致病菌毒力因子分析-VFDB 2012 update

VFDB 2012update:toward the genetic diversity and molecular evolution of bacterial virulence factors
Lihong Chen,Zhaohui Xiong,Lilian Sun,Jian Yang*and Qi Jin*
State Key Laboratory for Molecular Virology and Genetic Engineering,Institute of Pathogen Biology,Chinese Academy Medical Sciences and Peking Union Medical College,Beijing 100176,China
Received September 15,2011;Accepted October 17,2011
ABSTRACT
The virulence factor database (VFDB,http://www https://www.360docs.net/doc/7b2557457.html,/VFs/)has served as a comprehensive repository of bacterial virulence factors (VFs)for >7years.Bacterial virulence is an exciting and dynamic field,due to the availability of complete se-quences of bacterial genomes and increasing sophisticated technologies for manipulating bacteria and bacterial genomes.The intricacy of virulence mechanisms offers a challenge,and there exists a clear need to decipher the ‘language’used by VFs more effectively.In this article,we present the recent major updates of VFDB in an attempt to summarize some of the most important virulence mechanisms by comparing different compositions and organiza-tions of VFs from various bacterial pathogens,iden-tifying core components and phylogenetic clades and shedding new light on the forces that shape the evolutionary history of bacterial pathogenesis.In addition,the 2012release of VFDB provides an improved user interface.INTRODUCTION
Bacterial virulence factors (VFs)are fascinating for a number of reasons.First,the ability of successful patho-gens to establish infections,produce disease and survive in a hostile environment is provided by a large armamentar-ium of virulence mechanisms.Elucidating the molecular mechanisms of VFs can improve understanding of the cellular and molecular basis of pathogenesis.Second,many important virulence factors interact with host cells and modulate their functions.Investigating the complex and ?nely balanced interactions between hosts and patho-gens can uncover useful tools for studying normal host cellular processes.Third,a much deeper understanding
of the mechanisms of action of VFs will inform new avenues for identifying promising approaches to disease prevention and therapy.Fueled by recent technological innovations in the life sciences,the ?eld of microbial viru-lence has expanded rapidly over the past decade.
Since its inception in 2004,the virulence factor database (VFDB,https://www.360docs.net/doc/7b2557457.html,/VFs/)has provided the broadest and most comprehensive up-to-date information regarding experimentally validated bacterial virulence factors (e.g.extracellular products,such as enzymes and toxins and secreted effectors or cell-associated products,such as capsular polysaccharides and outer membrane proteins),and has further explored plasticity in the reper-toire of VFs on an intra-genera level since its second release (1,2).To summarize the common themes in bac-terial virulence and to re?ect the diversity of genomic encoding,structural architecture and functional original-ity,we recently updated VFDB with an enhanced user interface and new contents dedicated to inter-genera com-parative analysis of VFs involved in host cell attachment and invasion,bacterial secretion systems and effectors,toxins,and iron-acquisition systems (Table 1).DATABASE UPDATES Data sources and processing
The core dataset of VFDB only covers experimentally demonstrated VFs from 24genera of medically important bacterial pathogens.Several predicted VFs from complete genomes were also included for comparative analyses in the second release (2),but this information is still far from suf?cient for a comprehensive study of the genetic diver-sity and molecular evolution of VFs.Many VFs found in human pathogens have homologues present in animal or plant pathogens,and,sometimes,even in non-pathogens.Additionally,the genomic sequences encoding most func-tionally validated VFs are fragmentary,rather than complete genomes in the public domain.Therefore,via exhaustive literature screening and expert review,the
*To whom correspondence should be addressed.Tel:+861067877732;Fax:+861067877736;Email:zdsys@https://www.360docs.net/doc/7b2557457.html,
Correspondence may also be addressed to Jian Yang.Tel:+861067877735;Fax:+861067877736;Email:yangj@https://www.360docs.net/doc/7b2557457.html, The authors wish it to be known that,in their opinion,the ?rst two authors should be regarded as joint First Authors.
Published online 8November 2011
Nucleic Acids Research,2012,Vol.40,Database issue D641–D645
doi:10.1093/nar/gkr989
?The Author(s)2011.Published by Oxford University Press.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://www.360docs.net/doc/7b2557457.html,/licenses/by-nc/3.0),which permits unrestricted non-commercial use,distribution,and reproduction in any medium,provided the original work is properly cited.
basic information on >1200VFs was collected from over 1100original research papers (Table 1).These collected VFs,derived from 75genera of bacteria,were organized into four super-families and 31subclasses in VFDB (Table 1).Nevertheless,we do not intend to discuss the biological diversity of certain VFs;therefore,only experi-mentally veri?ed VFs were included.In addition,if more than one sequence was available for an individual species,only the representative one was collected into the database for the sake of brevity.
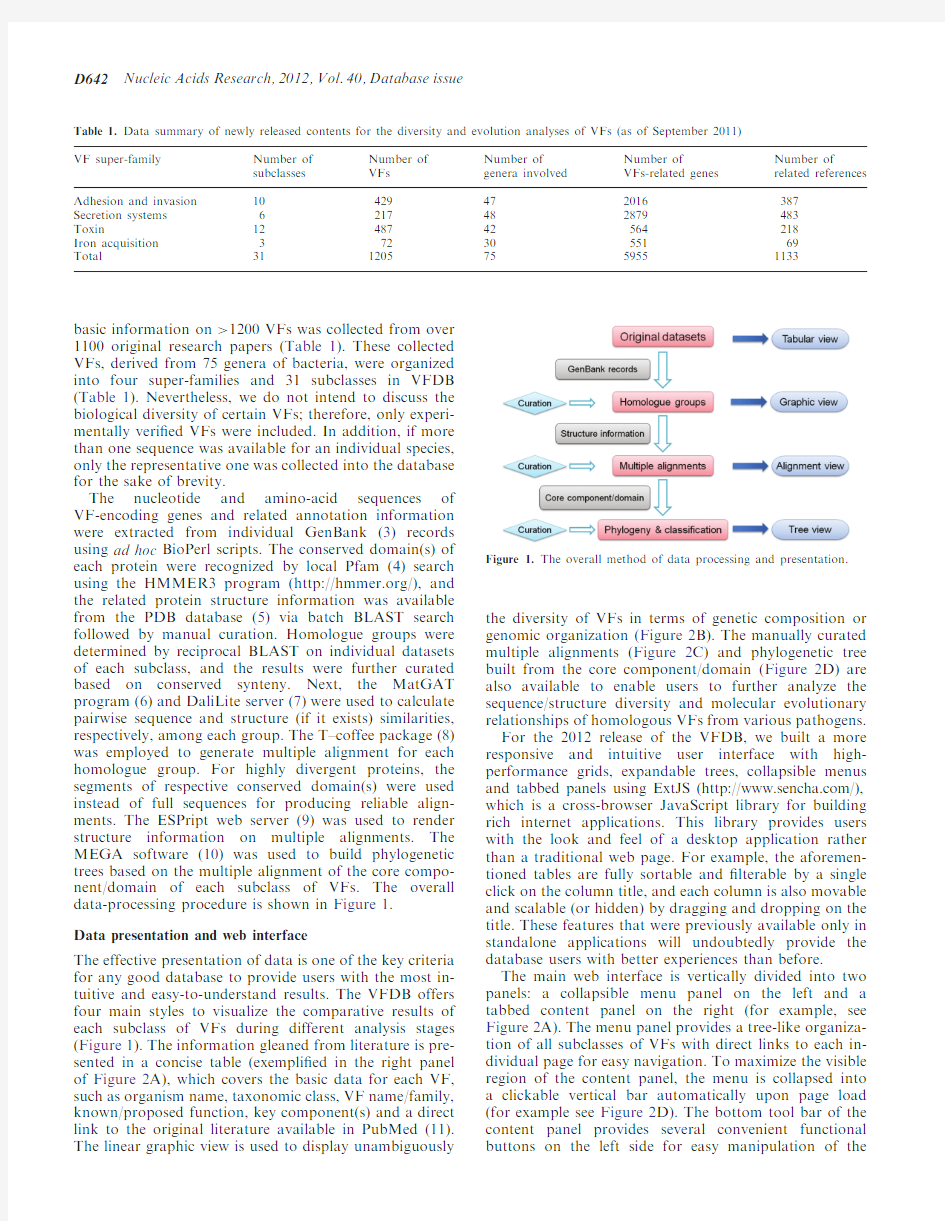
