第七章 配合物反应动力学
第七章配合物反应动力学

第七章配合物反应动力学部门: xxx时间: xxx整理范文,仅供参考,可下载自行编辑第七章配合物反应动力学研究范围:取代、氧化还原、异构化、加成与消除、配体上进行的反应第一节配合物的反应类型1、取代反应[Cu(H2O>6]2+ + NH3 [Cu(NH3>4(H2O>2]2+ +H2Ob5E2RGbCAP[Mo(CO>6] + bipy [Mo(CO>4bipy][Cr(H2O>6]3+ + Cl− [Cr(H2O>5Cl]2+ + H2O2、氧化还原反应[Os(bipy>3]2++ [Mo(CN>6]3−[Os(bipy>3]3+ +[Mo(CN>6]4−p1EanqFDPw3、异构化反应cis-[CoCl2(en>2]+ trans-[CoCl2(en>2]+[Co(-ONO>(NH3>5]2+ [Co(-NO2>(NH3>5]2+4、加成和消除反应[IrICl(CO>(PPh3>2] + H2 [IrIIIClH2(CO>(PPh3>2]DXDiTa9E3d[PtIICl2(NH3>2] + Cl2 [PtIVCl4(NH3>2]cis-[PtIVHCl2Me(PEt3>2] cis-[PtIICl2(PEt3>2] +CH4RTCrpUDGiT5、配体的反应第二节取代反应动力学定义:配离子中一个配体被另一个自由配体取代的反应。
例:L5M-X+Y L5M-Y+X一、取代的反应机理1、SN1和SN2机理<1)离解机理<SN1机理)慢a.L5M-X = L5M+X<配位数下降6 5)b.L5M+Y=L5M-Y速率方程:d[L5M-Y]/dt= k[L5M-X]速率与Y浓度无关,是对[L5M-X]的一级反应。
<2)缔合机理<SN2机理)慢a、L5M-X+Y = L5MXY<配位数升高6 7)b、L5MXY = L5M-Y + Xd[L5M-Y]/dt=k[L5M-X][Y]属于二级反应。
第七章配合物反应动力学
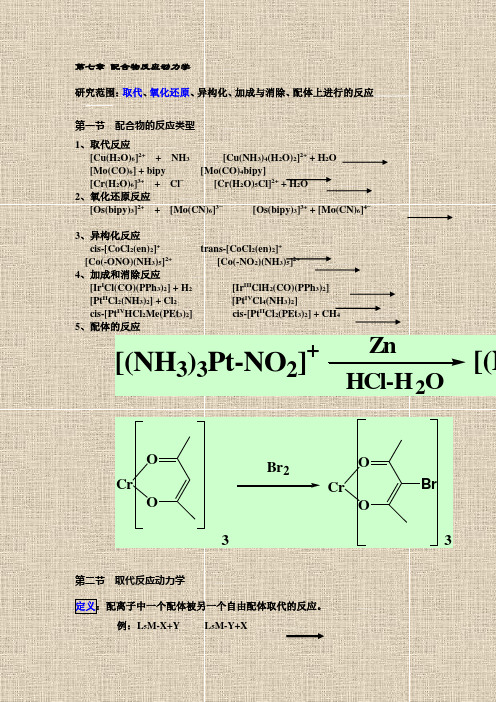
第七章配合物反应动力学研究范围:取代、氧化还原、异构化、加成与消除、配体上进行的反应第一节配合物的反应类型1、取代反应[Cu(H2O)6]2++ NH3 [Cu(NH3)4(H2O)2]2+ + H2O[Mo(CO)6] + bipy [Mo(CO)4bipy][Cr(H2O)6]3++ Cl−[Cr(H2O)5Cl]2+ + H2O2、氧化还原反应[Os(bipy)3]2++ [Mo(CN)6]3−[Os(bipy)3]3+ + [Mo(CN)6]4−3、异构化反应cis-[CoCl2(en)2]+trans-[CoCl2(en)2]+[Co(-ONO)(NH3)5]2+[Co(-NO2)(NH3)5]2+4、加成和消除反应[Ir I Cl(CO)(PPh3)2] + H2[Ir III ClH2(CO)(PPh3)2][Pt II Cl2(NH3)2] + Cl2[Pt IV Cl4(NH3)2]cis-[Pt IV HCl2Me(PEt3)2] cis-[Pt II Cl2(PEt3)2] + CH45、配体的反应ZnHCl-H2O [(NH3)3Pt-NO2]+[(NCrOOCrOOBrBr233第二节取代反应动力学定义:配离子中一个配体被另一个自由配体取代的反应。
