序列比对,构建进化树教学提纲
多重序列比对及系统发生树的构建

多重序列比对及系统发生树的构建【实验目的】1、熟悉构建分子系统发生树的基本过程,获得使用不同建树方法、建树材料和建树参数对建树结果影响的正确认识;2、掌握使用Clustalx进行序列多重比对的操作方法;3、掌握使用Phylip软件构建系统发生树的操作方法。
【实验原理】在现代分子进化研究中,根据现有生物基因或物种多样性来重建生物的进化史是一个非常重要的问题。
一个可靠的系统发生的推断,将揭示出有关生物进化过程的顺序,有助于我们了解生物进化的历史和进化机制。
对于一个完整的进化树分析需要以下几个步骤:⑴ 要对所分析的多序列目标进行比对(alignment)。
⑵ 要构建一个进化树(phyligenetic tree)。
构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。
而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。
进化树枝条的长度代表着进化距离。
独立元素法包括最大简约性法(M aximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。
⑶ 对进化树进行评估,主要采用Bootstraping法。
进化树的构建是一个统计学问题,我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。
如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的"进化树"。
植物基因家族进化树的构建

植物基因家族进化树的构建一、数据收集在构建植物基因家族进化树之前,需要收集相关的基因序列数据。
这些数据可以通过各种数据库,如NCBI、Ensembl等获取。
在收集数据时,需要注意以下几点:1. 选择具有代表性的物种,覆盖尽可能多的系统发育分支;2. 确保所收集的基因序列数据质量可靠,无测序错误和拼接错误;3. 对于每个基因家族,应尽可能收集多个成员的序列,以便进行多序列比对和树的构建。
二、序列比对在获得基因序列数据后,需要进行多序列比对。
比对的目的是为了找到不同物种间基因序列的相似性和差异性,从而确定它们之间的系统发育关系。
常用的多序列比对软件有MUSCLE、CLUSTAL W等。
在进行多序列比对时,需要注意以下几点:1. 选择合适的比对参数,以保证比对结果的准确性和可靠性;2. 在比对过程中,需要注意保持基因序列的原始阅读框,避免引入不必要的拼接错误;3. 对于较长的基因序列,可以分段进行比对,以提高计算效率和准确性。
三、距离矩阵计算在多序列比对的基础上,需要计算不同物种间基因序列之间的距离。
距离矩阵的计算是树构建的重要步骤之一。
常用的距离矩阵计算方法有:1. 欧氏距离法:直接计算不同物种间基因序列的差异数目,得到距离矩阵;2. Kimura距离法:基于Kimura模型计算不同物种间基因序列的差异概率,得到距离矩阵;3. Jukes-Cantor距离法:考虑基因序列的突变率和进化速率,计算不同物种间基因序列的差异概率,得到距离矩阵。
在选择距离矩阵计算方法时,需要根据具体情况选择适合的方法。
如果数据量较大或序列较短时,可以考虑使用欧氏距离法;如果数据量较小或序列较长时,可以考虑使用Kimura或Jukes-Cantor距离法。
四、树构建方法选择在获得距离矩阵后,需要选择合适的树构建方法来构建进化树。
常用的树构建方法有:1. UPGMA(Unweighted Pair Group Method with Arithmetic Mean):将距离矩阵中的行或列进行聚类分析,根据聚类结果构建树;2. Neighbor Joining:基于距离矩阵中的最近邻关系构建树;3. Maximum Parsimony:基于树的构建准则函数(如最小改变数、最小代价等)构建树。
系统进化树的构建方法

