医 疗 器 械 产 品 分 类 界 定
医疗器械分类目录(2002版)

关于编制《医疗器械分类目录》的通知药监械函[2001]13号各医疗器械标准化技术归口单位或医疗器械质量检测中心:根据《医疗器械监督管理条例》第五条国家对医疗器械实行分类管理的规定,经研究决定,将委托并请你中心依据国家药品监督管理局令(第15号)《医疗器械分类规则》的要求,对原1998版《中国医疗器械产品分类目录》进行全面修订调整。
现将修订调整的范围及有关要求通知如下:一、请按国家药品监督管理局令(第15号)《医疗器械分类规则》的要求,对原1998版《中国医疗器械分类目录》中的内容进行全面修订调整,同时对修订调整的内容应附有详细的文字说明。
二、关于分类与代码的编写:原《目录》是按GB7635—87《全国工农业(商品、物资)分类与代码》标准等要求进行编排的,即68xx。
目前考虑该标准正在修订过程中,现暂不标注医疗器械行业代号(68),只要求标注医疗器械产品类代号(01—99)。
三、在编写《医疗器械分类目录》时,原《目录》中的基本格式要求不变,应将产品类代号和标题名称、序号、名称、品名举例、管理类别详细列出,并具体予以明确。
四、请你中心在本归口的医疗器械标准范围内对原《目录》进行修订调整,遇有归口范围不明确的,应附文字说明。
五、请各中心将修订调整后的《医疗器械分类目录》(部分)及文字说明于5月15日前函报我司标准处。
特此通知国家药品监督管理局医疗器械司二OO一年四月四日关于印发《医疗器械分类目录》的通知国药监械[2002]302号各省、自治区、直辖市药品监督管理局:根据《医疗器械监督管理条例》第五条的规定,国家药品监督管理局组织制定了《医疗器械分类目录》,现予印发,自印发之日起施行。
该《目录》不包含按医疗器械管理的体外诊断试剂产品,国家药品监督管理局将另行印发体外诊断试剂产品分类目录。
原国家医药管理局发布的《医疗器械分类目录》即行废止。
特此通知附件:《医疗器械分类目录》国家药品监督管理局二○○二年八月二十八日关于2002版《医疗器械分类目录》有关问题的批复国药监械[2002]410号北京市药品监督管理局:你局《关于新版〈医疗器械分类目录〉中几点问题的请示》(京药监械〔2002〕29号)收悉,现批复如下:一、97版《目录》包括而2002版《目录》未包括的产品,原则上不再作为医疗器械管理;二、按97版《目录》已注册的产品,如该产品上市后无不良事件,可待该产品注册证到期时,再按2002版《目录》类别要求换证;三、接骨螺钉见骨钉(6846-1),功能辅助装置见器官辅助装置(6846-5),医用夹板为I类产品,序号为6864-3;四、使用已注册的义齿材料生产的义齿为Ⅱ类医疗器械,序号为6863-16;五、一次性使用活检针为Ⅲ类医疗器械,可重复使用活检针为Ⅱ类医疗器械,序号为6815;六、无创监护仪器为Ⅱ类医疗器械;七、医用卫生材料及敷料(无菌或非无菌)均执行2002版《目录》;八、在没有制定新的体外诊断试剂分类规定前,体外诊断试剂暂按Ⅲ类产品管理;九、2002版《目录》中煮沸消毒设备的序号为6857-7,牙周塞治剂与根管充填材料使用相同的序号(6863-3)。
2022年医疗器械安全知识竞赛试题之简答题(附含答案)

