心脏发育异常相关罕见病的分子遗传学进展
表观遗传学上的新进展

表观遗传学上的新进展表观遗传学是研究基因表达和调控的一门学科。
近年来,随着技术的不断进步,表观遗传学研究取得了许多新的进展,为人类的健康和疾病诊治提供了新的思路和方法。
一、表观遗传学的基本概念表观遗传学是指影响基因表达和继承的非DNA序列遗传信息传递。
表观遗传学的研究对象包括DNA甲基化、组蛋白修饰、染色质重塑和非编码RNA等。
DNA甲基化是指甲基化酶将甲基基团添加到DNA分子的胞嘧啶残基上,从而影响基因的表达。
组蛋白修饰是指通过化学修饰组蛋白蛋白质表面上的氨基酸残基来改变染色质的结构和功能,进而影响基因表达。
染色质重塑是指通过重塑染色质的结构和构象来影响基因表达和调控。
非编码RNA是指不能被翻译成蛋白质的RNA分子,它们可以通过多种机制来影响基因表达和调控。
二、表观遗传学的新进展1、单细胞表观遗传学传统的表观遗传学研究往往受到细胞异质性的限制,无法全面揭示基因表达和调控的复杂性。
但是,随着单细胞技术的逐步成熟,研究者已经可以对单个细胞进行表观遗传学的研究。
这种单细胞表观遗传学的方法可以更准确地描绘不同细胞类型和状态下基因表达和调控的差异,为精准医学的发展提供了新的理论和方法支持。
2、表观遗传学与遗传性疾病的关系表观遗传学的异常会导致一系列遗传性疾病的发生。
例如,DNA甲基化异常与许多遗传性疾病密切相关,如罕见病Gyrate atrophy和某些细胞周期异常等。
同时,许多复杂疾病如癌症、自闭症、肥胖症和心血管疾病等也受到表观遗传学的影响。
3、表观遗传学在肿瘤治疗中的应用表观遗传学的研究已经为肿瘤诊治提供了新的指导和思路。
例如,一些表观遗传学的药物如DNA甲基转移酶抑制剂和组蛋白去乙酰化酶抑制剂等已被广泛应用于癌症治疗中。
此外,表观遗传学研究也发现了许多肿瘤驱动基因的表观遗传机制,为肿瘤特异性治疗和药物研发提供了新的思路。
4、表观遗传学与环境因素的互动人类在不断变化的环境中生存,环境因素对人类健康和疾病的发生有着重要的影响。
家族性高胆固醇血症,细说这种绝对吃心的遗传病

家族性高胆固醇血症,细说这种绝对吃心的遗传病家族性高胆固醇血症(FH)作为一种常染色体(共)显性遗传病,其主要特征表现为血清低密度脂蛋白胆固醇(LDL-C)水平明显升高,多部位皮肤黄色瘤形成,以及早发动脉粥样硬化性心血管疾病(ASCVD)。
该病其实并不罕见,但患病率却一直被低估;虽然降脂治疗在不断发展,但该病的治疗率却非常不乐观。
FH患者终生受累于高水平LDL-C的困扰。
早筛查、早治疗可以降低FH患者早发ASCVD 的风险,改善存活率。
本期将从临床表现、筛查诊断以及治疗等各个方面带你认识这种绝对'吃'心的遗传代谢性疾病:FH。
一.概况FH是由于低密度脂蛋白在肝脏代谢有关的基因发生致病性突变所致,可以分为杂合子(HeFH)、纯合子(HoFH)、复合杂合子和双重杂合子这四种类型,以杂合子型最为常见。
杂合子型FH在总人群中的患病率约为1/137,患者的动脉粥样硬化进程加速5倍,较非FH患者的冠心病发病风险增加15倍;在早发心肌梗死患者中FH的患病率高达7.1%。
纯合子型FH则是一种严重的罕见病,患病率在1/100万到3/100万,患者的动脉粥样硬化进展更早更快,可在儿童及青年期发生心绞痛或心肌梗死,并于20~30岁之前死亡。
FH发病的主要机制在于基因突变导致低密度脂蛋白受体(LDLR)表达缺失或功能异常,使血清LDL无法在肝脏得到有效清除,进而导致血清总胆固醇和LDL-C浓度升高,并在组织内过度蓄积,最终出现动脉粥样硬化等临床症状。
目前已检测到的致病基因包括编码LDLR、载脂蛋白B(Apo B)、前蛋白转换酶枯草溶菌素9(PCSK9)、LDL 受体衔接蛋白1(LDLRAP1)等多个基因。
其中以LDLR基因突变最为常见,该基因突变直接导致LDLR表达缺失及功能障碍。
Apo B是LDLR的主要配体,该基因突变将阻碍LDLR与血清LDL正常结合。
PCSK9基因突变会导致LDLR被过度降解以及肝细胞内胆固醇的加速合成。
法布里病教学演示课件

介绍适用于法布里病家庭的辅助生殖技术,如胚 胎植入前遗传学诊断(PGD)等。
3
遗传咨询在生育过程中的作用
强调遗传咨询在生育决策中的重要性,帮助家庭 理解并接受相关风险。
家系调查和遗传资源保护
家系调查
01
开展法布里病家系调查,收集患者及其家庭成员的临床和遗传
信息。
遗传资源保护
02
建立法布里病遗传资源库,保存患者及其家庭成员的生物样本
遗传咨询服务内容
01
疾病遗传方式解释
详细解释法布里病的遗传方式 ,包括X染色体连锁隐性遗传
的特点。
02
风险评估
根据家族史和遗传背景,评估 个体及家庭成员患病风险。
03
遗传检测建议
提供遗传检测相关信息,如检 测时机、方法及意义等。
生育建议及辅助生殖技术介绍
1 2
生育建议
针对法布里病患者及其家庭成员的生育需求,提 供个性化生育建议。
多学科协作诊疗