The nucleotide and amino-acid sequences of VF-encoding genes and related annotation information were extracted from individual GenBank (3)records using ad hoc BioPerl scripts.The conserved domain(s)of each protein were recognized by local Pfam (4)search using the HMMER3program (https://www.360docs.net/doc/7b2557457.html,/),and the related protein structure information was available from the PDB database (5)via batch BLAST search followed by manual curation.Homologue groups were determined by reciprocal BLAST on individual datasets of each subclass,and the results were further curated based on conserved synteny.Next,the MatGAT program (6)and DaliLite server (7)were used to calculate pairwise sequence and structure (if it exists)similarities,respectively,among each group.The T–coffee package (8)was employed to generate multiple alignment for each homologue group.For highly divergent proteins,the segments of respective conserved domain(s)were used instead of full sequences for producing reliable align-ments.The ESPript web server (9)was used to render structure information on multiple alignments.The MEGA software (10)was used to build phylogenetic trees based on the multiple alignment of the core compo-nent/domain of each subclass of VFs.The overall data-processing procedure is shown in Figure 1.Data presentation and web interface
The effective presentation of data is one of the key criteria for any good database to provide users with the most in-tuitive and easy-to-understand results.The VFDB offers four main styles to visualize the comparative results of each subclass of VFs during different analysis stages (Figure 1).The information gleaned from literature is pre-sented in a concise table (exempli?ed in the right panel of Figure 2A),which covers the basic data for each VF,such as organism name,taxonomic class,VF name/family,known/proposed function,key component(s)and a direct link to the original literature available in PubMed (11).The linear graphic view is used to display unambiguously
the diversity of VFs in terms of genetic composition or genomic organization (Figure 2B).The manually curated multiple alignments (Figure 2C)and phylogenetic tree built from the core component/domain (Figure 2D)are also available to enable users to further analyze the sequence/structure diversity and molecular evolutionary relationships of homologous VFs from various pathogens.For the 2012release of the VFDB,we built a more responsive and intuitive user interface with high-performance grids,expandable trees,collapsible menus and tabbed panels using ExtJS (https://www.360docs.net/doc/7b2557457.html,/),which is a cross-browser JavaScript library for building rich internet applications.This library provides users with the look and feel of a desktop application rather than a traditional web page.For example,the aforemen-tioned tables are fully sortable and ?lterable by a single click on the column title,and each column is also movable and scalable (or hidden)by dragging and dropping on the title.These features that were previously available only in standalone applications will undoubtedly provide the database users with better experiences than before.
The main web interface is vertically divided into two panels:a collapsible menu panel on the left and a tabbed content panel on the right (for example,see Figure 2A).The menu panel provides a tree-like organiza-tion of all subclasses of VFs with direct links to each in-dividual page for easy navigation.To maximize the visible region of the content panel,the menu is collapsed into a clickable vertical bar automatically upon page load (for example see Figure 2D).The bottom tool bar of the content panel provides several convenient functional buttons on the left side for easy manipulation of the
Table 1.Data summary of newly released contents for the diversity and evolution analyses of VFs (as of September 2011)VF super-family Number of subclasses Number of VFs Number of genera involved Number of
VFs-related genes Number of
related references Adhesion and invasion 10429472016387Secretion systems 6217482879483Toxin
1248742564218Iron acquisition 3723055169Total
31
1205
75
5955
1133
Figure 1.The overall method of data processing and presentation.
D642Nucleic Acids Research,2012,Vol.40,Database issue
Figure 2.The updated VFDB web interface.(A )Menu panel (left)and tabular view of basic data sets (right).(B )Graphic view for multiple-component VFs,color-coded by homologue groups.(C )Structure-based multiple alignment of homologous VFs.(D )Deduced phylo-genetic tree based on key component/domain (the menu panel is collapsed as a vertical bar in the left).(E )Color-shaded matrix of pairwise sequence similarities.(F )Popup window with detailed gene information along with a graphic illustration of conserved domains and a 3D structure preview.
Nucleic Acids Research,2012,Vol.40,Database issue D643
tables and for saving the web contents as a local?le(Excel table,PNG?gure or FASTA sequences).In addition, there are icon buttons on the right side of the tool bar for rapid switching between the aforementioned different data presentation styles.
Genetic diversity of VFs
The diversity of genetic composition or genomic organiza-tion of homologous VFs from different pathogens may re?ect the evolutionary relationships of the bacteria in terms of virulence.To facilitate future studies on the di-versity of VFs,the composition and organization of VFs are highlighted in the graphic view for easy comparison. For single-gene-encoded VFs,domain architectures are shown as colored bars with a direct link to the respective protein family information.As for multiple-component VFs,all genes are depicted as clickable arrows in the linear map and are color-coded by homologue groups (Figure2B).Therefore,it becomes straightforward to ?nd out whether those VF-related genes are clustered or scattered on the genomes of various pathogens.For example,the genes encoding synthesis of type IVa pili are generally dispersed throughout the bacterial genome while those of type IVb pili are arranged in a contiguous cluster.We endeavor herein to provide a framework for further investigations into whether these genes were acquired separately or whether all genes were previously in a single cluster that was disrupted by genomic rearrangements.