2022年医疗器械安全知识竞赛试题之简答题(附含答案)1、什么是医疗器械?答:国务院2000年1月4日颁布的《医疗器械监督管理条例》对医疗器械有这样一个定义,即医疗器械是指单独或者组合使用于人体的仪器、设备、器具、材料或者其他物品,包括所需要的软件;其用于人体体表及体内的作用不是用药理学、免疫学或者代谢的手段获得,但是可能有这些手段参与并起一定的辅助作用;其使用旨在达到下列预期目的: (一)对疾病的预防、诊断、治疗、监护、缓解;(二)对损伤或者残疾的诊断、治疗、监护、缓解、补偿;(三)对解剖或者生理过程的研究、替代、调节;(四)妊娠控制。
2、医疗器械产品分为哪几类?答:医疗器械产品分为3类。
第一类医疗器械是指,通过常规管理足以保证其安全性、有效性的医疗器械。
“今日有、明日无”的优惠报价,如游说成功,业务员必趁热打铁,紧跟老人回家取钱,只收现金且多方推托不开发票。
(2)门面“贴金”,甚至公然宣称“经合法批准”、“国家重点扶持项目”等。
为了骗取老人的信任,不法经营者不惜在市内繁华地带租用较高级的写字楼,向老人吹嘘经营者的实力如何雄厚、如何得到国家有关部门批准和扶持,某某领导人曾亲临指导等等,冠冕堂皇地打着“合法”的旗号, 使人深信不疑。
(3)卖点新奇,打所谓“绿色、环保、健康、高科技”的旗号。
抓住老人对健康的特殊心理要求,派发精美的宣传广告资料,将不具备应有功能的产品虚假夸大成“包治百病”的“灵丹妙药”。
24、选购医疗器械要考虑哪几点问题?答:选购医疗器械要注意以下几点:一是因人而异,因时制宜。
应根据自己的现状,想一想自己的身体状况(或病情)是否需要一台医疗器械?选用药品还是器械更恰当?眼前的这款产品是否适合自己的身体情况(或病情)?产品的性能价格比是否合理?结合自己的实际情况将上述问题排一排队,这样就会比较清醒地认识自己所要选购的产品是不是自己所需要的。
二是购买医疗器械时,应到有医疗器械生产许可证或医疗器械经营许可证的单位(其生产或经营的产品应在许可证核定范围内)选购。
医疗器械划分.
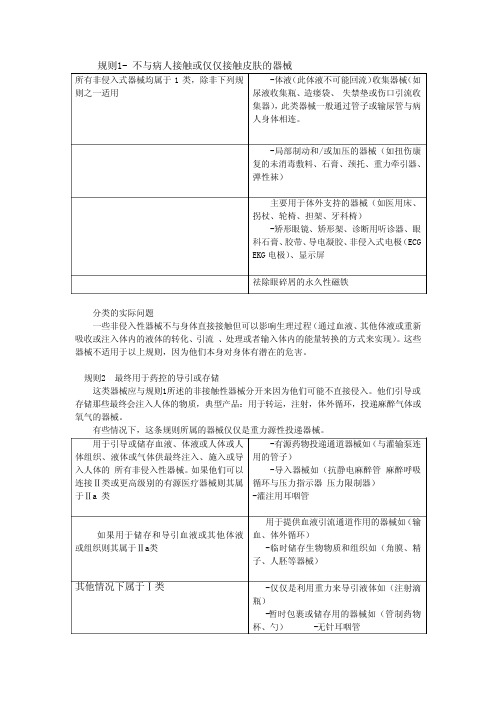
规则1- 不与病人接触或仅仅接触皮肤的器械所有非侵入式器械均属于1类,除非下列规则之一适用-体液(此体液不可能回流)收集器械(如尿液收集瓶、造瘘袋、 失禁垫或伤口引流收集器),此类器械一般通过管子或输尿管与病人身体相连。
-局部制动和/或加压的器械(如扭伤康复的未消毒敷料、石膏、颈托、重力牵引器、弹性袜)主要用于体外支持的器械(如医用床、拐杖、轮椅、担架、牙科椅)-矫形眼镜、矫形架、诊断用听诊器、眼科石膏、胶带、导电凝胶、非侵入式电极(ECG EKG电极)、显示屏祛除眼碎屑的永久性磁铁分类的实际问题一些非侵入性器械不与身体直接接触但可以影响生理过程(通过血液、其他体液或重新吸收或注入体内的液体的转化、引流 、处理或者输入体内的能量转换的方式来实现)。
这些器械不适用于以上规则,因为他们本身对身体有潜在的危害。
规则2 最终用于药控的导引或存储这类器械应与规则1所述的非接触性器械分开来因为他们可能不直接侵入。
他们引导或存储那些最终会注入人体的物质,典型产品:用于转运,注射,体外循环,投递麻醉气体或氧气的器械。
有些情况下,这条规则所属的器械仅仅是重力源性投递器械。
用于引导或储存血液、体液或人体或人体组织、液体或气体供最终注入、施入或导入人体的 所有非侵入性器械。