例:L5M-X+Y L5M-Y+X一、取代的反应机理1、SN1和SN2机理(1)离解机理(SN1机理)慢a.L5M-X = L5M + X(配位数下降6 5)b.L5M + Y = L5M-Y速率方程:d[L5M-Y]/dt = k[L5M-X]速率与Y浓度无关,是对[L5M-X]的一级反应。
(2)缔合机理(SN2机理)慢a、L5M-X + Y = L5MXY(配位数升高6 7)b、L5MXY = L5M-Y + Xd[L5M-Y]/dt = k[L5M-X][Y]属于二级反应。
* SN1和SN2是两种极限情況。
二.活性与惰性配合物及理论解释1、活性与惰性配合物1)定义:配体可被快速取代的配合物,称为活性配合物;配体取代缓慢的配合物,称为惰性配合物划分标准:配合物与反应试剂(浓度均为0.1M)在25℃时反应,t1/2>1min,称为惰性配合物;t1/2<1min,称为活性配合物。
配合物的反应

第六页,共140页。
活化配合物和过渡态的区别:过渡态是一个能态, 而活化配合物是设想在这一能态下存在的一个化合物
假一级反应的速率常数用k观表示:
k观 = ks + ky[Y]
k观~ [Y]作图,得一直线,直线的斜率为ky,截距为ks。
第二十五页,共140页。
例如对于下列反应
[Pt(diPy)Cl2] + Py 甲醇[Pt(diPy)PyCl]+ + Cl-
控制[Pt(diPy)Cl2] = 1×10-5~1×10-4
过渡态
E
过渡态
E a
反应能
产物
反应物
中间物
产物
时间(或反应坐标)
时间(或反应坐标)
(a)
(b)
,共140页。
2.活性配合物和惰性配合物
定义:在取代反应中,凡配体可以快速地被其它 配体所取代的配合物称为活性配合物。而配体取代 缓慢的配合物称为惰性配合物。
目前国际上采用陶比(H·Taube)所建议的标准:在 25℃,各反应物浓度均为0.1mol·L-1条件下,取代反
即[Co(NH3)6]3+是热力学上不稳定的。但实际上在室温 下[Co(NH3)6]3+的酸性溶液可以保持几天而无显著的分解。 这说明反应的速率是非常慢的,所以[Co(NH3)6]3+是动力 学上的惰性配合物,而在热力学上却是不稳定配合
物。
第十二页,共140页。
3.离解机理、缔合机理及交换机理 配合物的取代反应的类型
配合物的反应动力学及反应机理

SN1速度
减小
增加
(zēngjiā)
不影响
不影响
减小
增加 增加 增加
SN2速度
增加
增加
增加 减小 减小
减小 减小 减小
* 近似地适用于半满或全满d壳层情况,不适用于具有共价键或键配合物
共八十四页
2. 配体的取代(qǔdài)反应
2.3 八面体配合(pèihé)物的取代反应的速率方程
1. 离解(D)机理
共八十四页
1. 基本概念
1.6 势能(shìnéng)曲线 活化(自由)能
过渡态:瞬间存在的不稳定物种;
中间体:相对稳定的过渡(guòdù)化合物,在慢反应中能检测其存在;
共八十四页
1. 基本概念
1.7 活化(huóhuà)参数
通常反应机理(jī lǐ)不能够完全由速率方程决定,还需从实验获得的活化参数予以证实。
例:[Fe(H2O)6]3+ + Y 比 [Fe(H2O)5(OH)]2+ + Y 反应速率大2个数量级