Detailed information on each gene,including genomic location,coding strand,scienti?c name,product and se-quences,as well as a graphic illustration of conserved domains and a preview of3D structure(s)(if they exist) are available from a popup window upon clicking on the linear map(Figure2F).By default,the linear maps are ordered on the basis of the phylogenetic tree(see below)to emphasize potential correlations between genetic vari-ations and molecular evolution of VFs.In addition,the linear maps of each VF are also organized in a highly scalable grid,which enables users to sort and?lter the VFs easily to construct customized graphic comparisons. Sequence/structure variations and phylogenetic analysis We explore the sequence and structure similarity of each homologue group in order to provide insight into how VFs may have evolved from common ancestors or may have exploited different mechanisms to arrive at similar biological activities.The multiple-alignment of homolo-gous VFs is displayed by superimposing the crystal struc-ture of the representative protein,and secondary structural elements are highlighted on top of the alignment (Figure2C).It will be helpful to disclose possible similar structures deduced from homologous sequences.Color-shaded matrices summarizing the pairwise sequence/struc-ture similarities among each homologue group are also provided in an attempt to illustrate sequence variations within each group and reveal potential protein pairs that share low sequence similarities but produce highly similar 3D structures.For example,within the a-hemolysin sub-family of b-barrel pore-forming toxins,the overall sequence identities of the core leukocidin domain from Vibrio cholerae cytolysin(VCC)and most of other members are<30%(Figure2E),but their structure com-parison scores are notably high,indicating clear similarities at the structural level.However,it should be noted that protein pairs displaying signi?cant sequence homology and similar enzymatic activities might still differ in host cell targets,thereby playing different roles in bacterial pathogenesis,such as Escherichia coli SopE and SopE2,Pseudomonas ExoS and ExoT,and the Shigella IpaH proteins.
The growing diversity of VFs has prompted numerous efforts to develop classi?cation schemas and unravel the evolutionary origins of VFs.For example,six major ?mbrial clades of chaperone/usher systems and seven dif-ferent families of T3SS are already well-established (12,13).Therefore,we performed extensive phylogenetic analysis of subclass or subfamily in the VFDB. Phylogenetic trees are labeled by species and color-coded by bacterial taxonomy(for example,see Figure2D).This analysis may not only provide insights into the evolution-ary history of VFs but may also facilitate future classi?-cation of newly identi?ed VFs using the existing schemas. As a preliminary result,we found aerolysin-like toxin family and a-hemolysin family each might be further divided into two groups(Figure2D),though additional investigations are needed.
DISCUSSION
Bacterial pathogenicity is one of the most important sub-jects in microbiology.The pathogenicity of bacteria depends on the ability to employ virulence factors, which are localized to the cell surface,released into the extracellular milieu or injected directly into host cells.Obviously,much is yet to be learned from the sophisticated virulence strategies posed by bacterial pathogens.There is an increasing need to review the entire?eld and perform bioinformatic mining of the ex-plosively growing data regarding bacterial VFs.VFDB is dedicated to meeting these demands by providing up-to-date,thought-provoking information and various analytical tools.Nevertheless,we acknowledge that our work represents only a preliminary characterization of VFs.The increasingly rapid expansion of knowledge con-cerning the multifaceted aspects of VFs will continue to challenge our capacity to compile the latest and most relevant information for the scienti?c community. FUNDING
National Basic Research Program from the Ministry of Science and Technology of China(grants2009CB522603 and2011CB504904to J.Y.and Q.J.,respectively);Beijing Nova Program(grant2009A67to J.Y.).Funding for open access charge:Beijing Nova Program.
Con?ict of interest statement.None declared.
D644Nucleic Acids Research,2012,Vol.40,Database issue
REFERENCES
1.Chen,L.,Yang,J.,Yu,J.,Yao,Z.,Sun,L.,Shen,Y.and Jin,Q.
(2005)VFDB:a reference database for bacterial virulence factors.
Nucleic Acids Res.,33,D325–D328.
2.Yang,J.,Chen,L.,Sun,L.,Yu,J.and Jin,Q.(2008)VFDB2008
release:an enhanced web-based resource for comparative
pathogenomics.Nucleic Acids Res.,36,D539–D542.
3.Benson,D.A.,Karsch-Mizrachi,I.,Lipman,D.J.,Ostell,J.and
Sayers,E.W.(2011)GenBank.Nucleic Acids Res.,39,D32–D37. 4.Finn,R.D.,Mistry,J.,Tate,J.,Coggill,P.,Heger,A.,Pollington,J.E.,
Gavin,O.L.,Gunasekaran,P.,Ceric,G.,Forslund,K.et al.(2010) The Pfam protein families database.Nucleic Acids Res.,38,
D211–D222.
5.Rose,P.W.,Beran,B.,Bi,C.,Bluhm,W.F.,Dimitropoulos,D.,
Goodsell,D.S.,Prlic,A.,Quesada,M.,Quinn,G.B.,Westbrook,J.D.
et al.(2011)The RCSB Protein Data Bank:redesigned web site and web services.Nucleic Acids Res.,39,D392–D401.
6.Campanella,J.J.,Bitincka,L.and Smalley,J.(2003)MatGAT:
an application that generates similarity/identity matrices using
protein or DNA sequences.BMC Bioinformatics,4,29.
7.Holm,L.and Park,J.(2000)DaliLite workbench for protein
structure comparison.Bioinformatics,16,566–567.
8.Di Tommaso,P.,Moretti,S.,Xenarios,I.,Orobitg,M.,
Montanyola,A.,Chang,J.M.,Taly,J.F.and Notredame,C.(2011)
T-Coffee:a web server for the multiple sequence alignment of
protein and RNA sequences using structural information and
homology extension.Nucleic Acids Res.,39,W13–W17.
9.Gouet,P.,Robert,X.and Courcelle,E.(2003)ESPript/ENDscript:
extracting and rendering sequence and3D information
from atomic structures of proteins.Nucleic Acids Res.,31,
3320–3323.
10.Tamura,K.,Dudley,J.,Nei,M.and Kumar,S.(2007)MEGA4:
Molecular Evolutionary Genetics Analysis(MEGA)software
version4.0.Mol.Biol.Evol.,24,1596–1599.
11.Sayers,E.W.,Barrett,T.,Benson,D.A.,Bolton,E.,Bryant,S.H.,
Canese,K.,Chetvernin,V.,Church,D.M.,DiCuccio,M.,
Federhen,S.et al.(2011)Database resources of the National
Center for Biotechnology Information.Nucleic Acids Res.,39,
D38–D51.
12.Nuccio,S.P.and Baumler,A.J.(2007)Evolution of the chaperone/
usher assembly pathway:?mbrial classi?cation goes Greek.
Microbiol.Mol.Biol.Rev.,71,551–575.
13.Troisfontaines,P.and Cornelis,G.R.(2005)Type III secretion:
more systems than you think.Physiology,20,326–339.