如果他们可以连接Ⅱ类或更高级别的有源医疗器械则其属于Ⅱa 类-有源药物投递通道器械如(与灌输泵连用的管子)-导入器械如(抗静电麻醉管 麻醉呼吸循环与压力指示器 压力限制器)-灌注用耳咽管如果用于储存和导引血液或其他体液或组织则其属于Ⅱa类用于提供血液引流通道作用的器械如(输血、体外循环)-临时储存生物物质和组织如(角膜、精子、人胚等器械)其他情况下属于Ⅰ类-仅仅是利用重力来导引液体如(注射滴瓶)-暂时包裹或储存用的器械如(管制药物杯、勺) -无针耳咽管如果一种器械比如管子与有源器械相连而发挥作用,则这种器械自动归于Ⅱa类,除非制造商明确规定其不与Ⅱa类或者更高级别的器械连用。
医疗器械与生物医学工程作业指导书

医疗器械与生物医学工程作业指导书第1章医疗器械概述 (3)1.1 医疗器械的定义与分类 (3)1.2 医疗器械的发展历程与现状 (4)1.3 医疗器械的监管体系 (4)第2章生物医学工程基础 (4)2.1 生物医学工程的定义与范畴 (4)2.2 生物医学工程的技术分支 (4)2.3 生物医学工程的应用领域 (5)第3章医疗器械设计与开发 (5)3.1 医疗器械设计原则与流程 (5)3.1.1 设计原则 (6)3.1.2 设计流程 (6)3.2 医疗器械的人因工程 (6)3.2.1 人因工程概述 (6)3.2.2 人因工程在医疗器械设计中的应用 (6)3.3 医疗器械的可靠性评价 (7)3.3.1 可靠性评价方法 (7)3.3.2 可靠性评价在医疗器械中的应用 (7)第4章医疗器械制造与工艺 (7)4.1 医疗器械制造材料 (7)4.1.1 金属材料 (7)4.1.2 陶瓷材料 (8)4.1.3 聚合物材料 (8)4.1.4 复合材料 (8)4.2 常见医疗器械制造工艺 (8)4.2.1 注塑成型 (8)4.2.2 挤出成型 (8)4.2.3 精密铸造 (8)4.2.4 机加工 (8)4.2.5 焊接 (8)4.3 医疗器械质量控制与检测 (8)4.3.1 材料检验 (9)4.3.2 加工过程控制 (9)4.3.3 成品检验 (9)4.3.4 质量管理体系 (9)第5章医疗器械注册与认证 (9)5.1 医疗器械注册流程与要求 (9)5.1.1 注册概述 (9)5.1.2 注册流程 (9)5.1.3 注册要求 (9)5.2 医疗器械临床试验 (10)5.2.2 临床试验要求 (10)5.2.3 临床试验流程 (10)5.3 国际医疗器械认证简介 (10)5.3.1 国际医疗器械认证概述 (10)5.3.2 常见国际医疗器械认证 (10)5.3.3 认证流程及要求 (10)第6章医疗器械市场营销 (11)6.1 医疗器械市场分析 (11)6.1.1 市场规模及增长 (11)6.1.2 市场竞争格局 (11)6.1.3 市场需求分析 (11)6.2 医疗器械营销策略 (11)6.2.1 产品策略 (11)6.2.2 价格策略 (11)6.2.3 渠道策略 (11)6.2.4 推广策略 (12)6.3 医疗器械市场前景与发展趋势 (12)6.3.1 创新技术驱动发展 (12)6.3.2 跨界融合加速 (12)6.3.3 智能化、个性化发展 (12)6.3.4 国际化竞争加剧 (12)第7章医疗器械使用与维护 (12)7.1 医疗器械的正确使用 (12)7.1.1 使用前的准备工作 (12)7.1.2 操作流程 (12)7.1.3 使用过程中的注意事项 (12)7.2 医疗器械的维护与保养 (13)7.2.1 日常保养 (13)7.2.2 定期维护 (13)7.3 医疗器械故障分析与处理 (13)7.3.1 故障诊断 (13)7.3.2 故障处理 (13)7.3.3 预防措施 (13)第8章生物医学成像技术 (14)8.1 X射线成像技术 (14)8.1.1 X射线成像原理 (14)8.1.2 X射线成像设备 (14)8.1.3 X射线成像应用 (14)8.2 核磁共振成像技术 (14)8.2.1 核磁共振成像原理 (14)8.2.2 核磁共振成像设备 (14)8.2.3 核磁共振成像应用 (14)8.3 超声成像技术 (15)8.3.2 超声成像设备 (15)8.3.3 超声成像应用 (15)第9章生物医学信号检测与处理 (15)9.1 生物医学信号概述 (15)9.2 生物医学信号检测技术 (15)9.2.1 信号检测原理 (15)9.2.2 信号检测传感器 (15)9.2.3 信号检测系统设计 (15)9.3 生物医学信号处理方法 (16)9.3.1 信号预处理 (16)9.3.2 信号特征分析 (16)9.3.3 信号模式识别 (16)9.3.4 信号源分离与重构 (16)第10章医疗器械与生物医学工程创新与发展 (16)10.1 医疗器械创新趋势 (16)10.1.1 智能化发展 (16)10.1.2 微创化技术 (16)10.1.3 个性化定制 (16)10.2 生物医学工程新技术 (17)10.2.1 生物材料 (17)10.2.2 纳米技术 (17)10.2.3 基因测序与基因编辑 (17)10.3 医疗器械与生物医学工程的未来发展展望 (17)10.3.1 医疗器械的创新与整合 (17)10.3.2 生物医学工程技术的临床应用 (17)10.3.3 医疗器械监管与产业发展 (17)第1章医疗器械概述1.1 医疗器械的定义与分类医疗器械是指在医学诊断、治疗、康复和预防疾病过程中,单独或组合使用,用于人体的仪器、设备、器具、材料或其他类似物品。
最新版《医疗器械分类目录》2022年1月1日起施行

最新版《医疗器械分类⽬录》2022年1⽉1⽇起施⾏最新版《医疗器械分类⽬录》2022年12⽉1⽇起施⾏01有源⼿术器械说明⼀、范围本⼦⽬录包括以⼿术治疗为⽬的与有源相关的医疗器械,包括超声、激光、⾼频/射频、微波、冷冻、冲击波、⼿术导航及控制系统、⼿术照明设备、内窥镜⼿术⽤有源设备等医疗器械。
⼆、框架结构本⼦⽬录按照产品预期⽤途和专业技术及功能特点进⾏层级排序,共划分为10个⼀级产品类别,在⼀级产品类别的基础上根据先设备后附件的形式设⽴⼆级产品类别共25个,列举120个品名举例。
本⼦⽬录包括2002版医疗器械分类⽬录中《6821医⽤电⼦仪器设备》《6822医⽤光学器具仪器及内窥镜设备》《6824医⽤激光仪器设备》《6825医⽤⾼频仪器设备》《6854⼿术室急救室诊疗室设备及器具》《6858医⽤冷疗低温冷藏设备及器具》和《〈6816烧伤(整形)科⼿术器械〉(部分)》,还包括了2012版医疗器械分类⽬录中《〈6823医⽤超声仪器及有关设备〉(部分)》。
三、其他说明(⼀)医⽤激光光纤与激光治疗仪配套应⽤,传输激光器产⽣的能量,⽤于激光⼿术治疗。
依据《关于⼀次性前列腺治疗套件等产品分类界定的通知》(国⾷药监械〔2008〕587号)和《国家⾷品药品监督管理局关于超声肿瘤治疗系统等17个产品分类界定的通知》(国⾷药监械〔2012〕36号)分类界定⽂件规定管理类别为⼆类,分类编码6824。
因此将医⽤激光光纤纳⼊《01有源⼿术器械》⽬录中。
(⼆)射频消融设备⽤灌注泵,管理类别由第三类降为第⼆类。
(三)发光⼆极管(LED)⼿术照明灯,管理类别由第⼆类降为第⼀类。
02⽆源⼿术器械说明⼀、范围本⼦⽬录包括通⽤⼑、剪、钳等各类⽆源⼿术医疗器械,不包括神经和⼼⾎管⼿术器械、⾻科⼿术器械、眼科器械、⼝腔科器本⼦⽬录按照⽆源⼿术器械的功能⽤途及产品特性分为15个⼀级产品类别。