反应速率只与水合金属离子溶度有关,与外来配体L无关。
反应机理:
r k[M(H2O)x ]
[M(H2O)x ]n slow[M(H2O)x1]n H2O [M(H2O)x1]n Lm fast[M(H2O)x1L]nm
键的断裂是反应速率的决定步骤
共八十四页
2. 配体的取代(qǔdài)反应
(2)缔合(置换(zhìhuàn))机理(A机理,SN2机理)
1. 中心原子的电子结构
d0-d2电子:有空的d轨道,易接受外来配体的电子,生成 配位数为7 的活化配合物。
Cr3 (d3 ) :
d3-d6电子:惰性。(需要较高活化能)
普通化学第七章 配位化合物

间是以配位键结合的。这种特殊的结合方式使得配合
物具有特殊的稳定性和空间构型。下面我们就来介绍
一下配合物的价键理论如何来解释配位键和配离子的
空间构型。 中心离子——提供空轨道:电子对接受体 Lewis酸
配位体——提供孤对电子:电子对给予体 Lewis碱
§ 7.2 配合物的结构 ——价键理论和空间构型
构的配合物。 ( © 螯合物) 最常见的螯合剂:en、EDTA
(三)金属有机化合物:金属原子直接与有机配体中的 碳原子结合的配合物。
如:二茂铁 [Fe(C5H5)2]
根据成环原子数目,可以分成四元环、五元环、 六元环、七元环,其中五元环和六元环最稳定,而且,
环的数目越多越稳定,[Cu(en)2]2+中有两个五元环,
一般来说,内轨型配合物因为有内层d轨道参与杂化,
能量较低,所以比结构相似的外轨型配合物要稳定。 稳定性:内轨型 > 外轨型 配离子的稳定性可以用稳定常数来衡量。在配离子 中,中心离子和配体通过配位键紧密结合在一起,但在 溶液中,并不是完全以配离子形式存在的,仍然有少部 分发生了解离:
[Cu(NH3)4](OH)2
[Co (NH3)5Cl]Cl2 氯化一氯· 五氨合钴(Ⅲ) c、中性配合物 [Pt(NH3)2Cl2] 二氯· 二氨合铂(Ⅱ) 四羰基合镍
[Ni(CO)4]
(4) 某些配位体具有相同的化学式,但由于配位原子不 同而有不同的命名,使用时要加以注意: -NO2-(以氮原子为配位原子) -ONO-(以氧原子为配位原子) 硝基 亚硝酸根
习惯上,配合物和配离子没有严格区分, 配离子也可以叫配合物。
二、组成
( © 配位化合物的组成)
内界是整个配合物的中心,由中心离子(或原子) 和配体构成。它们在溶液中不以简单离子存在,而是一 个整体。 (一)中心离子: 配离子的核心,一般是带正电荷的金属离子, 也有的是原子。过渡金属的离子最适合做中心离 子(ⅢB~ⅡB):Fe2+、Fe3+、Co2+、Ni2+、Cu2+、 Zn2+、Ag+;也有少数高氧化态的非金属元素离子: Si(Ⅳ)、P(Ⅴ)。
配合物的结构及异构现象

[Ni(CN)4]2- (d8) , [PdCl4]2- (d8), [Pt(NH3)4]2+ (d8), [Cu(NH3)4]2+ (d9)
一般地,当4个配体与不含有d8电子构型的过渡金属离子或原子配位时可形成四面体构型配合物。
而d8组态的过渡金属离子或原子一般是形成平面正方形配合物, 但具有d8组态的金属若因原子太小, 或配位体原子太大, 以致不可能形成平面正方形时, 也可能形成四面体的构型。
双帽四方反棱柱体 双帽12面体
配位数为10的配位多面体是复杂的, 通常遇到的有双帽四方反棱柱体和双帽12面体。