法布里病涉及多系统、多器官受累,未来多学科协作诊疗模式将得 到更广泛应用,提高患者生存质量。
社会支持与关注
随着社会对罕见病认知的提高和政策的不断完善,法布里病患者将获 得更多社会支持和关注,推动疾病的诊疗和管理水平不断提升。
THANKS
和遗传信息。
家系调查和遗传资源保护的意义
03
阐述家系调查和遗传资源保护在疾病研究、药物研发和临床试
验等方面的重要性。
06
研究进展与未来展望
国内外研究现状概述
基因突变研究
法布里病是一种由X染色体上基 因突变引起的遗传性疾病,国内 外学者在基因突变筛查、基因型 与表型关系等方面取得了重要进
展。
临床表现与诊断
一例Cornelia de Lange综合征患儿的致病基因突变分析
·60·《发育医学电子杂志 》2021年1月 第9卷 第1期 J Dev Med (Electronic Version),Jan 2021,V ol.9,No.1患儿,男,4岁2个月,因智力低下,身材矮小就诊。
患儿为孕1产1,足月顺产,出生时有窒息史。
婴儿期喂养困难,易吐奶(呈喷射状),进食少。
身高、体重增长缓慢,头较小。
2岁多才能独立行走、开始叫爸妈,现只会说单个字。
查体:身高92 cm(-3.48标准差),体重12.5 kg(-2.56标准差),头围46.2 cm(-3.15标准差)。
特殊面容:睫毛长,双侧眉毛浓密,弓形眉,连眉,鼻头小而翘,人中长略显外凸,嘴唇薄。
血常规未见异常,血生化提示血清25-羟基维生素D 含量偏低,生长激素激发试验正常。
腹部彩色超声肝、脾、肾未见异常,头颅MRI 平扫未见异常,垂体高径约3 mm,其内信号欠均,形态未见异常。
数字X 线胸部正侧位片和脊柱正位片未见异常,左手正位片示左侧桡骨远端、尺骨近端、诸腕骨及第1~2掌骨、第2~5掌指骨发育程度与标准骨龄片比较,符合2岁男孩标准。
染色体核型未见染色体数目结构异常。
智能发育筛查测试(developmental screening test,DST )结果显示发育商<49(异常)和智力指数<44(异常)。
父母体健,否认近亲结婚,家族中无类似患者。
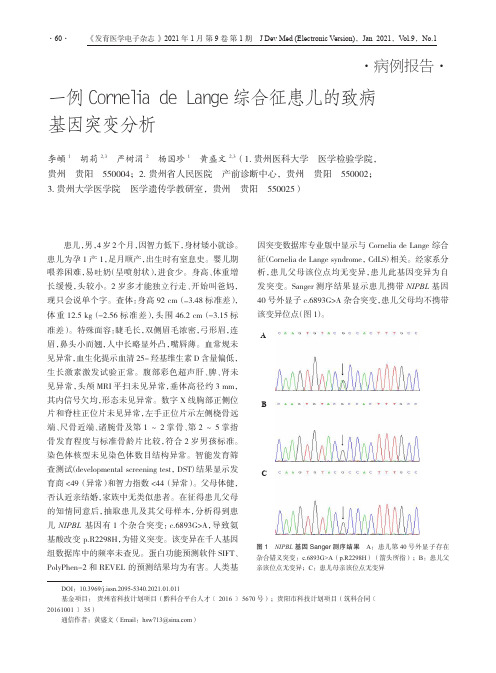
在征得患儿父母的知情同意后,抽取患儿及其父母样本,分析得到患儿 NIPBL 基因有1个杂合突变:c.6893G>A,导致氨基酸改变 p.R2298H,为错义突变。
该变异在千人基因组数据库中的频率未查见。
蛋白功能预测软件SIFT、PolyPhen-2和REVEL 的预测结果均为有害。
人类基一例Cornelia de Lange 综合征患儿的致病基因突变分析李頔1 胡莉2,3 严树涓2 杨国珍1 黄盛文2,3(1.贵州医科大学 医学检验学院,贵州 贵阳 550004;2. 贵州省人民医院 产前诊断中心,贵州 贵阳 550002;3. 贵州大学医学院 医学遗传学教研室,贵州 贵阳 550025)·病例报告·DOI :10.3969/j.issn.2095-5340.2021.01.011基金项目: 贵州省科技计划项目(黔科合平台人才﹝2016﹞5670号);贵阳市科技计划项目(筑科合同﹝20161001﹞35)通信作者:黄盛文(Email :***************)因突变数据库专业版中显示与 Cornelia de Lange 综合征(Cornelia de Lange syndrome,CdLS )相关。
微缺失和微重复综合征ppt课件

胸腺发育不良: 60% 胸腺不发育: 15-20% 特点: 数量上:22q11DS患者的平均T细胞数在婴儿期仅为正常值的50%,在成人期为正常值的80%。 质量上:减少的T细胞主要为CD4/CD25 T细胞,其数量大大减少,但是单个T细胞的功能并没有受太多影响。 对体液免疫的影响:2-4%的患者出现IgA的减少,约10%的患者IgG生成延迟,但是患者体内T细胞的分化过程和其他抗体的生成过程均正常。
22q11微缺失综合征:2主要临床表现 免疫缺陷
*
17-60%的患者伴有低钙血症: 原因:甲状旁腺发育不良、甲状旁腺素分泌功能低下 表现:该症多见于1年内的新生儿,患儿多出现惊厥、喉痉挛、手足抽搐等低钙表现。 注意:由于导致低钙血症的原因较少,故如出现新生儿低钙血症需高度怀疑22q11DS。有报道在成年患者中也发现有低钙血症者。 将近4%的患者发现有生长激素的缺乏,患者往往表现为身 材矮小。另有报道发现有甲状腺功能低下及垂体功能低下 者,这是导致患者生长发育迟缓的主要原因之一。
*
22q11微缺失综合征:3相关综合征
DiGeorge 综合征 ,DGS 腭心面综合征,VCFS 椎干异常面容综合征,CAFS/CTAF Cayler心面综合征 Opitz 综合征
相关综合征
*
DiGeorge综合征又称 家族性三、四咽囊综合征 胸腺甲状旁腺发育不良综合征 DiGeorge畸形
22q11微缺失综合征:2主要临床表现 免疫缺陷
3细胞免疫功能缺陷
*