Nucleic Acids Research,2012,Vol.40,Database issue D645
化脓隐秘杆菌毒力因子的研究进展
化脓隐秘杆菌毒力因子的研究进展 郭文洁,赵敬翠,刘耀川,朱竟赫,刘明春 (沈阳农业大学畜牧兽医学院,辽宁沈阳110161) 中图分类号:S852.61 文献标识码:A 文章编号:052926005(2010)0120052202 化脓隐秘杆菌(A rcanobacterium pyogenes)又名化脓放线菌或化脓棒状杆菌,为革兰染色阳性的短棒状杆菌,普遍存在于牛、羊、猪和其他重要经济动物的黏膜上[1],是一种条件性致病菌。化脓隐秘杆菌可在家畜间广泛传播,导致广泛多样的皮肤、内脏器官和关节的非特异性化脓性感染,如急性化脓性乳房炎、慢性脓肿性乳房炎、关节炎、心内膜炎、肝脓肿、子宫炎以及流产和不孕等,给养殖业带来较大的经济损失。 目前已发现的化脓隐秘杆菌毒力因子有4类,分别为化脓隐秘杆菌溶血素(PLO)、胶原结合蛋白(CbpA)、神经氨酸酶(Nan)及菌毛合成蛋白(Fim)。以下分别对各类毒力因子的研究现状做一综述,期望对化脓隐秘杆菌引起的感染性疾病的防治有所帮助。1 化脓隐秘杆菌的毒力因子 1.1 溶血素 化脓隐秘杆菌能产生一种溶血素(Pyol ysi n)即PLO,分子量为57.9kDa,由plo基因编码。plo基因含1605个碱基。若plo基因发生插入失活,将导致PLO的溶血活性丧失。 化脓隐秘杆菌溶血素是一种外毒素,能够溶解多种动物的红细胞、免疫细胞,并引起试验动物的皮肤坏死以及动物的死亡。PLO还呈现出对牛多形核粒细胞和袋鼠肾细胞的细胞毒性效应。PLO被认为具有氧化稳定性,并且具有胆固醇依赖性。由于在自然感染和人工感染动物的血浆中都发现了抗溶血素抗体,说明它能够在活体内表达并具有免疫原性。Billington S J等[2]于1997年研究发现,假设组织产生单独的溶血素,重组PLO的未纯化特异性抗体能完全中和化脓隐秘杆菌的溶血活性;此外,这些抗体还能够被动地保护小鼠防御化脓隐秘杆菌的致命性攻击。Jo st B H等[3]于1999年应用经甲醛灭活的重组体PLO对小鼠进行预防接种,发现其可保护小鼠免受腹膜内化脓隐秘杆菌的攻击;并于2003年发现了3种类毒素即H IS2PLO.F(497)、 收稿日期:2009201212 基金项目:国家自然科学基金(30972214);辽宁省自然科学基金(20082126);辽宁省教育厅科学研究计划(2008641) 作者简介:郭文洁(19832),女,硕士生,研究方向为兽医药理学与毒理学,E2mail:gwj0825@https://www.360docs.net/doc/7b2557457.html, 通讯作者:刘明春,E2mail:liumingchun@https://www.360docs.net/doc/7b2557457.html, HIS2PLO.Delta P(499)和HIS2PLO.A(522),均可用于小鼠的被动免疫,且这3种物质不具有溶血活性,因此无需灭活,进而实现了对化脓隐秘杆菌病的免疫预防[4]。 研究还发现PLO是巯基活化溶细胞素家族的成员,其结构有30%~40%与许多革兰阳性菌产生的巯基活化溶血素相同。但PLO与高度保守的巯基活化溶细胞素有所不同,特别是巯基活化必需的半胱氨酸残基被丙氨酸所取代。在诱变作用方面, PLO中的丙氨酸残基与半胱氨酸残基相比不具有巯基活化作用,进而导致毒素局部构象的差异。 1.2 胶原结合蛋白 胶原结合蛋白(collagen2 binding protein,CbpA)是在化脓隐秘杆菌中发现的第一种粘附素,它是一种蛋白,存在于细菌表面。Paula A E等[5]研究发现化脓隐秘杆菌的神经氨酸酶缺乏型突变体只是减少了对宿主细胞的粘附,并没有丧失粘附活性,进而应用Far Western blotting方法研究发现了CbpA蛋白。编码CbpA的基因为cbpA,含有3500个碱基,表达产物为124.7kDa。经克隆和序列分析证实CbpA为典型的细胞表面粘附素家族成员。CbpA蛋白包含有一个N2末端的配体结合区域(即A区域)和一个C2末端的重复区域(即B区域)。 CbpA的主要作用是连接胶原蛋白,促进细菌对宿主细胞的粘附,从而增强细菌的侵袭力,提高其毒力作用。6个组氨酸标记的重组体CbpA(HIS2 CbpA)能够与I、II和IV型胶原蛋白结合,但不表现纤维蛋白连接活性。另外,CbpA还可促进化脓隐秘杆菌与HeLa细胞株、3T6细胞系的粘附,敲除cbpA基因后其粘附力分别为原来的38.2%和57.0%。HIS2CbpA对化脓隐秘杆菌粘附性的调节具有剂量依赖性,且cbpA基因仅存在于48%的化脓隐秘杆菌中。因此,CbpA虽然对化脓隐秘杆菌的粘附性产生一定的影响,但并不如神经氨酸酶强[5]。最新研究表明,抗Cbp A的抗体能够有效抑制CbpA的聚集及其对胶原蛋白的粘附[6]。 1.3 神经氨酸酶 目前,在化脓隐秘杆菌中发现的神经氨酸酶(neuraminidase,Nan)有两种,即Nan H 和NanP,分别由nan H和nan P编码。Nan具有多种毒力作用,能够分解宿主细胞的唾液酸作为碳源,促进其在低养分条件下的生长;降低膜表面黏液的 25中国兽医杂志2010年(第46卷)第1期 Chinese Journal of Veterinary Medicine
大肠杆菌的研究与应用
大肠杆菌的研究与应用 中文摘要:大肠埃希氏菌(E.coli)通常称为大肠杆菌,是Escherich在1885年发现的,在相当长的一段时间内,一直被当作正常肠道菌群的组成部分,认为是非致病菌。直到20世纪中叶,才认识到一些特殊血清型的大肠杆菌对人和动物有病原性,尤其对婴儿和幼畜(禽),常引起严重腹泻和败血症。本文通过对大肠杆菌的结构及其致病机理等进行分析描述,以供大家参考学习。 关键词:大肠杆菌;致病性;危害;预防 The English abstract:Escherichia coli (E.c oli) are usually called escherichia coli, Escherich is found in 1885, in a long period of time, has been regarded as the normal bowel flora, that is part of the pathogen. Until the 20th century, realized some special type of escherichia coli serum of people and animals, especially for the infants and young (birds), often cause severe diarrhea and sepsis. Based on the structure and pathogenic escherichia coli mechanism analysis of reference, the study. Keywords:escherichia coli;The pathogenicity;Hazards;prevent 一、结构特征 大肠杆菌是人和许多动物肠道中最主要且数量最多的一种细菌,周身鞭毛,能运动,无芽孢。主要生活在大肠内。能发酵多种糖类产酸、产气,是人和动物肠道中的正常栖居菌,婴儿出生后即随哺乳进入肠道,与人终身相伴,其代谢活动能抑制肠道内分解蛋白质的微生物生长,减少蛋白质分解产物对人体的危害,还能合成维生素b和k,以及有杀菌作用的大肠杆菌素。正常栖居条件下不致病。它侵入人体一些部位时,可引起感染,如腹膜炎、胆囊炎、膀胱炎及腹泻等。人在感染大肠杆菌后的症状为胃痛、呕吐、腹泻和发热。感染可能是致命性的,尤其是对孩子及老人。其主要具有以下一些特征: 1、大肠杆菌是细菌,属于原核生物;具有由肽聚糖组成的细胞壁,只含有核糖体简单的细胞器,没有细胞核有拟核;细胞质中的质粒常用作基因工程中的运载体。 2、大肠杆菌的代谢类型是异养兼性厌氧型。 3、人体与大肠杆菌的关系:在不致病的情况下(正常状况下),可认为是互利共生(一般高中阶段认为是这种关系);在致病的情况下,可认为是寄生。 4、培养基中加入伊红美蓝遇大肠杆菌,菌落呈深紫色,并有金属光泽,可鉴别大肠杆菌是否存在。 5、大肠杆菌在生物技术中的应用:大肠杆菌作为外源基因表达的宿主,遗传背景清楚,技术操作简单,培养条件简单,大规模发酵经济,倍受遗传工程专家的重视。目前大肠杆菌是应用最广泛,最成功的表达体系,常做高效表达的首选体系。 6、大肠杆菌在生态系统中的地位,假如它生活在大肠内,属于消费者,假如生活在体外则属于分解者。[1]