十一配位的化合物极少, 理论上计算表明, 配位数为11的配合物很难具有某个理想的配位多面体。 可能为单帽五角棱柱体或 单帽五角反棱柱体, 常见于 大环配位体和体积很小的 双齿硝酸根组成的配合物中。
早在1893年维尔纳(瑞士)建立配位理论时,就已经提出了使中心离子周围配体之间的静电斥力最小,配合物最稳定,即配体间应尽力远离,从而采取对称性分布,而实际测定结构的结果证实了这种设想。
配合物的配位数与其空间结构有一定的联系,但配位数相同时,由于配体的不同,与中心离子的作用不同,而空间结构也会不同。
配位数3 (D3h)三角形
配位数5 (D3h,T4v )主要为三角双锥和四方锥
配位数8 (D4d , D2d )四方反棱柱和十二面体
配合物的配位数与几何构形
价键理论顺利地解释了配合物的分子构型。
显然, 分子构型决定于杂化轨道的类型:
配 位 数 2 3 4 4 杂化轨道 sp sp2 sp3 dsp2 分子构型 直线 三角形 正四面体 正方形 配 位 数 5 5 6 杂化轨道 sp3d d2sp2, d4s sp3d2, d2sp3 分子构型 三角双锥 四方锥 正八面体
配合物反应动力学
x
y
y _x fast
速率方程与进入基团y的浓度有关: r = k [ML5X][Y] (SN2双分子亲核取代)
(3).交换机理(Interchange), I机理 (Ia, Id)
X X Y Y
过渡态,不能检出
r k[M0] I机理和A机理的判断: 中间产物(intermediate)存在足够长的时间,能否被分离 或检出 例:Pt(SnCl3)53- 和 Ni(CN)53-五配位的中间体被光谱检出
旁位基团(spectator ligands) (例如, 对位效应)
空间效应(steric effects)
1. 八面体配合物的取代反应
x +y y +x
(1)离解机理(dissociative), D机理
x rate determing slow +y fast y
中间体 可测定
Y的浓度和性质无关,只与起始配合物的浓度有关,因此 速率方程为: 反应速率= k[ML5 X] ( 速率方程与进入基团y的浓度无关,SN1单分子亲核) Co(CN)5H2 O2
H2O OH F RNH2 py NH3 Cl Br SCN I
NO2 C6H5 SC(NH2)2 CH3 NO H PR3 C2H4 CN CO
Cl Pt Cl
H3N Pt H3N
Cl Cl
NH3
Cl Pt Cl
k1 k2
Co(CN)52 + H2O
k3 k4
Co(CN)52 + Y
Co(CN)5Y3
Y分别为Br, I, SCN, N3时, k1值均为1.6103s1 说明反应与进入基团无关
无机化学第七章化学动力学基础
反应历程
H2O2+2Br-+2H+2H2O+Br2是下列基元反应构成 H++H2O2H3O2+ H3O2+H++H2O2 H3O2++Br-H2O+HOBr(慢反应) HOBr+H++Br-H2O+Br2 因速度决定步骤为慢反应,即v=k[H3O2+][Br-] 但初态时并没有H3O2+只有H2O2、Br-、H+,我们需要变换一下H3O2+ 因H++H2O2H3O2+为快反应,在溶液中立刻就达到了平衡
求该反应的反应级数m+n和速度常数k?
