欧盟医疗器械灭菌产品要求的标准清单
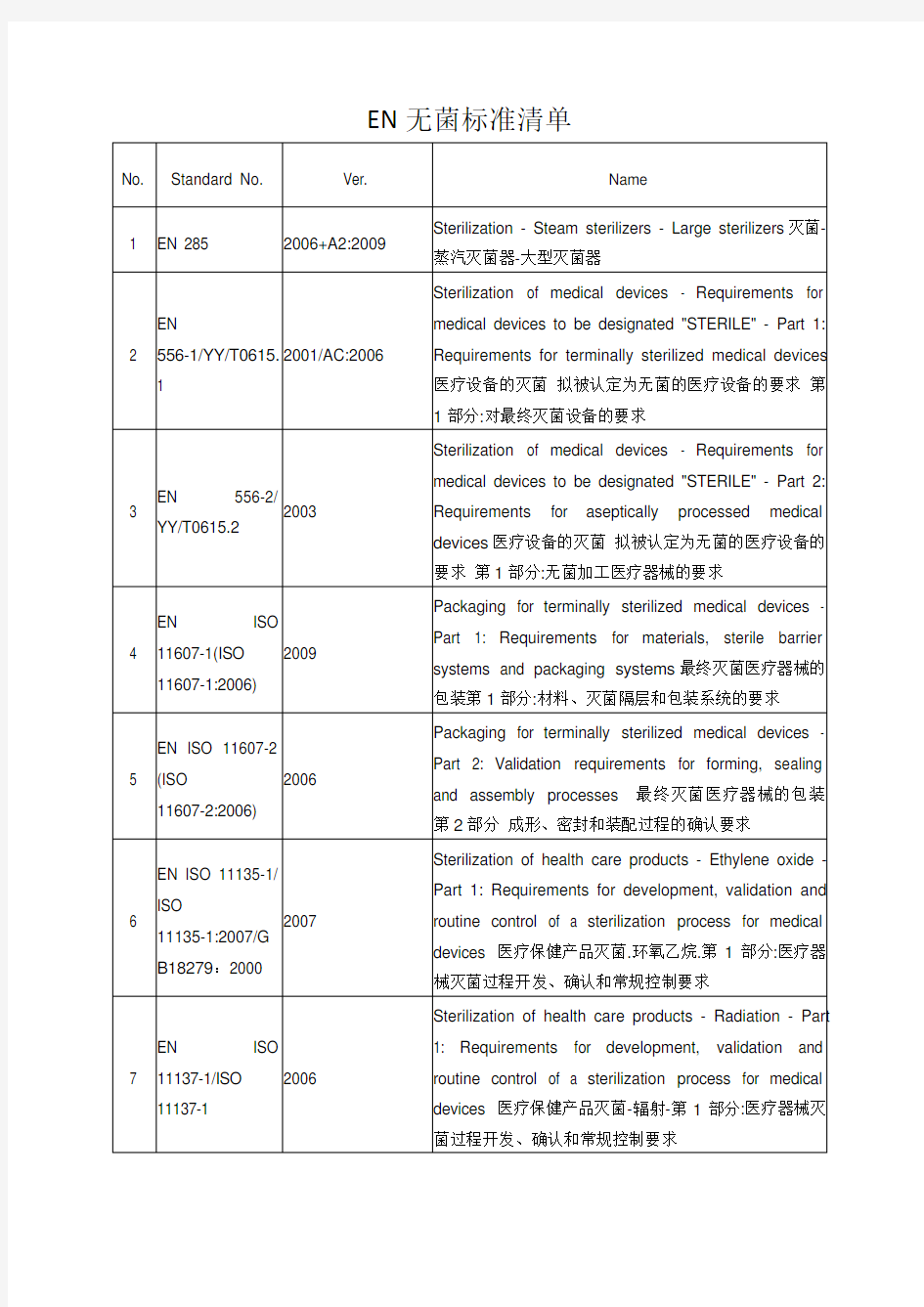

EN无菌标准清单
欧洲有源医疗器械相关法规
COMMISSION REGULATION (EU) No 207/2012 of 9 March 2012 on electronic instructions for use of medical devices (Text with EEA relevance) THE EUROPEAN COMMISSION, Having regard to the Treaty on the Functioning of the European Union, Having regard to Council Directive 90/385/EEC of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices ( 1 ), and in particular Article 9(10) thereof, Having regard to Council Directive 93/42/EEC of 14 June 1993 concerning medical devices ( 2 ), and in particular Article 11(14) thereof, Whereas: (1) For some medical devices the provision of instructions for use in electronic form instead of in paper form can be beneficial for professional users. It can reduce the environmental burden and improve the competitiveness of the medical devices industry by reducing costs, while maintaining or improving the level of safety. (2) Such possibility of providing instructions for use in elec - tronic form instead of in paper form should be limited to certain medical devices and accessories intended to be used in specific conditions. In any case, for reasons of safety and efficiency users should always have the possi -bility to obtain those instructions for use in paper form on request. (3) In order to reduce potential risks as far as possible, the appropriateness of the provision of instructions for use in electronic form should be subject to a specific risk assessment by the manufacturer. (4) In order to ensure that users have access to the instructions for use, appropriate information about access to those instructions for use in electronic form and about the right to request the instructions for use in paper form, should be provided. (5) To ensure unconditional access to the instructions for use in electronic form and to facilitate the communication of updates and of product alerts, the instructions for use in electronic form should also be available through a website. (6) Regardless of the language obligations imposed on manufacturers by the law of the Member States, manu -facturers who provide instructions for use in electronic form should indicate on their website in which Union languages those instructions are available. (7) Except for medical devices of Class I, as defined in Annex IX to Directive 93/42/EEC, the fulfilment of the obligations laid down in this Regulation should be reviewed by a notified body during the procedure applicable for conformity assessment based on a specific sampling method. (8) As the