克雷伯菌致病因子的研究进展_沈定树
?综述?克雷伯菌致病因子的研究进展 沈定树综述 施致远审校 【摘要】 荚膜是克雷伯菌的重要毒力因子,荚膜中K抗原的毒力程度与其所含的荚膜脂多糖 (CPS)中甘露糖的量有关,肺炎克雷伯菌荚膜可以抑制宿主的免疫反应。克雷伯菌有两种主要的菌 毛,均能使细菌与粘膜或泌尿生殖道、呼吸道和肠道的上皮细胞相结合。克雷伯菌抗血清杀菌活性主 要是通过补体介导所形成的连锁反应,在菌体的表面聚集成复合物,在防御机制中具有重要作用。铁 在细菌的生长繁殖中具有重要作用,含铁细胞主要是作为蛋白质的氧化还原催化剂,参与氧和电子的 转运过程。 【关键词】 克雷伯菌属; 致病因子 医源性克雷伯菌最常见的感染部位是泌尿道与呼吸道,由于这两个部位的免疫机理有很大的不同,因此引起泌尿道感染(urinary t ract infections,U TI)的克雷伯菌株毒力因子与呼吸道感染所分离出的菌株毒力因子亦不相同。 荚膜抗原 克雷伯菌的荚膜成分主要是由复合酸性脂多糖组成,其亚单位含有4~6个糖分子,常见的糖醛酸为负电荷,能分类成77个血清型。荚膜是克雷伯菌的重要毒力因子,由荚膜形成的纤维结构的厚包裹以多层方式覆盖在菌体的表面,从而保护细菌免受多形核中性粒细胞的吞噬。荚膜作用的分子机制是抑制补体的活性,特别是补体C3b。Y okochi等[1]报道,荚膜除了抗吞噬功能外,荚膜多糖还能抑制巨噬细胞的分化及功能。对鼠的研究表明,注入大剂量克雷伯菌荚膜脂多糖(cap sular polysaccharide,CPS),甚至可以产生免疫力的停顿,使抗特异性荚膜抗体的产生呈现剂量依赖性减少。不同的荚膜类型其毒力有很大的不同,表达有荚膜抗原K1和K2的菌株在鼠腹膜炎时毒力最强,而分离的其他血清型菌株则很少或没有毒力。在诱导鼠的皮肤损害时,克雷伯菌血清型K1、K2、K4和K5所表达的毒力比其他荚膜类型更强。 荚膜中K抗原的毒力程度与其所含的荚膜脂多糖(CPS)的甘露糖的量有关,低毒力的荚膜型,如K7或K21a抗原,含有甘露糖2α22/32甘露糖或L2鼠李糖2α22/32L2鼠李糖的重复序列,这种序列通过吞噬细胞表面植物血凝素来识别,植物血凝素介导调理素为依赖性吞噬作用,也称为植物血凝素吞噬作用。此种作用既可以由细菌表面的植物血凝素介导,也可以是用作受体的吞噬细胞植物血凝素介导。含有甘露糖2α2 2/32甘露糖2特异植物血凝素或甘露糖受体的巨噬细 胞能识别、消化随之破坏含有CPS重复序列甘露糖2α22/3甘露糖或L2鼠李糖2α22/3L2鼠李糖的分子结构。相反,缺乏这种重复序列的菌株则不被巨噬细胞所识别,因此也不会被吞噬。可见,不含有甘露糖或鼠李糖序列的血清型菌株与感染性疾病的关系更密切[2]。 荚膜抗原K2血清型在U TI、肺炎或菌血症患者分离出的菌株中是最常见的血清型,也是临床分离株中主要的血清型,但在自然环境中却很难碰到。有关荚膜甘露糖2α22/32甘露糖序列对清除宿主体内的克雷伯菌的意义,Kabha等[3]观察到,肺表面A蛋白(surfactant protein,SP2A)通过调理素作用和激活肺泡巨噬细胞而增强对克雷伯菌K1型菌株的吞噬作用,但对K2型菌株则不能。这种反应受甘露聚糖的抑制,因此也受吞噬细胞甘露糖受体的调节。Ofek 等[4]研究了肺表面蛋白2D(surfactant protein,SP2D)与肺炎克雷伯菌感染的肺泡巨噬细胞之间的关系,表明SP2D与克雷伯菌CPS的相互作用增加了肺泡巨噬细胞的免疫活性,对呼吸道病菌而言,SP2D在介导肺炎克雷伯菌的凝集和/或调理中具有重要作用。Sahly等[5,6]提出SP2D与O2多糖的相互作用对清除肺部的革兰阴性致病菌有重要作用。研究表明[7],血清型K21a、K26、K36和K50克雷伯菌株对回盲肠上皮细胞株(HC T28)和膀胱上皮细胞株(T24)的粘附性能比无荚膜的K21a/3、K36/3、K50/3的克雷伯菌变异株要明显降低。虽然荚膜型菌株在粘附水平上没有差别,但细菌通过膀胱细胞的内化比通过盲肠细胞的内化要明显低下,表明膀胱细胞缺乏对克雷伯菌内化所需的成分。Y oshida等[8]的动物试验结果提示,肺炎克雷伯菌荚膜可以抑制宿主的免疫反应,有利于细菌的生长,并导致肺炎、败血症乃至死亡。 菌 毛 在感染过程中,细菌必须尽可能与宿主黏膜表面 作者单位:317000浙江省台州医院检验科
细菌毒力岛的研究进展
细菌毒力岛的研究进展 1 毒力岛基本特征及分类 1.1基本特征 毒力岛(virulenceisland)又称致病性岛(pathogenicity island),是近年来在细菌分子学研究领域出现的新概念。1997年Hacker等对毒力岛下了较为精确的定义:即毒力岛是编码细菌毒力基因簇的一分子量相对较大的染色体DNA片段。毒力岛具有下列基本特征[1~4]:(1)编码细菌毒力基因簇的一个相对分子质量较大的(20~100k左右)染色体DNA片段。(2)一些毒力岛的两侧具有重复序列和插入元件,但是也可以没有。(3)毒力岛往往位于细菌染色体的tRNA基因位点内或附近,或者位于与噬菌体整合有关的位点,肠致病性大肠杆菌(EPEC)的LEE毒力岛就位于转运RNAselC位点[2,3]。(4)毒力岛DNA片段的G+Cmol%、密码使用和宿主细菌染色体有明显差异,有的比宿主细胞的G+Cmol%明显高,有的明显低。(5)毒力岛编码的基因产物许多是分泌性蛋白和细胞表面蛋白,如溶血素、菌毛和血红素结合因子,一些毒力岛编码细菌的分泌系统(如Ⅲ型分泌系统)、信息传导系统和调节系统。(6)一种病原菌可以有一个或几个毒力岛。(7)一部分学者认为,细菌的毒力岛应该包括位于噬菌体和质粒上的、与细菌的毒力有关的、其G+C 百分比和密码使用与宿主细胞明显不同的DNA片段。(8)毒力岛可能与新发现的病原性细菌有关。 1.2 分类 目前发现的毒力岛根据其G+C百分比与宿主菌的差异,可分成两类:即高G+C 毒力岛,如小肠结肠炎耶尔森菌的毒力岛;低G+C毒力岛,如大肠杆菌、沙门氏菌以及幽门螺杆菌中的毒力岛。根据毒力岛编码的产物性质可分为致病性岛和共生岛两大类。 2 结构与功能 2.1 结构 毒力岛是由独特的DNA片段构成,其不同来源的毒力岛的分子量、密码使用、G+C百分比各异。毒力岛主要含有与细菌毒力有关的基因,此外,RS和IR在毒力岛上也比较常见,而且,IR的类型也多种多样。大多数毒力岛在染色体上的位置
磷酸盐调节因子与大肠杆菌的的发病机制
磷酸盐调节因子与大肠杆菌的的发病机制摘要: 在感染过程中,细菌必须与调节基因表达相协调,以应对环境的刺激。磷酸盐调节因子被PhoBR的两个组件的调节系统所控制。PhoBR是在机体饥饿和盐酸盐调节基因处于稳态的时候被激活的调节基因,许多研究突出显示了Pho 调节因子在细菌的发病机制中起的重要作用,研究出了PhoBR基因是怎么被诱导表达的,另外调节基因参与盐酸盐的代谢系统,引导了许多细胞进程的调节。Pho调节因子有多种多样的功能,减弱毒性和改变许多病毒的特性,包括对宿主细胞的粘附力和对抗菌表位的抵抗力、酸度和氧化性的控制。这篇综述概述了Pho调节因子和大肠杆菌的致病性之间的关系,并举例说明,另外调节了磷酸盐的稳态,Pho调节因子在调节应激和毒力反应时起关键作用。 目录: 1.前言 2.Pho调节因子的诱导 3.Pho调节因子的活性和EXPEC的毒力 4.解剖对PhoBR和Pst系统有关的在特殊组织 5.氧化应激反应 6.细菌细胞表面的修饰 7.粘附素的产生和粘附 8.肠致病性大肠杆菌 9.Pst系统和肠致病性大肠杆菌菌株的粘附 10.在适应环境中出血性大肠杆菌时,Pho调节因子也被激活。 11.出血性大肠杆菌的毒力因子被PhoBR调节 12.结论 致谢 参考文献 1.前言 为了适应和在不同的微生物环境中生存,细菌必须感觉和回应细胞外的信号,对环境刺激的适当反应,可以被双组分调控的系统转导,这包括趋化现象的调节,渗透调节,新陈代谢和运输。一个典型的双组分调控系统(TCRS)由内膜组氨酸激酶传感器蛋白(HK)和一个反应调节器(RR),它作为DNA 粘合蛋白发挥作用,激活或基因表达复压。 磷,是一种细胞内容物,是细胞中第三丰富的元素,它存在于许多分子中,包括细胞膜脂,多糖和核酸。磷酸盐与能量的新陈代谢有关,也是一种转导信号,由TCRS介导。在细胞外集中的磷酸盐,由PhoR编码的HK和PhoB编码的RR双组分调控系统PhoBR运送,当细胞外聚集的磷酸盐低于4μM时PhoBR表达磷酸盐受限制,在磷酸盐受限制的条件下,PhoBR诱导基因属于磷酸盐调节子,它包含与获取能量和新陈代谢有关的不同的磷酸盐组基因。Pho调节因子的控制和跨膜信号转导很大程度上与大肠杆菌和芽孢杆菌环境中的无机磷有关,在大肠杆菌K-12中,Pho调节因子包括31个基因,除了参与磷酸盐的动态平衡,也与作为诱导结果,减弱细菌的毒性有关。 应对不同环境条件下入侵的病原体,宿主病原体相互作用是一个动态过程。病原体在宿主不同的部位生存,需要对环境中直接的不同刺激有适当的反应力。专门的调控系统控制毒力因子的表达,在许多调节水平相互作用中