浓度对化学反应速率的影响
浓度对化学反应速率的影响
解:由速度方程v=k[CO]m·[Cl2]n 得:v1=k[CO]m·[Cl2]1n v2=k[CO]m·[Cl2]2n
v=k[CO]·[Cl2]3/2 m+n=2.5 即 对CO为一级
对Cl2为1.5级
基元反应的速度方程
恒温下,基元反应的反应速度与各反应物浓度系数次方的乘积成正比。也称为质量作用定律
对: aA + bB dD+eE
则: v=k[A]a·[B]b
如:
对于反应 H2O2+2Br-+2H+2H2O+Br2的速度方程不能写成v=k[H2O2][H+]2[Br-]2,因其不是一个五元反应。
一步完成的化学反应称基元反应,由一个基元反应构成的化学反应称为简单反应;由两个或三个基元反应构成的化学反应称为非基元反应或复杂反应。
7-4 反应历程
反应历程
如:H2O2+2Br-+2H+2H2O+Br2 是由下列一系列基元反应构成
配位化学反应动力学和机理
配合物的活性和惰性 与热力学的稳定性不应混淆。 由下边的图可以看出, 配合物 的热力学稳定性取决于反应 物和产物的能量差,而配合物 的活性、惰性则决定于反应 物与活化配合物之间的能量差。
配合物热力学稳定性与动力学活泼性处于矛盾情 况的两个典型例子是Co(NH3)63+和[Ni(CN)4]2-: 六氨合钴(III)离子在室温酸性水溶液中的H2O取代 NH3的反应, 在数日内无显著变化, 说明配合物是惰 性的但反应4Co(NH3)63++20H3O++ 6H2O 4Co(H2O)62++24NH4++O2的平衡常数K=1025, 说明 Co(NH3)63+在热力学上是不稳定的。这意味着反应自 由能变负值很大, 而反应活化能正值很大。 而反应 [Ni(CN)4]2-+4H2O [Ni(H2O)4]2++4CN-的平 衡常数K=10-22, 说明[Ni(CN)4]2-是热力学稳定的。 然而研究表明, [Ni(CN)4]2-在动力学上却是活泼的。
对于离解机理(D)反应 中心离子或离去配体的半径越小, 电荷越高, 则金属-配体键越牢固, 金属-配体键越不容易断 裂, 越不利于离解机理反应, 意味着配合物的惰性越大。 对于缔合机理(A)反应 若进入配体的半径越小, 负电荷 越大, 越有利于缔合机理反应, 意味着配合物的活性越大。但 中心离子的电荷的增加有着相反的影响:一方面使M-L键不 容易断裂, 另一方面又使M-Y键更易形成。究竟怎样影响要 看两因素的相对大小。一般来说, 中心离子半径和电荷的增加 使得缔合机理反应易于进行。进入配体半径很大时, 由于空间 位阻, 缔合机理反应不易进行
配位化合物的反应十分广泛, 有取代反应, 电子转移 反应, 分子重排反应和配体上的反应等等, 本章具体地讨论 配体的取代反应和涉及电子转移的氧化还原反应。
04 配位化合物的反应机理与动力学性质
反应物: [Co(NH3)5Cl]2+: Co3+ d6,LS, 惰性, 在酸性溶液中稳定; [Cr(H2O)6]2+: Cr2+ d4,HS, 活性, 水交换反应快, t1/2<10-9s
内界机理
Co Cl
III
H2O Cr
II
eCo
III
Cl
Cr
II
Co
II
Cl
Cr
III
内界机理
电子转移之后,配合物电子结构、状态发生变化 生成物:Co2+,d7,活性的,水交换反应,
1. 外界机理
两配合物的配位内界保持不变,电子通过外界传递。
*Fe(H2O)62+ + Fe(H2O)63+ *Fe(H2O)63+ + Fe(H2O)62+
在轨道能量相近,半径相近的过渡状态直接传递。
电子转移反应的机理:
1. 外界机理
*Fe(H2O)62+ + Fe(H2O)63+ *Fe(H2O)63+ + Fe(H2O)62+