患者常常出现反复的细菌、病毒感染,而且感染好转的时间延长,最常见的有 鼻窦炎(27%) 中耳炎(25%) 支气管炎(7%) 肺炎(4%) 9%的患者有自身免疫性疾病 青少年类风湿性关节炎 自身免疫性甲状腺炎 特发性血小板减少性紫癜 炎性肠病。
马方综合征演示课件

利用基因编辑技术建立马方综合征的动物模型,以模拟人 类疾病的发生和发展过程,为深入研究发病机制提供有力 工具。
诊断技术改进方向
1 2 3
基因检测技术的优化
提高基因检测的准确性和敏感性,降低假阳性和 假阴性率,为马方综合征的早期诊断和精准治疗 提供依据。
影像学诊断技术的创新
开发高分辨率、高灵敏度的影像学诊断技术,如 超声心动图、MRI等,以实现对马方综合征心血 管系统病变的精确评估。
05
并发症与风险
Chapter
心血管并发扩张,增加主动脉夹层或破裂
的风险。
心脏瓣膜病变
02
二尖瓣脱垂和主动脉瓣狭窄是常见的并发症,可能导致心脏功
能下降。
心律失常
03
患者可能出现各种心律失常,如心房颤动、室性心动过速等,
增加心脏性猝死的风险。
眼部并发症
晶状体脱位
马方综合征患者晶状体容易脱位,可能导致视力下降或复视。
高度近视
患者往往伴有高度近视,增加视网膜脱落的风险。
青光眼
马方综合征患者发生青光眼的风险增加,可能导致视力丧失。
其他器官受累风险
骨骼系统
患者可能出现脊柱侧凸、鸡胸等骨骼畸形,以及关节过度伸展和 韧带松弛等问题。
肺部
马方综合征患者肺部受累可能导致自发性气胸或肺大泡等问题。
推动科研与临床转化
鼓励科研机构和企业加大投入力度,推动马方综合征相关科研成果的 临床转化和应用,为患者提供更多有效的治疗选择。
THANKS
感谢观看
患者应定期接受眼科检查,并采 取必要的措施保护眼睛,如佩戴 合适的眼镜或隐形眼镜等。
06
研究与展望
Chapter
发病机制研究进展
OTC基因新变异致鸟氨酸氨甲酰基转移酶缺陷病1例报告
·论著·OTC基因新变异致鸟氨酸氨甲酰基转移酶缺陷病1例报告*闫红芳1 李 蒙2 蔡香然3 邓 梅1 宋元宗11.暨南大学附属第一医院儿科 (广东 广州 510630)2.广东省妇幼保健院新生儿科 (广东 广州 511400)3.暨南大学附属第一医院医学影像中心 (广东 广州 510630)【摘要】目的 报告1例鸟氨酸氨甲酰基转移酶缺陷病(ornithine transcarbamylase deficiency,OTCD)患儿的临床及分子遗传学特点,为本病诊疗提供参考。
方法 回顾性分析一例OTCD患儿临床和实验室资料。
结果 患儿女性,1岁6月,因“反复呕吐伴烦躁不安4月余”就诊。
查体发现双下肢张力减退,双侧膝关节反射亢进,双侧踝阵挛阳性。
血生化谷丙转氨酶、谷草转氨酶及血氨升高,尿液有机酸分析显示尿嘧啶、乳清酸和4-羟基苯乳酸水平升高。
遗传学分析在患儿OTC基因检出c.612_614del (p.Ile204del)新生变异,结合 ACMG标准判断该变异具有致病性。
经限制蛋白质摄入、精氨酸、瓜氨酸和苯甲酸钠等治疗后患儿病情控制仍不理想,于2岁时进行肝移植治疗,移植后患儿肝功能和血氨恢复正常。
结论 本文通过临床和遗传学研究,发现1个OTC新变异c.612_614del,确诊了一例OTCD患儿,为本病确诊和遗传咨询提供了遗传学标记物,同时为临床和实验室特征的科学认识积累了资料。
【关键词】尿素循环障碍;新变异;鸟氨酸氨甲酰基转移酶;X连锁遗传;高氨血症【中图分类号】R72【文献标识码】A【基金项目】广州市科技计划项目(202201020088)DOI:10.3969/j.issn.1009-3257.2024.5.001Late-onset Ornithine Transcarbamylase Deficiency Caused by a Novel Variant of the OTC Gene: A Case Report*Yan Hong-fang1, Li Meng2, Cai Xiang-ran3, Deng Mei1, Song Yuan-zong1.1.Department of Pediatrics,the First affiliated Hospital of Jinan University,guangzhou 510630,guangdong Province,China2.Department of neonatology, guangdong Women and Children Hospital,guangzhou 511400,guangdong Province,China3.Department of Medical imaging Center, the First affiliated Hospital of Jinan University,guangzhou 510630,guangdong Province,Chinaabstract: objectivedeficiency (oTCD), so as to help reference for the