根据产品的具体⽤途的不同,分为83个⼆级产品类别,列举597个品名举例。
医疗器械分类_按1_2_3类解读

二类
电动貉故质跆?/
td>
无影灯接美涔庀宋脊馐质醯?/td>
宫腔冷冻治疗仪、冷冻低温治疗机、低温变速降温仪旱淞苹⒀顾跏嚼涠
持瘟埔?/td>
止血海绵接猛阎蕖II接猛阎床?/
td>
皮肤缝合钉接美
一次性使用导尿管淮涡允褂玫デ坏寄导管鞴?/td>
蚬堋⑺黄业寄蚬堋⒌ü芤鞴?/td>
体外震波碎石机、病人有创监护系统、颅内压监护仪写葱氖涑隽考啤III写炊嗟忌砑锹家恰⑿哪谙J鲜缤蓟⑿哪谕饽け瓴馔家恰III写葱缘缱友辜?/t
d>
眼人工晶体、角膜接触镜(软性残浴⑺苄谓悄そ哟ゾ担┘盎だ碛靡骸⒀勰谔畛湮铮úA宓龋⒄车镏省⒐嘧⒁海
ㄖ厮⒐栌停?/td>
心及血管写础⑶荒谑质跤媚诳
/td> 有创内窥镜(腹腔镜、关节镜、肾镜、胰腺镜、椎间盘镜、脑窦镜、胆道镜)、心及血管内窥镜(心内窥镜、血管内窥镜)、腔内手术用内窥镜(经尿道电切镜糜诟咂档缜惺质跤玫南宋诳导坝补苣
诳担?/td>
一次性使用输液器、输血器、静脉输液(血)针、血袋、采血器、血液成分分离器材、连接管路胙方哟サ目亍⒀郝送⒁┮汗寺四ぁ⒖掌寺四ぁ⒙樽淼脊堋⒁淮涡允褂醚汗似?/t
d>。
一二三类医疗器械分类目录

一二三类医疗器械分类目录一、医疗器械的分类:第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
二、医疗器械许可范围及生产条件:1、第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。
2、医疗器械注册人、备案人应当加强医疗器械全生命周期质量管理,对研制、生产、经营、使用全过程中医疗器械的安全性、有效性依法承担责任。
3、第一类医疗器械产品备案和申请第二类、第三类医疗器械产品注册,应当提交下列资料:(一)产品风险分析资料;(二)产品技术要求;(三)产品检验报告;(四)临床评价资料;(五)产品说明书以及标签样稿;(六)与产品研制、生产有关的质量管理体系文件;(七)证明产品安全、有效所需的其他资料。
4、申请第二类医疗器械产品注册,注册申请人应当向所在地省、自治区、直辖市人民政府药品监督管理部门提交注册申请资料。
申请第三类医疗器械产品注册,注册申请人应当向国务院药品监督管理部门提交注册申请资料。
三、一类、二类、三类医疗器械目录:一类,二类和三类术语管理类别,看医疗器械监督管理条例有相关的规定。
管理由低到高。
医疗器械,是指单独或者组合使用于人体的仪器、设备、器具、材料或者其他物品,包括所需要的软件;其用于人体体表及体内的作用不是用药理学、免疫学或者代谢的手段获得,但是可能有这些手段参与并起一定的辅助作用;其使用旨在达到下列预期目的:(1)对疾病的预防、诊断、治疗、监护、缓解;(2)对损伤或者残疾的诊断、治疗、监护、缓解、补偿;(3)对解剖或者生理过程的研究、替代、调节;(4)妊娠控制。