反位效应 (trans effect)
通过对Pt(II)配合物取代反应的系列研究,得出
反位效应的强弱次序:
H2O < OH- < F- ~ RNH2 ~ Py ~ NH3 < Cl- < Br- <
SCN- ~ I- ~NO2- ~C6H3- < SC(NH2)2 ~ CH3- <NO- ~
H- ~ PR3 < C2H4 ~ ON- ~CO 反位效应是平面正方形配合物发生取代反应的 一个重要特征,用以指导合成一系列的几何异构体。
0.1M反应物溶液混合,1分钟内能否完成反应,
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第七章配合物反应动力学研究范围:取代、氧化还原、异构化、加成与消除、配体上进行的反应第一节配合物的反应类型1、取代反应[Cu(H2O)6]2++ NH3 [Cu(NH3)4(H2O)2]2+ + H2O[Mo(CO)6] + bipy [Mo(CO)4bipy][Cr(H2O)6]3++ Cl−[Cr(H2O)5Cl]2+ + H2O2、氧化还原反应[Os(bipy)3]2++ [Mo(CN)6]3−[Os(bipy)3]3+ + [Mo(CN)6]4−3、异构化反应cis-[CoCl2(en)2]+trans-[CoCl2(en)2]+[Co(-ONO)(NH3)5]2+[Co(-NO2)(NH3)5]2+4、加成和消除反应[Ir I Cl(CO)(PPh3)2] + H2[Ir III ClH2(CO)(PPh3)2][Pt II Cl2(NH3)2] + Cl2[Pt IV Cl4(NH3)2]cis-[Pt IV HCl2Me(PEt3)2] cis-[Pt II Cl2(PEt3)2] + CH45、配体的反应ZnHCl-H2O [(NH3)3Pt-NO2]+[(NCrOOCrOOBrBr233第二节取代反应动力学定义:配离子中一个配体被另一个自由配体取代的反应。
例:L5M-X+Y L5M-Y+X一、取代的反应机理1、SN1和SN2机理(1)离解机理(SN1机理)慢a.L5M-X = L5M + X(配位数下降6 5)b.L5M + Y = L5M-Y速率方程:d[L5M-Y]/dt = k[L5M-X]速率与Y浓度无关,是对[L5M-X]的一级反应。
(2)缔合机理(SN2机理)慢a、L5M-X + Y = L5MXY(配位数升高6 7)b、L5MXY = L5M-Y + Xd[L5M-Y]/dt = k[L5M-X][Y]属于二级反应。
* SN1和SN2是两种极限情況。
二.活性与惰性配合物及理论解释1、活性与惰性配合物1)定义:配体可被快速取代的配合物,称为活性配合物;配体取代缓慢的配合物,称为惰性配合物划分标准:配合物与反应试剂(浓度均为0.1M)在25℃时反应,t1/2>1min,称为惰性配合物;t1/2<1min,称为活性配合物。
2)与热力学稳定常数的关系活性与惰性是动力学上的概念,不可与稳定性混为一谈。
惰性配合物也可能是热力学不稳定的配合物。
过渡态Ea反应物H产物反应坐标2、理论解释1)价键理论A、外轨型配合物是活性的(如sp3d2杂化配合物);B、内轨型配合物,如(n-1)d轨道中有空轨道,则是活性的,否则是惰性的。
d2sp3内轨型的配合物:(n-1)d ns np活性:d0Sc3+, Y3+, La3+活性:d1Ti3+活性:d2V3+惰性:d3Cr3+惰性:d4Cr2+惰性:d5Mn2+, Fe3+惰性:d6Fe2+, Co3+sp3d2外轨型配合物:ns np nd解释:若按SN2机理反应,容易理解。
外轨型配合物:空nd 轨道与sp3d2轨道能量接近。