猪链球菌2型毒力因子研究进展
猪链球菌2型毒力因子研究进展 摘要:猪链球菌是重要的人兽共患病,尤以猪链球菌2型最为流行。通过对猪链球菌2型毒力因子的研究,已确定几种主要毒力因子,研究中又发现几种新型的毒力因子,期望从这些毒力因子当中发现它们的功能及其相互关系,揭开猪链球菌感染的神秘面纱,进而控制猪链球菌病的发生。 关键词:猪链球菌2型;主要毒力因子;新型毒力因子 猪链球菌(Streptococcus suis,SS)是世界范围内引起猪链球菌病最主要的病原,也是重要的人畜共患病病原体,根据菌体荚膜多糖抗原性的不同,分为35个血清型(1—34型,1/2型)[1,2]。可以引起猪的脑膜炎、关节炎、败血症、心内膜炎、肺炎、流产、多浆膜炎及人的急性脑膜炎、永久性耳聋、感染性中毒性休克综合症等疾病,严重的可导致死亡[1,3,4]。主要致病血清型为SSl、SS2、SSl/2、SS7、SS9和SSl4,其中以SS2流行最广、致病性最强[5]。我国发生的猪链球菌病主要由C群和D群引起。由猪链球菌2型引起的猪链球菌病,过去在欧洲、南亚、北美一些国家流行比较多,但近些年在我国也时常发生。国内1991年在广东省首次分离鉴定到SS2;1998年我国江苏发生的猪链球菌2型引起的链球菌病造成几十人感染,十余人死亡;2005年在四川发生的猪链球菌2型引起的链球菌病导致206人感染,38人死亡;2006年9月,广西某猪场发生由猪链球菌2型引起的链球菌病,导致多头猪只死亡,所幸无人感染。由于猪链球菌2型引起的链球菌病属人兽共患病,死亡率较高,不仅可造成巨大的经济损失,而且给公共卫生带来严重威胁。从2007年至2010年,每年暑假我都去猪场实习,发现我所到的几个猪场都将链球菌病列入免疫程序当中,可见人们对链球菌的恐惧并未消除。 SS2存在着强致病力、弱致病力和无致病力菌株。强致病株引起猪严重的临床症状,并能从中枢神经系统分离出病菌。弱致病力菌株仅引起温和的临床症状,偶尔能从中枢神经系统分离出该菌。无致病菌力株不引起临床症状。其致病力的差异与各菌株的毒力因子直接相关,猪链球菌的毒力因子较为复杂,已知猪链球菌的主要毒力因子有溶菌酶释放蛋白(MRP)、细胞外蛋白因子(EPF)、溶血素(SLY)、荚膜多糖(CPS)、纤连蛋白/血纤蛋白原结合蛋白(FPBS)、毒力相关序列ORF2、谷氨酸脱氢酶(GDH)等,不同地区分离的猪链球菌菌株,其毒力因子出现的概率也不一样,尚缺乏统一的评价标准[6-9]。引起脑膜炎、败血症和关节炎综合症的猪链球菌大约只有一半能用目前发现的毒力因子作为检测指标。因此,探索新的毒力因子已成为该领域的研究热点和重点[3]。 1 主要毒力因子 MRP和EPF在致病性菌株检出率很高,在非致病性菌株中检出率极少,常作为判断致病性的指标之一;SLY分不具有很强的地域性,且在猪链球菌粘附和裂解Hep-2细胞的过程中发挥了重要的作用,纯化的SLY能导致人脑微血管内皮单层细胞产生病变;FPBS在细菌定植靶器官的过程中起到一定的作用;毒力相关序列ORF2在强弱毒株之间的序列存在差异,这种差异可能导致菌株致病性不同;GDH和CPS与猪链球菌的血清型分型有关,同时又都是毒力因子。 2 新型毒力因子 2.1 反应调节因子RevS基因 反应调节因子RevS基因是在2002年发现的SS2的第一个反应调节因子,并被证明是一个
构成细菌毒力的物质基础是---文本资料
一、填空题 1、构成细菌毒力的物质基础是和。 2、外毒素可分为、、三大类。 二、是非题 ()1、血桨凝固酶试验是检测金黄色葡萄球菌致病性的依据。 三、选择题 1、与细菌致病力有关的结构是: A、芽胞B、中介体C、异养菌D、菌毛 2、血浆凝固酶试验阳性,常见下列哪种菌 A、大肠杆菌 B、表皮葡萄球菌 C、淋球菌 D、金黄色葡萄球菌 3、外毒素的正确描述是 A、多数革兰阳性菌产生 B、为脂多糖 C、菌体死亡裂解后释放酶 D、多数革兰阴性菌产生 4、病原菌在血液中大量生长繁殖并引起严重临床症状 A、败血症 B、菌血症 C、毒血症 D、脓毒血症 5、与鉴别细菌有关的细菌代谢产物是 A、维生素 B、抗生素 C、细菌素 D、色素 6、构成细菌毒力的物质基础是 A、侵袭力和毒素 B、毒力 C、毒素 D、芽胞 四、名词解释 1、败血症
2、菌血症 3、毒血症 4、类毒素 五、问答题 1、简述构成细菌致病性的物质有哪些? 2、试述细菌外毒素与内毒素的主要区别点? 正确答案: 一、填空题 1.侵袭力、毒力 2. 神经毒、细胞毒、肠毒素 二、是非题 1.√ 三、选择题 1.D、 2.D、 3.A、 4.A、 5.D、 6.D 四、名词解释 2.病原菌侵入血流后,在其中大量繁殖并产生毒性产物,引起全身中毒症 状。 3.病原菌由局部侵入血流,但未在血流中生长繁殖,只是暂时或一过性通 过。 4.产外毒素的病原菌在局部组织中生长繁殖,只有外毒素进入血循环,并 损害易感的组织细胞,引起特殊的毒性症状。 5.细菌的外毒素经甲醛处理后,脱毒保留其抗原性,称类毒素 五、问答题: 1. 一细菌的毒力:①侵袭力:荚膜、微荚膜,菌毛(粘附素),侵袭性酶(血 浆凝固酶、透明质酸酶等)②毒素:外毒素主要由革兰阳性菌产生,为蛋白 质,由细胞毒、神经毒素和肠毒素组成。内毒素主要由革兰阴性菌产生,为脂多糖。 二侵入数量
致病菌毒力因子分析-VFDB 2012 update
VFDB 2012update:toward the genetic diversity and molecular evolution of bacterial virulence factors Lihong Chen,Zhaohui Xiong,Lilian Sun,Jian Yang*and Qi Jin* State Key Laboratory for Molecular Virology and Genetic Engineering,Institute of Pathogen Biology,Chinese Academy Medical Sciences and Peking Union Medical College,Beijing 100176,China Received September 15,2011;Accepted October 17,2011 ABSTRACT The virulence factor database (VFDB,http://www https://www.360docs.net/doc/7b2557457.html,/VFs/)has served as a comprehensive repository of bacterial virulence factors (VFs)for >7years.Bacterial virulence is an exciting and dynamic field,due to the availability of complete se-quences of bacterial genomes and increasing sophisticated technologies for manipulating bacteria and bacterial genomes.The intricacy of virulence mechanisms offers a challenge,and there exists a clear need to decipher the ‘language’used by VFs more effectively.In this article,we present the recent major updates of VFDB in an attempt to summarize some of the most important virulence mechanisms by comparing different compositions and organiza-tions of VFs from various bacterial pathogens,iden-tifying core components