系统进化树的构建方法系统进化树(systematic phylogenetic tree)是用于描述不同物种之间进化关系的一种图形化表示方法,可以帮助我们理解物种的起源、演化和分类。
构建系统进化树主要涉及到物种的分类学和进化生物学知识,以及系统发育分析方法。
下面将介绍系统进化树的构建方法。
1.选择研究对象:确定研究的物种范围,通常会选择有代表性的物种,包括已知的和新发现的物种。
2.收集DNA序列数据:从每个研究对象中提取DNA样本,并通过PCR扩增得到所需的基因序列。
常用的基因包括线粒体基因COI、核基因ITS 等,根据具体研究目的和对象进行选择。
3.序列比对:将收集到的DNA序列进行比对,通常采用计算机程序进行全局比对,比对结果会显示序列之间的同源区域和差异。
4. 构建系统进化树:有多种方法可以构建系统进化树,其中最常用的是系统发育建模方法,如最大简约法(maximum parsimony)、最大似然法(maximum likelihood)和贝叶斯推断(Bayesian inference)等。
最大简约法是最简单和最常用的构建系统进化树的方法之一、它基于简约原则,认为进化过程中最少的演化步骤是最可能的。
方法将不同物种的序列进行比对,统计共有的字符以及不同的字符,根据最小化改变的原则,得到进化树。
最大似然法使用概率模型来计算物种之间的进化关系,根据序列数据的概率分布确定最可能的进化树。
这种方法考虑了不同序列字符的不同演化速率以及序列之间的相关性。
贝叶斯推断方法基于贝叶斯统计学原理,通过计算不同进化树的后验概率来确定最有可能的进化树。
该方法能够对不同进化模型和参数进行全面的推断,但计算复杂度较高。
5.进行分支长度调整和进化树根的定位:进化树的分支长度表示物种间的差异,可以根据各个物种间的差异大小进行调整。
进化树的根通常是已知的进化历史或已知的进化事件,如灭绝事件等,可以通过分析群体间的基因流动等信息进行推断。
序列比对,构建进化树
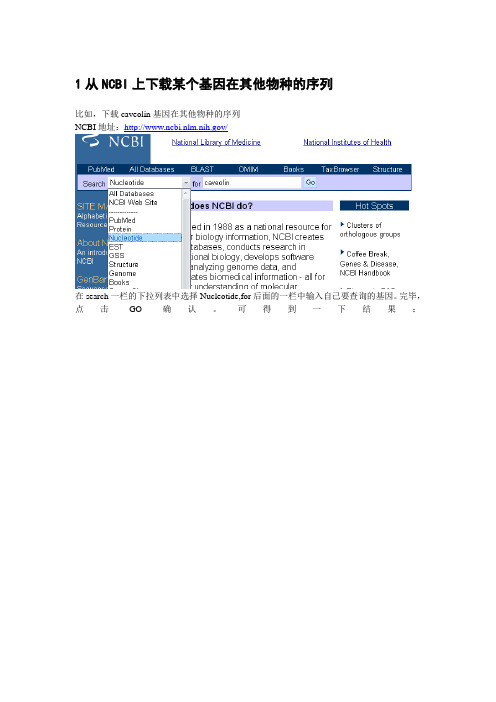
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
生物信息学中的序列比对与进化树构建算法研究

生物信息学中的序列比对与进化树构建算法研究序列比对是生物信息学中重要的分析方法之一,通过比对不同生物种类的DNA、RNA或蛋白质序列,可以揭示它们之间的相似性和差异性,并为分析进化关系、功能预测等提供基础。
序列比对的基本思想是将两个或多个序列进行比对,并找出它们之间的相似性。
在序列比对中,常用的方法有全局比对、局部比对和多序列比对。
全局比对方法是将整个序列进行比对,一般采用Needleman-Wunsch算法或Smith-Waterman算法。
这些算法根据序列间的单个碱基或氨基酸之间的匹配、错配和缺失情况,计算出序列的相似度得分。
全局比对方法适用于较短的序列,优点是能够找到完全匹配的区域,但是对长序列不适用,计算复杂度较高。
局部比对方法主要用于比对较长的序列或存在较大插入缺失的序列。
常用的算法有BLAST和FASTA算法。
这些算法采用快速搜索的策略,先找出序列间的高度相似的片段,然后再进行比对和分析。
局部比对方法能够找到较长序列内的相似片段,但可能无法找到全局的最优比对。
多序列比对方法用于比对三个或更多序列,揭示它们之间的共同特征和区别。
常用的方法有多重序列比对和进化树构建。
多重序列比对旨在将多个序列按照匹配和错配的原则进行比对,以找到共同的序列区域。
进化树构建方法基于序列的相似性和进化关系,将多个序列构建成进化树,以揭示它们之间的进化关系。
在序列比对的过程中,常用的比对算法还包括Pairwise比对、局部比对、多重比对等方法。
这些方法都有自己的特点和适用范围,根据具体的研究目的和数据特点选择合适的方法进行序列比对。
进化树构建是生物信息学中的重要研究方向之一,用于揭示不同生物种类之间的进化关系。
进化树是一种图形化的表示方式,能够清晰地展示物种间的分支关系、共同祖先以及进化时间。
进化树的构建主要基于序列的相似性和进化关系。