protection of the right to privacy of natural persons with respect to the processing of personal data should be ensured by manufacturers and notified bodies as well, it is appropriate to provide that websites containing instructions for use of a medical device fulfil the requirements of Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data ( 3 ). (9) In order to ensure safety and consistency, instructions for use in electronic form which are provided in addition to complete instructions for use in paper form should be covered by this Regulation as regards limited requirements in relation to their contents and websites. (10) It is appropriate to provide for a deferred application of this Regulation so as to facilitate the smooth transition to the new system and to allow all operators and Member States time to adapt to it. (11) The measures provided for in this Regulation are in accordance with the opinion of the Committee set up by Article 6(2) of Directive 90/385/EEC, HAS ADOPTED THIS REGULATION: Article 1 This Regulation establishes the conditions under which the instructions for use of medical devices referred to in point 15 of Annex 1 to Directive 90/385/EEC and in point 13 of Annex I to Directive 93/42/EEC may be provided in electronic form instead of in paper form. It also establishes certain requirements concerning instructions for use in electronic form which are provided in addition to complete instructions for use in paper form relating to their contents and websites. Article 2 For the purposes of this Regulation, the following definitions shall apply: (a) ‘instructions for use’ means information provided by the manufacturer to inform the user of the device of its safe and proper use, of its expected performances and of any ( 1 ) OJ L 189, 20.7.1990, p. 17. ( 2 ) OJ L 169, 12.7.1993, p. 1. ( 3 ) OJ L 281, 23.11.1995, p. 31.
欧盟医疗器械分类规则
欧盟医疗器械的分类规则(93/42 EEC ) 第1条: 不接触病人或仅接触未受损皮肤类器械 规则的一般解释 本规则是所有无更具体规则规范的器械的后备规则。 此规则通常适用于那些仅与未受损皮肤接触或与病人无接触的器械。 Rule 1 Examples 除非适用于以下列出的其他规则,所有非侵入 式器械都属于I 类 -用于防止体液回流的收集设备(例如用于收集人体废物的尿液收集瓶(urine collection bottles),造瘘袋(ostomy pouches),大小便失禁垫(incontinence pads)或伤口引流 设备(collectors used with wound drainage devices))。这 些装置可能通过导管和管道与患者连接。 - 用于固定身体部位或对身体部位施压的装置(如有助于扭伤愈合的非灭菌敷料,熟石膏,颈椎项圈,重力牵引装置,压缩针织品等)(non-sterile dressings used to aid the healing of a sprain, plaster of Paris, cervical collars, gravity traction devices, compression hosiery) - 一般用于患者外部支持的设备(如病床(hospital beds),病人吊(patient hoists),助行器(walking aids),轮椅(wheelchairs),担架(stretchers),牙科病人椅(dental patient chairs) - 矫正用眼镜和镜框(corrective glassed and frames) - 诊断用听诊器(stethoscopes for diagnosis) - 眼部闭塞膏药(eye occlusion plasters) - Incision drapes - 导电胶(incision drapes) - 非侵入性电极(用于脑电图或心电图的电极) Non-invasive electrodes (electrodes for EEG Or ECG) - 图像增感屏(image intensifying screens) - 用于清除眼部杂物的永磁体(permanent magnets for removal of ocular debris) 分类的实际问题 一些以间接方式与人体接触的非侵入性器械,通过血液,其他体液或重新进入人体的液体的存储、输送、处理或通过能量传递到人体的的方式影响人体内部的生理过程。这些器械不在此规则的适用范围之内。因这类器械以间接方式对人体内产生危害,其分类受另外规则的规范。 S G S
医疗器械法律法规整理
类型名称发布日期实施日期通用国务院令第650号《医疗器械监督管理条例》2014/3/72014/6/1通用主席令第22号 《中华人民共和国广告法》2015/4/242015/9/1通用卫生部令第82号《医疗器械召回管理办法(试行)》2011/5/202011/7/1通用局令第3号《食品药品行政处罚程序规定》2014/4/282014/6/1通用局令第14号《药品医疗器械飞行检查办法》2015/6/292015/9/1通用局令第15号《医疗器械分类规则》2015/7/142016/1/1通用局令6号《医疗器械说明书和标签管理规定》2014/7/302014/10/1通用总局令第19号《医疗器械通用名称命名规则》2015/12/21 通用国食药监械[2011]425号 《医疗器械不良事件监测工作指南》(试行)的通知 2011/9/16 通用食药监办械监函〔2015〕723号 征求医疗器械不良事件监测和再评价管理办法(征求意见稿)意见的函 2015/11/12 通用征求医疗器械分类目录(修订稿)意见的函 2016/9/30 通用食药监械管便函〔2014〕66号 征求《医疗器械标准管理办法》(修订草案征求意见稿)意见的通知2014/10/24 通用食药监办械监〔2016〕9进一步加强医疗器械抽验工作的通知2016/2/4 通用食药监办械监〔2016〕76号印发2016年国家医疗器械抽验产品检验方案的通知2016/6/3 通用食药监械监〔2014〕235号 印发国家重点监管医疗器械目录的通知 2014/9/30 通用征求《医疗器械监督检查员管理办法》意见的函 2014/12/30 类型名称发布日期实施日期临床总局令第25号《医疗器械临床试验质量管理规范》2016/3/232016/6/1临床食药监械管〔2014〕13号《创新医疗器械特别审批程序(试行)》2014/2/102014/3/1临床2014年第14号关于发布需进行临床试验审批的第三类医疗器械目录的通告2014/8/25 临床2014年 第12号 关于发布免于进行临床试验的第二类医疗器械目录的通告2014/8/21 临床2014年 第13号 关于发布免于进行临床试验的第三类医疗器械目录的通告2014/8/21 临床2016年第133号关于发布第二批免于进行临床试验医疗器械目录的通告(二类、三类)2016/9/30 临床2016年第98号关于开展医疗器械临床试验监督抽查工作的通告2016/6/8 临床2015年第117号关于开展药物临床试验数据自查核查工作的公告2015/7/22 临床征求《医疗器械临床试验机构资质认定管理办法》﹙征求意见稿﹚意见的通知2015/7/20 临床 临床食药监械管便函〔2014〕44号 关于《体外诊断试剂临床试验技术指导原则》和《体外诊断试剂说明书编写指导原则》征求意见的通知 临床2014年第16号关于发布体外诊断试剂临床试验技术指导原则的通告2014/9/11 临床体外诊断试剂临床试验技术指导原则 2014/9/11 临床发布医疗器械临床评价技术指导原则2015/6/12 临床医疗器械临床评价技术指导原则 2015/5/19 类型名称发布日期实施日期注册局令4号《医疗器械注册管理办法》2014/7/302014/10/1注册局令5号《体外诊断试剂注册办法》 2014/7/302014/10/1注册医疗器械产品技术要求编写指导原则 2014/5/30 注册医疗器械软件注册技术审查指导原则2015/8/5 注册总局2015年度医疗器械注册工作报告 2016/4/1
医疗器械进入欧盟医疗器械市场要求
入欧盟的相关知识 一、欧盟国家(共31 家) 1、2004 年5 月1 日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典; 2、2004 年5 月1 日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他; 3、欧洲自由贸易协会EFTA :挪威、列支敦士登、冰岛、瑞士; 4、新入欧盟:罗马利亚、保加利亚; 5、瑞士不要求产品携带CE 标志。 二、医疗器械CE 认证的通用要求 1、基本要求(总要求) (1 )安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析); (2 )风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统); ( 3 )性能符合性(产品的基本要求); (4 )器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。); (5 )器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。 2、基本要求的具体包括如下14 条: (1 )器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。 (2 )生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应: 首先应尽可能降低甚至避免危险。 其次,对无法避免的危险采取适当的防护措施,包括安装报警装置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。 ( 3 )器械必须取得生产者期望获得的功能。器械设计制造和包装应有利于第一条(2) (A)D 多规定的各项功能的发挥。 (4 )在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、 2 、 3 款所指的各项性 能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。 (5 )器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。 (6)副作用的大小同器械的使用性能相比较可以为人们所接受。 (7 )化学、物理和生物性能 (8)感染和微生物污染。 (9)组装和环境因素 (10 )检测器械 (11 )辐射防护 (12 )带有能源或与其他能源相连接的器械
医疗器械常用灭菌方法
灭菌是通过物理或化学方法清除器械、敷料等物体上的所有微生物(包括芽孢),使其达到无菌水平,以避免手术器械在使用过程中造成的感染发生及疾病传播。 无菌是指灭菌后单个微生物存活的概率,又称无菌保障水平(SAL),通常用10-n来表示。目前,一致认为SAL10-6为无菌水平。 手术器械的灭菌可有效预防医院感染,降低医院感染率。与人体组织、器官、破损皮肤、破损黏膜接触的医疗器械为高危险度物品,常见的高危险度物品包括:穿刺针、活检钳、腹腔镜、植入性医疗器械等。此类物品具有极高的感染风险,在使用前必须经过灭菌。 常见的灭菌方法包括:高压蒸汽灭菌、过氧化氢等离子灭菌、环氧乙烷灭菌、电离辐射灭菌和干热灭菌。 灭菌方式、原理、应用、优点及缺点对比 灭菌方式 原理 应用 优点 缺点
高压蒸汽 主要通过使微生物中的酶和蛋白质不可逆凝固及变性来杀灭微生物。 主要用来处理水、药剂、管制医疗废物和表面可直接接触蒸汽的无孔物品。对于带孔装载物 和器械,一般的灭菌温度和时间分别为132~135 ℃,持续3~4 min。常用的杀菌温度为121℃和132℃。 无毒、环保,过程易于控制和监测,杀菌快速有效,灭菌过程受有机或无机污物影响最小, 灭菌循环时间短,能有效穿透器械包和管腔型器械。 对热敏性器械有损坏,反复的灭菌暴露会损坏精密外科器具,可能会因湿包而造成器械生锈,潜在烫伤风险。 过氧化氢等离子 过氧化氢等离子体具有高氧化活性,通过破坏细胞的蛋白质,酶与核酸来杀灭微生物。 用于不耐热、不耐湿医疗用品的灭菌。 安全环保,无有毒物质残留,灭菌周期28—75 min,无须解析,适用于热和湿敏感的物品灭菌,易于操作、安装和监测,和大多数器械兼容。 不适用于纸纤维、棉麻和液体类物品,灭菌腔体尺寸较小(50~270 L),不适用于腔镜类或带 有细长管腔的器械,需要用合成类包装(如聚丙烯包装或聚烯烃管袋等),高于1 ppm计量的 过氧化氢可能会有毒。 环氧乙烷气体
(完整版)医疗器械全部法规汇总2018.1.8
目录 1. 医疗器械监督管理条例 (3) 2. 医疗器械注册管理办法 (28) 3. 体外诊断试剂注册管理办法 (47) 4. 体外诊断试剂注册管理办法修正案 (70) 5. 医疗器械说明书和标签管理规定 (71) 6. 医疗器械分类规则 (78) 7. 医疗器械通用名称命名规则 (87) 8. 医疗器械临床试验质量管理规范 (90) 9. 关于印发医疗器械应急审批程序的通知 (116) 10. 关于印发创新医疗器械特别审批程序(试行)的通知 (119) 11. 关于第一类医疗器械备案有关事项的公告 (133) 12. 医疗器械召回管理办法 (154) 13. 关于发布第一类医疗器械产品目录的通告 (165) 14. 关于发布医疗器械产品技术要求编写指导原则的通告 (231) 15. 关于实施《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》有关事项的通知235 16. 关于发布免于进行临床试验的第二类医疗器械目录的通告 (242) 17. 关于发布免于进行临床试验的第三类医疗器械目录的通告 (356) 18. 关于发布需进行临床试验审批的第三类医疗器械目录的通告 (378) 19. 关于印发医疗器械检验机构开展医疗器械产品技术要求预评价工作规定的通知 (382) 20. 关于公布医疗器械注册申报资料要求和批准证明文件格式的公告 (385) 21. 关于公布体外诊断试剂注册申报资料要求和批准证明文件格式的公告 (414) 22. 关于发布体外诊断试剂临床试验技术指导原则的通告 (435) 23. 关于发布体外诊断试剂说明书编写指导原则的通告 (450) 24. 关于实施第一类医疗器械备案有关事项的通知 (461) 25. 关于印发境内第三类和进口医疗器械注册审批操作规范的通知 (465) 26. 关于印发境内第二类医疗器械注册审批操作规范的通知 (476) 27. 关于发布医疗器械优先审批程序的公告 (492) 28. 关于发布创新医疗器械特别审批申报资料编写指南的通告 (499) 29. 关于发布医疗器械审评沟通交流管理办法(试行)的通告 (506) 30. 医疗器械生产监督管理办法 (517) 31. 医疗器械生产质量管理规范 (534) 32. 关于发布医疗器械生产质量管理规范附录无菌医疗器械的公告 (549) 33. 关于发布医疗器械生产质量管理规范附录植入性医疗器械的公告 (560) 34. 关于发布医疗器械生产质量管理规范附录体外诊断试剂的公告 (574) 35. 关于医疗器械生产日常监督现场检查工作指南的通知 (587) 36. 关于医疗器械生产经营备案有关事宜的公告 (596) 37. 关于实施《医疗器械生产监督管理办法》和《医疗器械经营监督管理办法》有关事项的通知 (607)