diagnosis and treatment of the disease. Methods The clinical and laboratory data of a pediatric patient with oTCD were retrospectively analyzed. Results: The patient was a female aged 1 year and 6 months with the complaint of "recurrent vomiting and restlessness over 4 months". Physical examination revealed bilateral hypotonia of the lower limbs, hyperreflexia of the knees, and clonus of the ankles. Laboratory analysis revealed elevated levels of serum ammonia as well as transaminases. on urinary organic acids analysis, a large quantity of uracil, orotate and 4-hydroxyphenyllactate was detected. The patient was heterozygous for the de novo variant c.612_614del(p.ile204del) in the oTC gene, which was evaluated “pathogenic” according to the aCMg standard and guideline. oTCD was diagnosed, and uptake restriction of proteins and oral administration of arginine, arginine citrulline and sodium benzoate were given. However, the response of the patient was not promising, and hence liver transplantation was underwent at her age of 2 years. as a result, the liver function and blood ammonia returned to normal. Conclusion Through clinical and genetic research, this study identified a novel oTC variant c.612_ 614del in a patient with oTCD. The findings provided genetic markers for the diagnosis of oTCD and genetic counseling in the affected family, and accumulated data for the scientific understanding of the clinical and laboratory characteristics of this disease.Keywords: Urea Cycle Disorder; Novel Variant; Ornithine Transcarbamylase; X -linked Inheritance; Hyperammonemia 尿素循环障碍(urea cycle disorders,UCD)指因参与尿素循环的酶和转运蛋白功能或结构缺陷引起的,导致氨无法通过尿素循环途径转化为尿素并排出体外,从而使血液中氨升高并引起一系列临床表现的一组遗传代谢病。
PRKAR1A基因突变导致肢端发育不全1型4例分析
基金项目:国家重点研发计划(2018YFC1002501);广西八桂学者专项(2016A20)作者简介:谌 飞,男,1992年生,硕士,技师,主要从事罕见病诊断及致病机制研究。
通信作者:罗静思,E-mail :。
PRKAR 1A 基因突变导致肢端发育不全1型4例分析谌 飞1, 覃再隆1, 陈少科2, 范 歆1, 李 川2, 易 赏1, 沈亦平1, 罗静思1(1.广西壮族自治区妇幼保健院遗传代谢中心实验室,广西 南宁 530002;2.广西壮族自治区妇幼保健院内分泌遗传病专科,广西 南宁 530002)摘要:目的 探讨4例由PRKAR 1A 基因突变导致的肢端发育不全1型患儿的临床表现和实验室检测结果。
方法 收集4例肢端发育不全1型患儿的临床资料和实验室检测结果,同时进行全外显子高通量测序,以临床表型为主要突变过滤依据,采用Sanger 测序验证突变来源,根据《遗传变异分类标准与指南》进行变异致病性分类。