内轨型配合物:(1)若(n-1)d有空轨道;(2)若无(n-1)d空轨道:价键理论之不足之处:a.只能作定性划分;b.认为d8组态Ni2+八面体配合物(外轨型)为活性,实验表明为惰性;c.无法解释Cr3+(d3)和Co3+(d6)八面体配合物比Mn3+(d4)和Fe3+(d5)更为惰性的实验事实。
2)晶体场理论反应物过渡态SN1 八面体-----四方锥(CN=5)SN2 八面体-----五角双锥(CN=7)晶体场活化能:CFAE=CFSE(过渡态)—CFSE(反应物)过渡态CFSE(过渡态)EaEa'反应物CFSE(反应物)产物反应坐标若CFAE≤0,活性;CFAE >0,惰性。
由于CFAE与过渡态构型有关,因此可用来判定SN1还是SN2机理(数据见下页表格)。
晶体场活化能(CFAE)(Dq)d n强场(低自旋)弱场(高自旋)四方锥(SN1)五角双锥(SN2)四方锥(SN1)五角双锥(SN2)d00 0 0 0d1-0.57 -1.28 -0.57 -1.28d2-1.14 -2.56 -1.14 -2.56d3 2.00 4.26 2.00 4.26d4 1.43 2.98 -3.14 1.07d50.86 1.70 0 0d6 4.00 8.52 -0.57 -1.28d7-1.14 5.34 -1.14 -2.56d8 2.00 4.26 2.00 4.26d9 -3.14 1.07 -3.14 1.07d100 0 0 0A、d0、d1、d2、d10及弱场下的d5、d6、d7八面体配合物的CFAE≤0,是活性配合物。
B、d8构型八面体配合物的CFAE>0,应为惰性配合物。
C、强场下,CFAE的顺序为:d6>d3>d4>d5,实际情况符合这一顺序。
D、判断反应机理。
应指出,CFAE只是活化能中的一小部分。
三、八面体配合物的取代反应。
1、酸性水解pH<3时:[Co(NH3)5X]2+ + H2O = [Co(NH3)5(H2O)]3+ + X-d[Co(NH3)5(H2O)]/dt=k A[Co(NH3)5X]*[H2O]=55.5M(1)发现[Co(NH3)5X]2+(X=Cl-、Br-、NO3-)的酸性水解反应过渡焓保持恒定。
过渡态反应物产物反应坐标H T(2) 发现下列反应[Co(LL)2Cl 2]+ + H 2O = [Co(LL)2(H 2O)Cl]2+ + Cl − * LL=H 2N-(CH 2)n -NH 2,(乙二胺、丙二胺、丁二胺)的速率随(LL)体积增大而增大,这支持SN1机理。
结论:八面体配合物的酸性水解大多为SN1机理,但SN2不能排除。
2、碱性水解[Co(NH 3)5Cl]2+ + OH − = [Co(NH 3)5OH]2+ + Cl − d[Co(NH 3)5(OH)]/dt = k B [Co(NH 3)5Cl][OH] 由此可推测为SN2机理。
但发现,[Co(py)4Cl 2]+碱性水解速率很小,且与OH -浓度无关。
这与SN2机理矛盾。
于是提出下列SN1 CB(conjugate base)机理:k 1 [Co(NH 3)5Cl]2+ + OH − [Co(NH 3)4(NH 2)Cl]+ + H 2O (快)k -1 k 2[Co(NH 3)4(NH 2)Cl]+ [Co(NH 3)4(NH 2)]2+ + Cl −(慢) [Co(NH 3)4(NH 2)]2+ + H 2O [Co(NH 3)5OH]2+(快) 采用稳态近似:d[Co(NH 3)4(NH 2)Cl]/dt = k 1[Co(NH 3)5Cl][OH]-k -1[Co(NH 3)4(NH 2)Cl][H 2O]-k 2[Co(NH 3)4(NH 2)Cl]=0[Co(NH 3)4(NH 2)Cl] = k 1[Co(NH 3)5Cl][OH]/(k -1[H 2O]+k 2) 反应速率:d[Co(NH 3)5(OH)]/dt = k 2[Co(NH 3)4(NH 2)Cl] = k 1k 2[Co(NH 3)5Cl][OH]/(k -1[H 2O] + k 2)= K[Co(NH 3)5Cl][OH]K = k 1k 2/(k -1[H 2O] + k 2)速率控制步骤是原始配合物共轭碱的离解,因此称为SN1 CB机理。