医疗器械分为三类:第一类医疗器械有:基础外科用刀《手术刀柄和刀片、皮片刀、疣体剥离刀、柳叶刀、铲刀、剃毛刀、皮屑刮刀、挑刀、锋刀、修脚刀、修甲刀、解剖刀等》第二类医疗器械:(a) 普通诊察器械类(6820):体温计、血压计;(b) 物理治疗及康复设备类(6826):磁疗器具;(c) 临床检验分析仪器类(6840):家庭用血糖分析仪及试纸;(d) 手术室、急救室、诊疗室设备及器具类(6854):医用小型制氧机手提式氧气发生器;(e) 医用卫生材料及敷料类(6864):匡用脱脂棉、医用脱脂纱布;(f) 医用高分子材料及制品类(6866):避孕套、避孕帽等第三类医疗器械:A、一次性使用无菌医疗器械1、一次性使用无菌注射器;2、一次性使用输液器;3、一次性使用输血器;4、一次性使用麻醉穿刺包;5、一次性使用静脉输液针;6、一次性使用无菌注射针;7、一次性使用塑料血袋;8、一次性使用采血器;9、一次性使用滴定管式输液器。
《医疗器械经营质量管理规范》培训试卷及答案

《医疗器械经营质量管理规范》培训试卷姓名:分数:一、填空题(每空2分,共50 分)1、《医疗器械经营质量管理规范》(2014年第58号),自公布起施行。
2、医疗器械经营企业应当在采购、验收、贮存、销售、运输、售后服务等环节采取有效的保障经营过程中产品的质量安全。
3、企业法定代表人或者负责人是医疗器械经营质量的要的条件,保证质量管理机构或者质量管理人员有效履行职责,确保企业按照本规范要求经营医疗器械。
4、企业质量负责人负责医疗器械质量管理工作,在企业内部对医疗器械质量管理具有。
5、进货查验记录和销售记录应当保存至医疗器械有效期后年;无有效期的,不得少于年。
植入类医疗器械进货查验记录和销售记录应当保存。
6、在库房贮存医疗器械,应当按质量状态采取控制措施,实行分区管理,包括待验区、合格品区、不合格品区、发货区等,并有明显区分(如可采用色标管理,设置待验区为、合格品区和发货区为、不合格品区为),退货产品应当单独存放。
7、库房温度、湿度应当符合所经营医疗器械说明书或者标签标示的要求。
对有特殊温湿度贮存要求的医疗器械,应当配备有效调控及监测温湿度的或者。
8、企业应当对冷库以及冷藏、保温等运输设施设备进行验证,并形成验证控制文件,包括验证方案、报告、评价和预防措施等,相关设施设备停用重新使用时应当进行验证。
9、经营第三类医疗器械的企业,应当具有符合医疗器械经营质量管理要求的经营的产品可追溯。
10、企业收货人员在接收医疗器械时,应当核实运输方式及产品是否符合要求,并对照相关采购记录和随货同行单与到货的医疗器械进行核对。
交货和收货双方应当对交运情况当场签字确认。
对不符合要求的货品应当立即报告质量负责人并。
11、收货人员对符合收货要求的医疗器械,应当按品种特性要求放于相应区域,或者设置状态标示,并通知验收人员进行验收。
需要冷藏、冷冻的医疗器械应当在内待验。
12、对需要冷藏、冷冻的医疗器械进行验收时,应当对其运输方式及运输过程的货温度等质量控制状况进行重点检查并记录,不符合温度要求的应当拒收。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
传真
签字
(盖章)
注:如填写内容可另附页
有源□
无源□
体外诊断试剂 □
预期用途(包括适用人群,禁忌症等)
结构特征
作用原理或机理
使用形式、状态、部位、期限及方法
材料特性
主要原材料、生产工艺及反应体系等相关特性的信息(体外诊断试剂)
产品主要风险点
其他需要说明的内容
申请息
联系人
电话
地址(邮编)
编号:
医疗器械 产 品 分 类 界 定
申请表
产品名称:
申请单位:
国家食品药品监督管理局制
填 表 说 明
1.本申请表应打印。填写内容应完整、清楚、整洁,不得涂改。
2.本申请表可从如下或免费下载。
以下栏目由申请单位填写
产品名称
(中/英)
产品类型