医疗器械进入欧盟医疗器械市场要求
入欧盟的相关知识 一、欧盟国家(共31家) 1、2004年5月1日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、 西班牙、葡萄牙、奥地利、芬兰、瑞典; 2、2004年5月1日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他; 3、欧洲自由贸易协会EFTA:挪威、列支敦士登、冰岛、瑞士; 4、新入欧盟:罗马利亚、保加利亚; 5、瑞士不要求产品携带CE标志。 二、医疗器械 CE认证的通用要求 1、基本要求(总要求) (1)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析); (2)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统); (3)性能符合性(产品的基本要求); (4)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。); (5)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。 2、基本要求的具体包括如下14条: (1)器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操 作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高 水平的防护办法。 (2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产 者应:首先应尽可能降低甚至避免危险。 其次,对无法避免的危险采取适当的防护措施,包括安装报警装 置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。 (3)器械必须取得生产者期望获得的功能。器械设计制造和包装应有利于第一条(2) (A)D多规定的各项功能的发挥。 (4)在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能 应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。 (5)器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会 发生根本逆变。 (6)副作用的大小同器械的使用性能相比较可以为人们所接受。 (7)化学、物理和生物性能 (8)感染和微生物污染。 (9)组装和环境因素 (10)检测器械 (11)辐射防护 (12)带有能源或与其他能源相连接的器械 (13)生产者提供的操作信息 (14)如果需要根据医疗数据确定器械是否满足基本要求,如第六款的情形,有关数据必须按照附录Ⅹ
医疗器械的灭菌方法以及灭菌效果的验证
医疗器械的灭菌方法以及灭菌效果的验证 [摘要]常用的灭菌方法有多种,如湿热灭菌法、干热灭菌法、辐射灭菌法、气体灭菌法和过滤除菌法。可根据被灭菌物品的特性采用—种或多种方法组合灭菌。灭菌后还要采取适当方法进行灭菌效果验证,这样才能保证产品无菌。 【关键词】灭菌方法;湿热;干热;辐射;灭菌效果;验证 一、灭菌方法 灭菌法系指用适当的物理或化学手段将物品中活的微生物杀灭或除去,从而使物品残存活微生物的概率下降至预期的无菌保证水平的方法。 无菌物品是指物品中不含任何活的微生物。对于任何一批灭菌物品而言,绝对无菌既无法保证也无法用试验来证实。一批物品的无菌特性只能相对地通过物品中活微生物的概率低至某个可接受的水平来表述,即无菌保证水平(Sterility assurance level,简称SAL)。实际生产过程中,灭菌是指将物品中污染微生物的概率下降至预期的无菌保证水平。最终灭菌的物品微生物存活概率,即无菌保证水平不得高于10-6。已灭菌物品达到的无菌保证水平可通过验证确定。 常用的灭菌方法有湿热灭菌法、干热灭菌法、辐射灭菌法、气体灭菌法和过滤除菌法。可根据被灭菌物品的特性采用—种或多种方法组合灭菌。只要物品允许,应尽可能选用最终灭菌法灭菌。若物品不适合采用最终灭菌法,可选用过滤除菌法或无菌生产工艺达到无菌保证要求,只要可能,应对非最终灭菌的物品作补充性灭菌处理(如流通蒸汽灭菌)。 1.湿热灭菌法 本法系指将物品置于灭菌柜内利用高压饱和蒸汽、过热水喷淋等手段使微生物菌体中的蛋白质、核酸发生变性而杀灭微生物的方法。该法灭菌能力强,为热力灭菌中最有效、应用最广泛的灭菌方法。药品、容器、培养基、胶塞以及其他遇高温和潮湿不发生变化或损坏的物品,均可采用本法灭菌。 采用湿热灭菌,被灭菌物品应有适当的装载方式,不能排列过密,以保证灭菌的有效性和均一性。 湿热灭菌法应确认灭菌柜在不同装载时可能存在的冷点。当用生物指示剂进一步确认灭菌效果时,应将其置于冷点处。本法常用的生物指示剂为嗜热脂肪芽孢杆菌孢子(Spores of Bacillus stearothermophilus)。 2.气体灭菌法
新版中国医疗器械法规清单