在进化树构建中,常见的方法包括距离法、最大简约法和最大似然法。
距离法基于序列间的距离矩阵,通过测量序列间的差异程度来构建进化树。
生物信息学中的序列比对与进化树构建

生物信息学中的序列比对与进化树构建生物信息学是一门涉及生命科学和计算科学的交叉学科,其应用在分子生物学、生物医学、生态学、进化论、生物技术等诸多领域中。
序列比对和进化树构建是生物信息学的重要组成部分,是理解生物学进化的重要途径之一。
一、序列比对序列比对是将两个或多个蛋白质或核酸序列究竟有多少相同、多少不同进行比较的过程。
序列比对在生物学中极其重要,因为它可以帮助科学家确定两个生物物种之间的相似性,进而推断它们之间的亲缘关系以及共同祖先的时间。
序列比对中最基础和常用的方法是全局比对和局部比对。
全局比对试图比较两个序列的完整长度,一般用于比较相似性较高的序列,它最先被应用于分析DNA和蛋白质,是序列比对过程中最古老、最经典的算法方法。
而局部比对则更注重比较两个序列中的相似区域,忽略其中任何间隔,通常用于比较两个较短的序列或者两个相对较不相关的序列。
例如,在核酸序列比对中,这种算法更适用于获取多个剪接变异或者重复序列之间的相似性。
另外,序列比对有一个关键问题,就是如何准确的衡量两条序列的相似性和相异性。
在这方面有很多方法,例如编辑距离、盒子型、PAM矩阵、BLOSUM 矩阵等等,其中都采用了不同的评分标准。
二、进化树构建进化树(Phylogenetic Tree)是用来表示生物物种间亲缘关系的结构,也称演化树或家谱树。
进化树是通过对基于DNA和RNA等生物分子序列进行分析,推导出各物种之间共同祖先的关系构建起来的,同时它也综合了形态、系统和分子信息等其他生物学数据。
进化树的构建过程中涉及许多算法,其中最基础的是贪心算法。
贪心法从序列的最初状态开始,一步步选择最佳的演化路径,最终得到最优的进化树;而Neighborhood-joining (NJ)算法则是以序列之间的 Jukes-Cantor 模型距离或 Kimura 二参数模型距离为基础,使用最小进化步骤(Minimum Evolution,ME)标准构建进化树,是目前应用比较广泛的算法。
序列比对,构建进化树
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:/在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
基因进化树的构建

基因进化树的构建
基因进化树(Phylogenetic tree)是用来描述不同物种或个体之间基因演化关系的一种图形表示方法。
构建基因进化树可以帮助我们了解物种之间的亲缘关系和演化历史。
以下是构建基因进化树的一般步骤:
1.收集基因序列数据:首先,需要收集感兴趣物种或个体的基因序列数据。
这些基因序列可以是DNA序列、蛋白质序列或其他分子标记。
2.序列比对:将收集到的基因序列进行比对,找出相同的区域。
这可以通过使用比对算法(如ClustalW、MAFFT等)来完成。
比对后的序列将有助于确定物种或个体之间的相似性。
3.构建进化模型:选择适合你的数据的进化模型。
进化模型描述了基因在演化过程中的变化方式。
常见的进化模型包括Jukes-Cantor模型、Kimur a模型、GTR模型等。
选择适当的模型可以提高进化树的准确性。
4.构建进化树:使用构建进化树的方法,如最大似然法(Maximum Li kelihood)、贝叶斯推断(Bayesian Inference)或距离法(Distance-based m ethods)来构建进化树。
这些方法基于序列的相似性和进化模型来计算物种或个体之间的进化距离或相似性。
5.进化树评估和解释:评估构建的进化树的可靠性和准确性。
可以使用统计方法(如Bootstrap分析)来评估节点的支持度。
解释进化树的结果,包括物种或个体之间的亲缘关系和演化历史。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