结果 4例肢端发育不全1型患儿的主要临床表现为身材矮小(-3s ~-6s )、低体质量(-2s ~ -4s )、短指、特殊面容及多激素抵抗等,但不同患儿的临床表现有一定差异,部分患儿存在生长激素部分缺乏。
4例患儿均检出PRKAR 1A 基因突变,其中3例患儿为同一突变c.1102C>T/p.Arg368*,1例患儿为c.1118A>G/p.Tyr373Cys ,均为致病性变异。
结论 检出PRKAR 1A 基因的2个新发突变,由此导致的肢端发育不全1型患儿的临床表现存在异质性。
关键词:PRKAR 1A 基因;肢端发育不全1型;全外显子高通量测序Analysis of 4 cases of acrodysostosis type 1 caused by PRKAR 1A gene mutation CHEN Fei 1,QIN Zailong 1,CHEN Shaoke 2,FAN Xin 1,LI Chuan 2,YI Shang 1,SHEN Yiping 1,LUO Jingsi 1.(1. Genetic and Metabolic Central Laboratory ,Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region ,Nanning 530002,Guangxi ,China ;2. Department of Endocrinologye and Genetic Diseases ,Maternal and Child Health Hospital of Guangxi Zhuang Autonomous Region ,Nanning 530002,Guangxi ,China )Abstract :Objective To study 4 cases of acrodysostosis 1 caused by PRKAR 1A gene mutation based on their clinical manifestations and laboratory parameters. Methods Clinical data and laboratory testing results of 4 cases of acrodysostosis 1 were collected ,and whole exome high-throughput sequencing was performed. Phenotype-driven variant filters were performed to identify candidate variants ,and Sanger sequencing was used to verify the parental origin of the variants. The pathogenicity of genetic variation was classified according to Standards and Guidelines for the Interpretation of Sequence Variants . Results The main clinical manifestations of the 4 cases were short stature (-3s - -6s ),low weight (-2s - -4s ),brachydactyly ,dysmorphic facial features and multi-hormone resistance. Different clinical manifestations were observed among individuals ,and some patients had partial growth hormone deficiency. Pathogenic mutations in PRKAR 1A were detected in the 4 cases. Of them ,3 cases had the mutation c.1102C>T/p.Arg368*,and the one had the mutation c.1118A>G/p.Tyr373Cys. Conclusion Totally ,2 new mutations in PRKAR 1A have been detected ,and there is heterogeneity in the clinical manifestations of patients with acrodysostosis 1.Key words :PRKAR 1A gene ;Acrodysostosis type 1;Whole exome high-throughput sequencing文章编号:1673-8640(2021)02-0147-06 中图分类号:Q754 文献标志码:A DOI :10.3969/j.issn.1673-8640.2021.02.006PRKAR 1A 基因主要指导蛋白激酶A (protein kinase A ,PKA )调节亚单位的合成,是PKA 四聚体(2个调节亚单位、2个催化亚单位)的关键组成部分之一。