四、平面正方形配合物的取代反应1、反应动力学与机理一般认为按SN2机理进行取代反应。
PtL3X + Y = PtL3Y + X所观测速率方程为d[PtL3Y]/dt = k Y[PtL3X][Y] + ks[PtL3X]反应途径:L3PtXL3PtXYL3PtXS L3PtS L3+Yky+Sks-X快快配合物取代反应速率(25℃)t1/2(min)[PtCl4]2−+ H2O = [PtCl3(H2O)]−+ Cl−300cis-[Pt(NH3)2Cl2] + H2O = [Pt(NH3)2(H2O)Cl]+ + Cl−3002、反位效应(trans effect)1)定义:反位效应是指离去基团反位上的配体对它的取代反应速率的影响,属于动力学范畴。
对Pt2+配合物取代反应,反位效应次序如下:CN−~ CO ~ C2H4 > H−~ [SC(NH2)] ~ PR3 ~ SR2 > CH3−> C6H5−~ NO2−~ I−~ SCN- > Br−> Cl−> 胺~ 氨> OH−> H2O*尚未找到一个对一切金属配合物通用的反位效应次序。
2)应用(制备配合物)PtCl ClClCl2-PtCl NH 3ClCl-PtCl NH 3NH 3Cl NH 3NH 3PtNH 3ClNH 3 NH 3+PtNH 3NH 3NH 3 NH 32+PtNNH 3Cl -Cl - Cl3)理论解释A 、极化作用理论++_+__+_++_ +__+_+_+ClClCl Cl Cl ClClBrPt* 由此理论,配体越易极化,反位效应越强。
I − > Br − > Cl − 与事实相符金属离子变形性越大,反位效应越明显,如 Pt 2+>Pd 2+,也与事实相符。
弱点:不能解释SN2机理。
B 、π键理论TLM LL*显然该理论不能解释不形成反馈π键的体系。
3、反位影响(trans influence)定义:基态配合物中,某配体对处于其反位的配体与中心原子间键的削弱程度,又称热力学反位效应。
反位影响表现为键长及键的伸缩振动频率等参数的变化。
反位影响与反位效应的顺序不完全一致。
PtCl Cl ClEt 3PPtClCl ClPtCl Cl0.238nm 0.2327nm 0.2317nm 反位影响Et 3P > C 2H 4 > Cl −, 但反位效应C 2H 4 > Et 3P > Cl −。
第三节配合物的氧化还原反应机理内容:讨论两个配合物之间的氧化还原反应。
1、外界反应机理。
1)生成前驱配合物。
Ox + Red = Ox || Red (前驱配合物) 快反应 两种反应物相互接近达到一定平衡距离。
2) 前驱配合物的化学活化、电子转移及后继配合物的形成。
Ox || Red = −Ox || Red + 慢反应化学活化:使两种反应物的内部结构(如M-L 键长)及中心原子的电子自旋状态相似,以便发生电子转移,生成后继配合物。
例:[Fe*(CN)6]3− ~ [Fe(CN)6] 4− 体系(二者均为低自旋) Fe 3+—CN 伸长; Fe 2+—CN 缩短再如: [Co*(NH 3)6]2+ ~ [Co(NH 3)6]3+高自旋 低自旋t 2g 5e g 2 t 2g 6 t 2g 4e g 23) 后继配合物离解为产物−Ox || Red + = Ox − + Red + 快反应第二步比较慢,是速率决定步骤。