和学生约瑟夫(Joseph)研究异戊二烯与苯醌反应产物的结构。
他们正确地提出了狄尔斯–阿尔德产物结构,也提出了反应可能经历的机理。
事实上他们离狄尔斯–阿尔德反应的发现已经非常近了。
但冯·欧拉并没有深入研究下去,因为他的主业是生物化学(后因研究发酵而获诺贝尔奖),对狄尔斯–阿尔德反应的研究纯属娱乐消遣性质的,所以狄尔斯-阿德尔反应再次沉没下去。
1921年,狄尔斯和其研究生巴克(Back)研究偶氮二羧酸二乙酯(半个世纪后因光延反应而在有机合成中大放光芒的试剂)与胺发生的酯变胺的反应,当他们用2-萘胺做反应的时候,根据元素分析,得到的产物是一个加成物而不是期待的取代物。
狄尔斯敏锐地意识到这个反应与十几年前阿尔布莱希特做过的古怪反应的共同之处。
这使他开始以为产物是类似阿尔布莱希特提出的双键加成产物。
狄尔斯很自然地仿造阿尔布莱希特用环戊二烯替代萘胺与偶氮二羧酸乙酯作用,结果又得到第三种加成物。
通过计量加氢实验,狄尔斯发现加成物中只含有一个双键。
如果产物的结构是如阿尔布莱希特提出的,那么势必要有两个双键才对。
这个现象深深地吸引了狄尔斯,他与另一个研究生阿尔德一起提出了正确的双烯加成物的结构。
1928年他们将结果发表。
这标志着狄尔斯-阿德尔反应的正式发现。
在他们的论文两个作者很深远地看到了这个反应对有机合成观念的颠覆作用,他们预言了该反应日后将在天然产物合成领域的重大意义。
当然两人在文章中也透露出地主恶霸的作风。
先是在文章开头把阿尔布莱希特提出的错误结构这件事用很恶毒的语言痛批一顿。
在文章最后又声明两人对该反应有专属权,不允许其他人使用(英译:We explicitly reserve for ourselves the application of the reaction discovered by us to the solution of such problems.)。
当然,科学界不把这些话当回事。
狄尔斯、阿尔德两人后来卷入该反应的发现权纷争中,分散了精力,没能实现他们预言的“在天然产物全合成中的应用”。
1950年,伍德沃德第一个开创了狄尔斯–阿尔德反应在全合成中的应用。
从此以后,合成大师们用睿智的大脑把狄尔斯–阿尔德反应的应用发挥到了炉火纯青的极致。
值得指出的是,在伍德沃德之前,中国化学家庄长恭曾经尝试过用狄尔斯–阿尔德反应来合成甾体化合物,但是由于当时缺乏对狄尔斯–阿尔德反应区域选择性的控制的知识而失败了。
机理[编辑]
这是一个一步完成的协同反应。
没有中间体存在,只有过渡态。
一般条件下是双烯的最高含电子轨道(HOMO)与亲双烯体的最低空轨道(LUMO)相互作用成键。
由于是不涉及离子的协同反应,故普通的酸碱对反应没有影响。
但是路易斯酸可以通过络合作用影响最低空轨道的能级,所以能催化该反应。
立体化学[编辑]
狄尔斯-阿尔德反应有如下规律:
1、区域选择性:反应产物往往以“假邻对位”产物为主。
即若把六元环产物比作苯环,那么环上官能团(假设有两个官能团)之间的相互位置以邻位(如1),或者对位为主(如3)。
2、立体选择性:反应产物以“内型(即5)”为主,即反应主产物是经过“内型”过渡态得到的。
3、立体专一性:加热条件下反应产物以“顺旋”产物为唯一产物;光照条件下以“对旋”产物为唯一产物。
比如以下两个热反应中,产物7、8的相对立体构型都是唯一的,两个烯烃原料原有的官能团A,B,C,D的顺反立体化学关系都在产物中得到忠实地翻译。
合成中的价值[编辑]
由于该反应一次生成两个碳碳键和最多四个相邻的手性中心,所以在合成中很受重视。
如果一个合成设计上使用了狄尔斯–阿尔德反应,则可以大大减少反应步骤,提高了合成的效率。
很多有名的合成大师都擅长运用狄尔斯–阿尔德反应于复杂天然产物的合成,比如罗伯特·伯恩斯·伍德沃德、艾里亚斯·詹姆斯·科里、丹尼谢夫斯基(Danishefsky)都是应用狄尔斯–阿尔德反应方面的高手。
据传伍德沃德在童年的时候就根据凯库勒苯环两种结构的不可辩性预测了狄尔斯–阿尔德反应的存在。
伍德沃德12岁的时候通过驻波士顿的德国外交官获得了一些德文化学期刊。
在其中一期上他读到了狄尔斯和阿尔德发表的文章见证了该反应的发现。
伍德沃德在其一生的合成实践中大量应用狄尔斯–阿尔德反应构建六元环。
伍德沃德于1960年代开始,与刚入哈佛大学做研究的理论化学家罗德·霍夫曼联手,结合大量的实验事实对狄尔斯–阿尔德反应和相关周环反应的立体化学做了透彻的理论研究,最终导致了在当时震撼了整个有机化学界的「分子轨道对称守恒原理」的诞生。
1979年伍德沃德逝世;1981年霍夫曼因该理论而获得当年度诺贝尔化学奖(与日
本人福井谦一分享)。
2004年,有机合成的另一位著名人物科里在伍德沃德逝世20多年后公开宣称伍德沃德剽窃了他思想而创立的对称守恒律。
这一切又使得狄尔斯-阿尔德反应充满了某种宿命的传奇色彩。
科里对狄尔斯–阿尔德反应也有很大的贡献,发明了一种路易斯酸催化的不对称狄尔斯–阿尔德反应。
在其合成前列腺素过程中,科里试图利用环戊二烯做狄尔斯–阿尔德反应来构筑前列腺素的母环,由此发明了不稳定烯酮的替代试剂。
丹尼谢夫斯基则以发明十分有用的“丹尼谢夫斯基双烯”用于狄尔斯–阿尔德反应而最为出名,在其全合成实践中狄尔斯–阿尔德反应也随处可见。
丹尼谢夫斯基双烯9与一个炔酸酯10反应,经酸化后得到一个苯衍生物11。