新版中国医疗器械法规清单 一、行政法规 1.《医疗器械监督管理条例》(国务院令第680号) 二、部门规章 1.医疗器械注册管理办法(CFDA局令第4号) 2.体外诊断试剂注册管理办法(CFDA局令第5号) 3.医疗器械说明书和标签管理规定(CFDA局令第6号) 4.医疗器械生产监督管理办法(CFDA局令第7号) 5.医疗器械经营监督管理办法(CFDA局令第8号) 6.药品医疗器械飞行检查办法(CFDA局令第14号) 7.医疗器械分类规则(CFDA局令第15号) 8.医疗器械使用质量监督管理办法(CFDA局令第18号) 9.医疗器械通用名称命名规则(CFDA局令第19号) 10.医疗器械临床试验质量管理规范(CFDA国家卫计委令第25号) 11.医疗器械召回管理办法(CFDA局令第29号) 12.体外诊断试剂注册管理办法修正案(CFDA局令第30号) 13.关于调整部分医疗器械行政审批事项审批程序的决定(CFDA局令第32号) 14.医疗器械标准管理办法(CFDA局令第33号) 15.医疗器械网络销售监督管理办法(CFDA局令第38号) 16.医疗器械不良事件监测和再评价管理办法(国家市场监督管理总局令第1号) 三、通告 1.关于发布第一类医疗器械产品目录的通告(CFDA通告2014年第8号) 2.关于发布医疗器械产品技术要求编写指导原则的通告(CFDA通告2014年第9号) 3.关于发布需进行临床试验审批的第三类医疗器械目录的通告(CFDA通告2014年第14 号) 4.关于发布体外诊断试剂临床试验技术指导原则的通告(CFDA通告2014年第16号) 5.关于发布体外诊断试剂说明书编写指导原则的通告(CFDA通告2014年第17号) 6.关于发布禁止委托生产医疗器械目录的通告(CFDA通告2014年第18号) 7.关于发布医疗器械生产企业供应商审核指南的通告(CFDA通告2015年第1号) 8.关于发布医疗器械临床评价技术指导原则的通告(CFDA通告2015年第14号) 9.关于发布医疗器械产品出口销售证明管理规定的通告(CFDA通告2015年第18号) 10.关于贯彻落实小微企业行政事业性收费优惠政策的通告(CFDA通告2015年第31号) 11.关于生产一次性使用无菌注、输器具产品有关事项的通告(CFDA通告2015年第71 号)
医疗器械进入欧盟医疗器械市场要求
入欧盟的相关知识一、欧盟国家(共31家) 1、2004年5月1日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典; 2、2004年5月1日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他; 3、欧洲自由贸易协会 EFTA:挪威、列支敦士登、冰岛、瑞士; 4、新入欧盟:罗马利亚、保加利亚; 5、瑞士不要求产品携带CE标志。 二、医疗器械CE认证的通用要求 1、基本要求(总要求) (1)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);(2)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统); (3)性能符合性(产品的基本要求); (4)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。); (5)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。 2、基本要求的具体包括如下14条: (1)器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。 (2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应: 首先应尽可能降低甚至避免危险。 其次,对无法避免的危险采取适当的防护措施,包括安装报警装置; 最后,告知用户所提供防护措施的弱点及其可能带来的危险。 (3)器械必须取得生产者期望获得的功能。器械设计制造和包装应有利于第一条(2) (A)D多规定的各项功能的发挥。 (4)在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。 (5)器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。 (6)副作用的大小同器械的使用性能相比较可以为人们所接受。 (7)化学、物理和生物性能 (8)感染和微生物污染。 (9)组装和环境因素 (10)检测器械 (11)辐射防护 (12)带有能源或与其他能源相连接的器械 (13)生产者提供的操作信息 )如果需要根据医疗数据确定器械是否满足基本要求,如第六款的情形,有关数据必须按照附录Ⅹ14(. 的规定取得 三、申请CE认证 (一)一般流程(所有的产品)
医疗器械进入欧盟医疗器械市场要求
医疗器械进入欧盟医疗器械市场要求 入欧盟的相关知识 一、欧盟国家(共31 家) 1、2004 年5 月1 日前:法国、德国、意大利、荷兰、比利时、卢森堡、丹麦、爱尔兰、英国、希腊、西班牙、葡萄牙、奥地利、芬兰、瑞典; 2、2004 年5 月1 日加入:波兰、匈牙利、捷克、斯洛伐克、斯洛文尼亚、爱沙尼亚、拉脱维亚、立陶宛、塞浦路斯、马耳他; 3、欧洲自由贸易协会EFTA :挪威、列支敦士登、冰岛、瑞士; 4、新入欧盟:罗马利亚、保加利亚; 5、瑞士不要求产品携带CE标志。 二、医疗器械CE认证的通用要求 1、基本要求(总要求) (1)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析); (2)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统); (3)性能符合性(产品的基本要求); (4)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。); (5)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。 2、基本要求的具体包括如下14 条: (1)器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。 (2)生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应: 首先应尽可能降低甚至避免危险。 其次,对无法避免的危险采取适当的防护措施,包括安装报警装置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。 (3)器械必须取得生产者期望获得的功能。器械设计制造和包装应有利于第一条(2) (A)D 多规定的各项功能的发挥。 (4)在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。 (5)器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。 (6)副作用的大小同器械的使用性能相比较可以为人们所接受。 (7)化学、物理和生物性能 (8)感染和微生物污染。