可治性罕见病—Silver-Russell综合征(SRS)
可治性罕见病—Silver-Russell综合征(Silver Russell Syndrome,SRS)一、疾病概述Silver-Russel综合征(Silver Russell Syndrome,SRS)是一种罕见的遗传异质性疾病,以严重的宫内及生后生长发育受限为主。
临床表现还有肢体不对称,颅面部畸形如三角脸,小下颌、牙列不齐等,一些还有第5小指向内弯曲等[1]。
在西方国家的发病率为1/30 000~1100 000[2],我国无发病率统计。
以往的研究表明该病主要由7号染色体母源性单亲二倍体[UPD7(mat)]和11p15区域母源性或父源性印记基因IGF2和H19表达缺陷所致,分别占7%~10%和38%~62%;另外约有1%的患儿在11p和7号染色体上有结构的微小改变,随着分子生物技术的进步,发现越来越多的印记基因表达异常可能与SRS有关,如CDKNIC和GRB10基因,我们发现的OSBPL5基因的低甲基化也可能会导致SRS的临床表现[3-7]。
另有近一半的患儿病因未明。
而北京儿童医院对25例病人研究发现llp15印记异常占50%[8]。
遗传学研究1. 第7号染色体的母源单亲二倍体已证实父亲表达的等位基因能够促进胎儿的生长友育,而母亲表达的等位基因则抑制生长发育。
UPD7( mat)时两条染色体上均表达母源性的等位基因,导致在胎儿期身材矮小[4]。
2. 11p15印记异常控制胎儿及出生后生长的2个主要印记区域ICRl和ICR2都位于llpl5区域,它们是由许多印记基因组成的基因簇:ICR1包含了母系表达非编码RNA 的H19基因和父系表达的编码胎儿生长因子IGF2基因;ICR2包含了母系表达的KCNQl和CDKNlC基因以及父系表达的KCNQl OT1基因。
印记异常会导致这些基因的表达异常。
IGF2位于H19的上游区域,两者之间有一甲基化差异区域(DMR)。
正常情况下,在母本是非甲基化的,而在父本是甲基化的。
可治性罕见病—范可尼贫血
可治性罕见病—范可尼贫血一、疾病概述范可尼贫血( Fanconi anemia,FA)是一常染色体隐性遗传和(或)X-连锁隐性遗传疾病,为先天性骨髓衰竭综合征中最常见的疾病,全球发病率为0. 3/10万[1]。
1927年Fanconi首次报道3例儿童再生障碍性贫血合并多发性先天畸形病例,故名为范可尼贫血[2]。
FA中位发病年龄为7岁,9%的FA患者直至成年期才被确诊,也有直至49岁才发病的患者,男:女约为2:1[3]。
目前,已经发现FANC -A、FANC -B、FANC -C、FANC - D1(BRCA2)、FANC -D2、FANC -E、FANC -F、FANC -G(XRCC9)、FANC -I( KIAA1794)、FANC -J(BRIP1)、FANC -L(PHF9/POG)、FANC -M(HEF)、FANC -N(PALB2)、FANC -O(RAD51C)、FANC -P (SLX4)、FANC -Q(XPF/ERCC4)、FANC -R( RAD51)、FANC -S(BRCA1)和FANC -T( UBE2T)等19个基因的异常可以造成FA,其中FANC -A突变最多见,约占70%,其次为FANC -C和FANC -G突变[4]。
这些基因编码的蛋白质参与了FA/BRCA功能网络,该功能网络与3条经典的基因损伤后DNA修复路径相关,分别是同源重组、核苷酸切除修复和经诱变的跨损伤DNA复制[5]。
在射线、有毒化学物质、细菌和病毒感染造成DNA损伤后,该损伤不能得到及时修复,造成突变的累积。
由于人体造血系统处于相对活跃的增殖状态,这些DNA损伤后易导致造血系统受累,表现为造血系统增殖低下,远期极易发生血液和其他系统的恶性转化[6]。
二、临床特征FA是一高度异质性的疾病,且疾病呈进展性,造成其临床表现多样化。
部分患者可仅有骨髓衰竭而无躯体畸形,或只有躯体畸形而无血液系统异常;患者常有阳性家族史,其自身和(或)家族成员恶性肿瘤发生率较高。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
・94・ 国际心血管病杂志2015年3月第42卷第2期 Int J CardiovascDis,M旦! , . , . 心脏发育异常相关罕见病的分子遗传学进展 盛伟黄国英 【摘要1心血管系统罕见病的发病率约为5/lo 000,由于其病因复杂,有效的治疗 药物较少,患者的死亡率较高。该文重点介绍心血管领域中与心脏发育异常相关罕见病 的研究进展。 【关键词1心血管疾病;罕见病;分子遗传机制 doi:10.3969/j.issn.1673—6583.2015.02.010