序列比对,构建进化树
1从NCBI上下载某个基因在其他物种的序列
比如,下载caveolin基因在其他物种的序列
NCBI地址:/
在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:
每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,
称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)
表示此序列为cDNA,不含内含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:
现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:
可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:
并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)
你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:
点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找范围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:
找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
注:此处比对得两个序列一个是你实验得到的基因序列,另一个是从NCBI下载的其他物种的序列,一般情况下你试验得到的测序序列会短于下载的其他物种的序列,所以要在这里进行下载的序列的编辑。
把你已经编辑好的序列按照上述比对方法(用你的实验得到的序列和同基因在其他物种的序列(并且是你已经掐头去尾编辑过的)两两比对,可以得出
某个基因在两两物种之间的同源性)如下图:
椭圆中的数字就是某基因在两个物种间的同源性。
另一种获得两物种某基因之间同源性的方法是:打开NCBI网站/
点击箭头指示的blast,得到:
点击箭头指示处
在1空白处黏贴上你的测序序列,2处选中,点击下面的blast
你将得到一个你测序序列与多个物种间某个基因的同源性。
如下图:
1为物种名称及描述,2为同源性
为了进行比对,要把序列格式转化成FASTA格式。
转化方法:新建TXT格式
文档,
第一排开始先写一个大于号(>),紧接着是写上物种名称。
如上图。
另起一行,把该物种的某基因的序列拷贝上。
注:这里的要拷贝过来的序列是经过上一步的掐头去尾的序列,得到:
然后保存。
方法:文件-另存为-保存类型选所有文件-文件名:物种名称.Fasta 3 序列比对与进化树构建,(可拷贝到你的论文中的图片)
打开Clustaxl软件,
单击在左上角的文件,载入序列,就可载入一条fasta格式的序列了。
载入完一条之后再点击添加序列就可载入第二条序列,后面要载入更多的序列也是点击添加序列。
载入完你要比对的序列后点击编辑下的选定全部序列,接着点击比对-进行完全比对。
得到:点击对齐
1是进化树文件的保存路径和名称,2是序列比对文件的路径和名称。
保存完这两个文件,可以用MEGA软件分别打开它们。
下图为序列比对文件打开后的界面:
1为选定的格式,2为文件保存的名称和路径。
用MEGA软件打开此文件。
得到这样一个界面:
1是指物种名称,2处的星号是指物种间同源的序列部分,星号空缺的地方是指不一样的碱基。
把全部序列比对的界面一次截屏黏贴到你论文中。
下面打开的是进化树的文件,界面如下:
可以通过截图把下面这个图拷贝到你论文上,
也可通过
点击1,2,两个下拉菜单,把改图拷贝到你的画图(系统工具,在程序-附件-画图,打开画图之后点击编辑黏贴,就可把改图拷贝到你的画图上,在把改图拷贝到你的论文中)上。
终于完事了~~~~~~~~~~·······。