欧盟医疗器械CE认证监管模式简介
欧盟医疗器械CE认证监管模式简介 一、欧盟简介 欧洲联盟(European Union),简称欧盟(EU),是由欧洲共同体(European Communities) 发展而来,是一个集政治实体和经济实体于一身、在世界上具有重要影响的区域一体化组织。1991年12月,欧洲共同体马斯特里赫特首脑会议通过《欧洲联盟条约》,通称《马斯特里赫特条约》(简称《马约》)。1993年11月1日,《马约》正式生效,欧盟正式诞生。欧盟的宗旨是―通过建立无内部边界的空间,加强经济、社会的协调发展和建立最终实行统一货币的经济货币联盟,促进成员国经济和社会的均衡发展‖,―通过实行共同外交和安全政策,在国际舞台上弘扬联盟的个性‖。目前欧盟有27个成员国(Member State),4个自由贸易区域(European Free Trade Area)。 欧盟的主要组织机构有: 欧洲理事会(European Council) ,即首脑会议,由欧盟成员国国家元首或政府首脑及欧盟委员会主席组成。是欧盟的最高权力机构。理事会下设有总秘书处。 欧盟委员会(Commission of European Union) ,是欧盟的常设执行机构。负责实施欧盟条约和欧盟理事会做出的决定,处理日常事务,代表欧盟对外联系和进行贸易等方面的谈判。 欧洲议会(European Parliament) ,是欧洲联盟的执行监督、咨询机构,在某些领域有立法职能,并有部分预算决定权。欧洲议会可以2/3多数弹劾欧盟委员会,迫其集体辞职。 二、医疗器械的相关法规文件 目前,欧盟已颁布实施的医疗器械指令有三个,包括: 1、有源植入医疗器械指令(EC-Directive 90/385/EEC)。该指令适用于心脏起搏器、可植入的胰岛素泵等有源植入医疗器械,于1993年1月1日生效,1995年1月1日强制实施。 2、医疗器械指令(EC-Directive 93/42/EEC)。该指令适用于除90/385 EEC指令和98/79 EEC指令规定以外的一般医疗器械,于1995年1月1日生效,并于1998年6月14日强制实施。 3、体外诊断医疗器械指令(EC-Directive 98/79/EEC)。该指令适用于血细胞计数器、妊娠检测装置等体外诊断用医疗器械,于1998年12月7日生效,2003年12月7日强制实施。 上述指令是欧盟范围内统一执行的医疗器械管理法规,其法律地位相当于中国的《医疗器械监督管理条例》和日本的药事法(The Pharmaceutical Affairs Law)。 三个医疗器械指令虽然颁布的时间不同,但相互关联。医疗器械指令(EC-Directive 93/42/EEC)是在有源植入医疗器械指令(EC-Directive 90/385/EEC)的基础上制订的,二者又同为体外诊断医疗器械指令(EC-Directive 98/79/EEC)的编写基础。三个指令的格式、内容、基本要求大致相同,并针对医疗器械的不同特点而规定了特殊条款。当新颁布的指令对已有指令的基本要求进行修改时,已有指令同时进行相应修订。 医疗器械指令(EC-Directive 93/42/EEC)由23项条款和12个附录组成,其主要内容为: 1、定义和范围(Definitions,scope) 2、上市与投入使用(Placing on the market and putting into service) 该条款中规定制造商需采取所有必要的措施,确保医疗器械在依照设计的目的安装、维护和使用时不会危及患者、使用者或相关人员的安全及健康。 3、基本要求(Essential requirements) 该条款中规定医疗器械必须符合指令附录Ⅰ中的基本要求。 4、医疗器械的自由流通和特殊用途的医疗器械(Free movement,devices intended for special purposes) 该条款中规定各成员国不能对符合指令规定的临床研究用器械、定制器械和带有CE标记的医疗器械产品设置流通障碍。同时规定定制器械、参展器械和临床研究用器械在使用时可无需带有CE标记。 5、可参考的标准(Reference to standards) 6、标准与技术法规委员会(Committee on Standards and Technical Regulations) 该条款规定依据83/189/EEC号指令第五条所设立的委员会应协助欧盟委员会工作。
医疗器械清洗消毒灭菌流程
医疗器械清洗消毒灭菌 流程 SANY标准化小组 #QS8QHH-HHGX8Q8-GNHHJ8-HHMHGN#
医疗器械的清洗、消毒、灭菌流程 1.盆、盘、碗、药杯 回收—→常水冲洗—→复合酶浸泡10分钟—→刷洗—→常水冲洗—→消毒液浸泡30分钟—→常水漂洗二遍—→纯水漂洗二遍—→控水—→干燥(90℃ 20分钟) 2.手术器械(除管腔器械外) 回收—→常水冲洗—→加酶超声清洗5分钟—→刷洗—→常水漂洗二遍—→纯水漂洗二遍—→水—→干燥(90℃ 20分钟)—→高压蒸汽灭菌 3.管腔器械 回收—→常水冲洗—→管腔注酶—→加酶超声清洗5分钟—→常水冲洗—→毛刷刷洗管腔—→加压水枪冲洗管腔—→常水冲洗—→消毒液浸泡30分钟—→常水漂洗二遍—→纯水漂洗二遍(纯水冲洗管腔)—→压力气枪吹干管腔—→干燥(90℃ 20分钟) 4.穿刺针 回收—→常水冲洗—→棉签清洁针座腔内—→针腔注酶—→加酶超声清洗5分钟—→常水冲洗—→加压水枪冲洗针腔内部—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍,并用纯水冲洗针腔内部—→压力气枪吹干管腔—→干燥(擦干)—→高压蒸汽灭菌 5.橡胶管 回收—→常水冲洗—→松节油擦洗外面污垢(胶布印)—→常水冲洗—→管腔注酶—→复合酶浸泡10分钟—→常水冲