心血管系统罕见病定义为发病率低于5/10 000 的疾病或病变l1],绝大多数属于遗传性疾病,在婴幼 儿时期即发生,主要包括先天性心脏缺陷、先天性 血管畸形和遗传性代谢病等。 1先天性心脏缺陷 1.1 Holt—Oram综合征 Holt—Oram综合征又称为心手综合征(heart- hand syndrome),是一种罕见的常染色体显性遗传 病,在新生儿中的发病率约1/lO万,男性和女性发 病率大致相同,主要表现为先天性心脏缺陷和上肢 骨骼畸形_2]。在HOS综合征患者中,房间隔缺损 约占患者总数的60 ,其次是室间隔缺损,而其他 类型的心脏缺陷报道很少,如法洛四联症l3]、室问隔 缺损合并肺动脉狭窄、动脉导管未闭等[4]。 遗传学研究证实,TBX5基因突变可能是引起 该疾病发生的重要因素[5]。在Holt—Oram综合征 患者中,由TBX5基因突变引起的疾病占35 9/6,已 有超过70个TBX5基因突变点被证实与该病发生 有关_l6]。通过对患者的临床表型分析发现,不同的 患者具有不同的表型特征,这可能与TBX5基因突 变的类型、突变位点不同,对基因功能损害程度不 同有关_7]。随着研究的不断深入,TBX5基因的一 些新突变也陆续被发现_8 ]。TFAP2B基因在心脏 发育和骨骼发育中起重要调控作用,该基因突变影 响骨形成蛋白(BMP)2和BMP4的表达,导致心脏 发育缺陷和骨骼畸形l1。]。Holt—Oram综合征具有 基金项目:国家自然科学基金面上项目(81370198);上海市科学 技术委员会科研计划连续资助项目(1 1 JC140140O) 作者单位:201102上海,复旦大学附属儿科医院上海市出生缺 陷防治重点实验室 通信作者:黄国英,Email:gyhuang@shmu.edu.cn 遗传异质性,这种表型的差异也有可能是其他不同 基因发生突变或表观遗传异常所引起,这需要在研 究中进一步的证实。 1.2马凡综合征 马凡综合征(Marfan S syndrome)是一种常染 色体显性遗传的结缔组织疾病,具有家族遗传性, 发病率约为0.2‰l1 。临床特征为心脏二尖瓣关闭 不全或脱落,主动脉瓣关闭不全,大动脉中层弹力 纤维发育不全,主动脉或腹总主动脉扩张形成主动 脉瘤或腹总主动脉瘤,当主动脉扩张到一定程度 时,将造成患者主动脉破裂死亡_1 。患者同时还伴 有骨骼畸形,全身管状骨细长、手指和脚趾细长呈 蜘蛛脚样,眼睛晶体半脱落、视网膜剥离等现象_l1 。 目前,马凡综合征是一种无法治愈且伴随终生的疾 病,大多数患者最终死于心血管疾病ll1 。 FBN1基因或TGFBR2基因突变被证实是引 起该病的主要原因l_1 。FBN1基因位于人染色 体的15ql 5-21.3区域,编码结缔组织的原纤维 蛋白,该基因的突变引起蛋白功能异常,使原纤 维蛋白微丝不能正常装配,导致马凡综合征_1 。 ADAMTSL613蛋白是细胞外基质中的微丝相关蛋 白,在结缔组织的形成和再生中起重要调控作用, 该蛋白能够通过促进微丝的装配从而改善症状,为 治疗提供了新策略r-”]。TGFBR2基因位于染色体 3p24.1区域,它编码一种转化生长因子,在细胞外 基质中通过和FBN1相互作用发挥功能。该基因的 杂合突变与MFS2的发生有关l_1 。已发现在马凡 综合征患者中有500多种FBN1变异类型。 TGFBR2基因的变异较少,Chen等_1 9_通过全外显 子测序,在患者的TGFBR2基因发现3个突变位 点,其中1个突变位点与疾病发生有关。TGFB信 国际心血管病杂志2015年3月第42卷第2期 Int J Cardiovasc Dis,March 2015,Vo1.42,No.2 号通路异常可能是马凡综合征发生的另一个重要 原因E。‘ 。 2先天性血管畸形 2.1 特发性婴儿动脉钙化 特发性婴儿动脉钙化(idiopathic infantile arterial calcification,IIAC)是一种常染色体隐性遗 传病,迄今为止文献报道过的病例不超过300例,其 中85 的患儿在6个月内死亡,其临床特征是大中 型动脉,包括主动脉、冠状动脉、肾动脉,由于内膜 纤维增生而发生硬化,出现广泛性的血管闭塞,导 致血管的弹性减少和血流量降低,表现为顽固性高 血压,伴随心脏、呼吸衰竭_2 。根据文献报道,在胚 胎期就诊断出有IIAC的占48 ,52 9/6的患者是在 出生或死亡后确诊,冠状动脉硬化是最坏的预后指 标,绝大多数婴儿因心肌梗死而死亡l2 。8O 的 I1AC患者发生了ENPP1基因突变,表明这个基因 可能是致病基因l_2 。ENPP1基因属于ENPP基 因家族,编码一种跨膜糖蛋白——外核苷酸焦磷酸 酶/磷酸二酯酶1(ENPP1),这种蛋白能够通过水解 底物产生无机焦磷酸盐(PPi),在调控磷酸盐水平、 骨的矿化和软组织钙化中起重要作用。