and phylogenetic clades and shedding new light on the forces that shape the evolutionary history of bacterial pathogenesis.In addition,the 2012release of VFDB provides an improved user interface.INTRODUCTION Bacterial virulence factors (VFs)are fascinating for a number of reasons.First,the ability of successful patho-gens to establish infections,produce disease and survive in a hostile environment is provided by a large armamentar-ium of virulence mechanisms.Elucidating the molecular mechanisms of VFs can improve understanding of the cellular and molecular basis of pathogenesis.Second,many important virulence factors interact with host cells and modulate their functions.Investigating the complex and ?nely balanced interactions between hosts and patho-gens can uncover useful tools for studying normal host cellular processes.Third,a much deeper understanding of the mechanisms of action of VFs will inform new avenues for identifying promising approaches to disease prevention and therapy.Fueled by recent technological innovations in the life sciences,the ?eld of microbial viru-lence has expanded rapidly over the past decade. Since its inception in 2004,the virulence factor database (VFDB,https://www.360docs.net/doc/7b2557457.html,/VFs/)has provided the broadest and most comprehensive up-to-date information regarding experimentally validated bacterial virulence factors (e.g.extracellular products,such as enzymes and toxins and secreted effectors or cell-associated products,such as capsular polysaccharides and outer membrane proteins),and has further explored plasticity in the reper-toire of VFs on an intra-genera level since its second release (1,2).To summarize the common themes in bac-terial virulence and to re?ect the diversity of genomic encoding,structural architecture and functional original-ity,we recently updated VFDB with an enhanced user interface and new contents dedicated to inter-genera com-parative analysis of VFs involved in host cell attachment and invasion,bacterial secretion systems and effectors,toxins,and iron-acquisition systems (Table 1).DATABASE UPDATES Data sources and processing The core dataset of VFDB only covers experimentally demonstrated VFs from 24genera of medically important bacterial pathogens.Several predicted VFs from complete genomes were also included for comparative analyses in the second release (2),but this information is still far from suf?cient for a comprehensive study of the genetic diver-sity and molecular evolution of VFs.Many VFs found in human pathogens have homologues present in animal or plant pathogens,and,sometimes,even in non-pathogens.Additionally,the genomic sequences encoding most func-tionally validated VFs are fragmentary,rather than complete genomes in the public domain.Therefore,via exhaustive literature screening and expert review,the *To whom correspondence should be addressed.Tel:+861067877732;Fax:+861067877736;Email:zdsys@https://www.360docs.net/doc/7b2557457.html, Correspondence may also be addressed to Jian Yang.Tel:+861067877735;Fax:+861067877736;Email:yangj@https://www.360docs.net/doc/7b2557457.html, The authors wish it to be known that,in their opinion,the ?rst two authors should be regarded as joint First Authors. Published online 8November 2011 Nucleic Acids Research,2012,Vol.40,Database issue D641–D645 doi:10.1093/nar/gkr989 ?The Author(s)2011.Published by Oxford University Press. This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://www.360docs.net/doc/7b2557457.html,/licenses/by-nc/3.0),which permits unrestricted non-commercial use,distribution,and reproduction in any medium,provided the original work is properly cited.