洗—→压力水枪冲洗管道内面—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍,并用纯水冲洗管腔—→压力气枪吹干管腔—→干燥(擦干) 6.筒、缸、瓶 回收—→常水冲洗—→刷洗—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍—→干燥(90℃ 20分钟)7.呼吸机管道 回收—→常水冲洗—→复合酶浸泡10分钟—→常水冲洗、刷洗—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍—→干燥(70℃ 20分钟) 8.湿化罐、集水杯、Y形管、接头 回收—→常水冲洗—→复合酶浸泡10分钟—→常水冲洗、刷洗—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍—→干燥(70℃ 20分钟) 9.氧气管、氧气湿化瓶 回收—→常水冲洗—→加压水枪冲洗管腔—→消毒液浸泡30分钟—→常水冲洗二遍—→纯水冲洗二遍—→擦干 附:特殊药液污染器具处理流程 1.碘污染器具:回收—→常水冲洗—→ 75﹪酒精浸泡20分 钟—→常水冲洗—→按常规器具处理 2.石蜡油污染器具:回收—→常水冲洗—→洗洁精刷洗—→ 常水冲洗—→按常规器具处理
欧盟医疗器械新法规对公告机构的管理情况简介_吕允凤
1 . China Medical Device Information | 中国医疗器械信息技术审评·专题 T echnical Review · Thematic Forum 欧洲议会和理事会于2017年4月5日通过了医疗器械法规(MDD )和体外诊断医疗器械法规(IVDD ),其中关于公告机构的规定均列于法规正文的第Ⅳ章,同时在附件Ⅶ列明了公告机构需满足的要求。整体来讲,欧盟新法规对公告机构的管理是以成员国主管机构为主体组织实施,欧盟委员会、欧盟级别的专家委员会参与并监管的模式开展的,对公告机构进行评估,并在委任后实施持续监管,持续审核的管理。现将其关于公告机构的相关内容归纳总结如下。 1. 公告机构的评估、委任及监管涉及的机构及主体 公告机构的管理涉及到以下机构及主体,首先明确其 概念及职责: 公告机构(Notified Body ):是根据本法规指定的合格评估机构。 专家委员会-医疗器械协调小组(MDCG ):由成员国根据其在医疗器械和体外诊断医疗器械领域的角色和专门技术指定的人员组成,应根据有关医疗器械的法规确定的条件和模式创建,以履行法规赋予的任务,向欧盟委员会提供意见并在确保统一执行法规方面协助委员会和各成员国。MDCG 可建立子组,以专业进行区分。职责包括促进信息交流和协调评估、协调当局提供各方面支持、适当的 法规授权以便补充或修正法规的某些非必要规定、以及法规执行权力。 负责公告机构的国家主管机构(本文中简称“国家主管机构”):成员国如计划评估合格评定机构为公告机构,或已委任一家公告机构,根据法规开展合格评定活动,则应任命一个主管机构,该机构根据国家法律由单独实体组成,负责建立和实施必要的程序,以进行公告机构的评估和通告以及公告机构的监管,包括其分包商和分支机构,此主管机构简称“负责公告机构的国家主管机构”。国家主管机构应保证其活动的客观性和公正性,并避免与公告机 收稿日期:2017-06-21 欧盟医疗器械新法规对公告机构的管理情况简介 吕允凤 国家食品药品监督管理总局医疗器械技术审评中心 (北京 100044) 文章编号:1006-6586(2017)15-0001-03 中图分类号:R197.39 文献标识码:A 内容提要: 2017年5月,欧盟官方期刊(Official Journal of the European Union )正式发布了医疗器械法规(MDR ,EU 2017/745)和体外诊断医疗器械法规(IVDR ,EU 2017/746),法规对公告机构提出了更为严格的要求。本文重点介绍欧盟法规对公告机构的管理要求。 关 键 词: 欧盟医疗器械法规 公告机构 A Brief Introduction to the Management of the Notify Body in the New Regulations on Medical Devices of the European Union LV Yun-feng Center for Medical Device Evaluation.CFDA (Beijing 100044)Abstract: May 2017, Official Journal of the European Union Officially issued the regulations on medical devices (MDR, EU 2017/745) and regulations on In vitro diagnostic medical devices(IVDR, EU 2017/746). The new regulations are more stringent on the requirements of the Notify Body. This article focuses on the EU regulatory requirements for the management of the Notify Body.Key words: the regulations on medical devices of the european union, notify body DOI:10.15971/https://www.360docs.net/doc/115000968.html,ki.cmdi.2017.15.001