PPi能够防 止羟基磷灰石晶体在动脉管中沉积,ENPP1基因突 变致使机体内PPi缺失,导致心脏中动脉血管出现 不同程度的钙化和内膜纤维化而引起IIAC疾病的 发生跚。 2.2弯刀综合征 弯刀综合征(scimitar syndrome)又称镰刀综合 征,发病率约是1/lo万--3/10万,女性患病率高于 男性,具有家族遗传性 2 。患者右侧肺部的全部或 部分肺静脉形成共同肺静脉,经右侧肺门前方或后 方,从心包右侧下降,在右心房与下腔静脉交界处 呈弯刀状向左侧行进,引流人下腔静脉,其主要特 征是右肺发育不全。由于静脉畸形,X线检查可发 现沿右心缘有弯刀状阴影,心脏向右移位,近似右 位心[ 。临床表现包括明显的心力衰竭、肺动脉高 压、不同程度的呼吸困难和右肺感染等症状,如不 能及时诊断和治疗,常导致患者死亡[2 。 弯刀综合征的发病机制仍不清楚,可能由于 胚胎肺芽形成期间肺部血管移位,引起血管引流 异常;也有可能是胚胎期的静脉丛、大静脉系统 以及脐卵黄静脉系之间的连接残留所致。心脏 发生右移可能与肺容积缩小牵拉有关,为继发性 改变 2 。 ・ 95 ・ 3遗传性代谢病 3.1 高同型半胱氨酸血症 高同型半胱氨酸血症属于常染色体隐性遗传, 因甲硫氨酸代谢障碍而引起血中同型半胱氨酸水 平异常增高,是多种心血管疾病的危险因素__3 。同 型半胱氨酸是甲硫氨酸的中间代谢产物,在人体内 有两条代谢途径:一是在胱硫醚缩合酶和胱硫醚酶 的催化下生成半胱氨酸,此过程中需要维生素B6的 参与,或经巯基氧化结合生成高胱氨酸;另一条途 径是在叶酸和维生素B 。的辅助作用下,由甲硫氨 酸合成酶催化发生甲基化重新合成甲硫氨酸,此过 程中甲基的供体是N5一甲基四氢叶酸,是四氢叶酸 经5,10一甲烯四氢叶酸还原酶催化而产生的 。 甲硫氨酸代谢异常的原因包括遗传因素和环 境营养因素。遗传因素主要是指编码甲烯四氢叶 酸还原酶(MTHFR)、胱硫醚缩合酶(CBS)、甲硫氨 酸合成酶(MS)的基因发生突变或缺失,而环境营养 因素是指代谢辅助因子如叶酸、维生素B6、B 缺 乏。Sukla等_3 ]研究发现,MTHFR C677T基因多 态性与高同型半胱氨酸血症的发生有显著的相关 性,MTHFR C677T突变使血浆中同型半胱氨酸水 平升高,通过补充维生素B1。和叶酸能够有效缓解 症状。胱硫醚缩合酶基因缺失小鼠通过添加维生 素B能够显著降低同型半胱氨酸水平,小鼠的动脉 粥样硬化得到一定程度改善l_3 。甲硫氨酸合成酶 是由MTR基因编码的蛋白,能够将血浆中同型半 胱氨酸再次甲基化生成甲硫氨酸,在维持血浆同型 半胱氨酸正常浓度中起重要作用。MTR基因突变 导致甲硫氨酸合成酶活性降低,使血浆中同型半胱 氨酸不能正常代谢降解,导致高同型半胱氨酸血 症口 。此外,表观遗传研究发现,高同型半胱氨酸 血症与基因的甲基化程度有关,血浆高同型半胱氨 酸能够降低DNA甲基转移酶1(DNMT1)活性,使 与心血管发育有关的基因甲基化异常,引起动脉粥 样硬化或心脏损伤l3 。应用去甲基化药物使高同 型半胱氨酸血症小鼠DNA甲基化程度降低,可以 改善主动脉重塑,缓解高血压_3 。 目前,对高同型半胱氨酸血症的治疗包括血液 透析、维生素治疗和联合血液净化疗法等。 3.2法布里病 法布里病(Fabry disease)是一种罕见的溶小体 储积症,属X_染色体连锁的隐性遗传病,在新生儿 中的发生率约为1/120 000 1/40 000_3 。研究表 ・96・ 际心血管病杂志2015年3月第42-? ̄第 nt』 rdi ! ! ! :丝! : 明,GLA基因突变是导致法布里病的主要因素。 GLA基因突变致使其编码的一种溶小体酵 素——A型a半乳糖苷酶(a—Gal A)功能异常,使糖 脂质受体(GL-3)无法正常分解而堆积在全身血管 或其他的组织器官中,最终导致组织或器官发生病 变,主要包括心脏、脑和肾脏疾病_3 。心脏病发生 风险随着年龄增长显著增加,最常见的是高血压和 心肌病,至疾病后期可能引发心绞痛、充血性心力 衰竭或心肌梗死,导致患者死亡l_3 。迄今为止,已 有超过600个GLA基因突变被证实与法布里病有 关,包括错义突变、无义突变和不同程度的缺失 等…。Wang等_41]发现,GLA基因的第二个外显 子在先证者中发生错义突变,精氨酸被替换成半胱 氨酸,导致 —Gal A的酶活性降低。 法布里病可以通过检测a—Gal A的酶活性进行 诊断。然而,女性患者由于X染色体失活的随机 性,单独用这种方法并不可靠。GLA基因的大部分 突变具有家族特异性,一旦确认患者的基因序列发 生突变,同样的突变也可能存在于其他未发病的家 族成员中_4 。随着基因工程技术的进步,酶替代疗 法目前已成功用于临床治疗,能够有效改善患者的 症状,延长患者寿命,提高生活质量L4 。 4展望 罕见病由于发病率低,现在面临着许多医学和 社会学问题。由于病例较少,样本难以获得,相关 病因学和发病机制等方面的研究也不多。目前,许 多国家或地区都颁布了罕见病的法律法规,通过在 药品生产、医疗保障、社会保险等多方面的强制规 定以提高罕见病的研究、诊断和治疗水平。