金黄色葡萄球菌毒力因子的研究进展
四综 述四 D O I :10.3760/c m a .j .i s s n .1673-436X.2015.16.009作者单位:530021南宁, 广西医科大学第一附属医院呼吸内科通信作者:陈一强,E m a i l :2667455027@q q .c o m 金黄色葡萄球菌毒力因子的研究进展 蔡双启 黄莹莹 陈一强 ?摘要? 金黄色葡萄球菌是社区获得性感染及医院获得性感染的常见致病菌三在金黄色葡萄球菌感染的发生二发展过程中,毒力因子起到了重要作用三本文就金黄色葡萄球菌的毒力因子作一综述,详细介绍杀白细胞素二溶血毒素二肠毒素二中毒休克综合征毒素1二耐热核酸酶二凝固酶以及表皮剥脱毒素的致病机制及研究现状三 ?关键词? 金黄色葡萄球菌; 毒力因子;研究进展R e s e a r c h p r o g r e s so nv i r u l e n c ef a c t o r so fS t a p h y l o c o c c u sa u r e u s C a iS h u a n g q i ,H u a n g Y i n g y i n g ,C h e n Y i q i a n g .D e p a r t m e n to f R e s p i r a t o r y M e d i c i n e ,t h eF i r s t A f f i l i a t e d H o s p i t a lo f G u a n g x i M e d i c a l U n i v e r s i t y ,N a n n i n g 530021,C h i n a C o r r e s p o n d i n g a u t h o r :C h e nY i q i a n g ,E m a i l :2667455027@q q . c o m ?A b s t r a c t ? S t a p h y l o c o c c u sa u r e u si sac o mm o n p a t h o g e n o fc o mm u n i t y - a c q u i r e di n f e c t i o na n d h o s p i t a l - a c q u i r e d i n f e c t i o n .V i r u l e n c e f a c t o r s p l a y a n i m p o r t a n t r o l e i n t h eo c c u r r e n c e a n dd e v e l o p m e n t o f s t a p h y l o c o c c a l i n f e c t i o n .T h i s a r t i c l e s u mm a r i z e s t h e v i r u l e n c e f a c t o r s o f S t a p h y l o c o c c u s a u r e u s ,i n t r o d u c e s l e u k o c i d i n ,h a e m o l y s i n ,s t a p h y l o c o c c a l e n t e r o t o x i n ,t o x i c s h o c k s y n d r o m e t o x i n -1,t h e r m o n u c l e a s e ,c o a g u l a s e a n de x f o l i a t i v e ,a n d t h e i r p a t h o g e n i cm e c h a n i s m s .?K e y w o r d s ? S t a p h y l o c o c c u s a u r e u s ;V i r u l e n c e f a c t o r s ;R e s e a r c h p r o g r e s s 金黄色葡萄球菌(S t a p h y l o c o c c u sa u r e u s ,S A )是皮肤二呼吸系统等感染的常见病原菌,其引起的坏死性肺炎及脓毒血症可直接威胁人类生命三S A 感染具有难治性和高病死率 的特点,与其可产生杀白细胞素(P a n t o n -V a l e n t i n e l e u k o c i d i n ,P V L )二溶血毒素(h a e m o l y s i n ,H L )[1] 等多种毒力因子密切相关三医学界针对S A 毒力因子在不同领域进行了多方面的研究,并取得了重大进展三本文拟对近年来S A 毒力因子的研究成果作一综述三 1 P V L P V L 是S A 产生的细胞外毒素,由V a n d eV e l d e 最早发 现三P V L 由S (L u k S -P V )和F (L u k F -P V )2类蛋白组成,相对分子质量分别为34000和33000,对应由l u k s - p v 和l u k f -p v 2个基因编码转录,此基因可通过噬菌体溶源性转换或质粒介导转入并整合至S A 的染色体上[2] 三S 组分能显著 增强人体巨噬细胞和中性粒细胞的趋化作用,它与细胞上特 异受体相结合后,启动钙离子通道,导致大量C a 2+ 内流,造成细胞裂解死亡三F 组分的特异性受体为卵磷脂,其可抑制环磷酸腺苷依赖性蛋白激酶的活性,导致细胞内大量环磷酸 腺苷蓄积,使细胞膜对C a 2+ 的通透性进一步增强,加剧细胞内外离子平衡紊乱三2种蛋白单独作用并不能促进白细胞 的坏死和凋亡,2种蛋白互相配对,形成环状结构的聚合体,插入靶细胞膜上,形成孔径大约为2n m 的孔道, 可选择性地允许二价阳离子如C a 2+二M g 2+等通过[3] , 进加速白细胞的坏死和凋亡三 P V L 早期被认为与皮肤感染如疖二 肿有关,社区获得性耐甲氧西林金黄色葡萄球菌(C A -M R S A )感染中,几乎均可检测出编码P V L 的l u k s -p v 和l u k f -p v 基因;而在大部分医院获得性耐甲氧西林金黄色葡萄球菌相关感染中,上述基因 并不能被检测出来[4] 三大多数学者认为P V L 增强了耐甲氧西林金黄色葡萄球菌的毒力[1] ,其与急性坏死性皮肤感染及肺部感染有相关性[5] 三但部分学者对P V L 的致病性提出了质疑,O t t o [6] 的研究为质疑提供了新的依据:①缺乏P V L 相 关基因的菌株仍具有显著的毒力;②在C A -M R S A 感染的动物实验中,P V L 相关基因敲除前后致病力没有太大改变三 这些不同的研究结论要求对P V L 在S A 的流行及发病机理中的作用进行更深层次的研究和探讨[ 7] 三2 H L H L 是S A 分泌的具有强致病性的穿孔素, 根据抗原性不同,将H L 分为α二β二γ二δ二ε5种类型三α-H L 为相对分子质量34000的不耐热蛋白质,在65?下30m i n 即可灭活,其由h l a 基因编码,受到a g r 二s a r A 等操纵子的调控;其作用方式为在细胞膜的疏水区形成微孔道,破坏细胞内外离子平 衡,致使细胞溶解[8] 三H L 可作用于红细胞二血小板和免疫细胞如淋巴细胞等;H L 可以改变血小板的形态, 被认为与S A 引起的败血症中发生的血栓事件有关[9] ;H L 可致多种 四 2421四国际呼吸杂志2015年8月第35卷第16期 I n t JR e s p i r ,A u g u s t 2015,V o l .35,N o .16
菌种毒力试验方法
菌种毒力试验方法 一、产毒液体培养基的制备 1.马铃薯-酵母膏-蔗糖培养基 取去皮马铃薯200-300g,切成小块,加水1000mL,煮沸20min,纱布过滤,制成马铃薯汁并补充水分至1000mL,加入10g酵母膏,100g蔗糖,分装后121℃高压灭菌15min。 2.麦芽汁-酵母膏培养基 1000mL麦芽汁中加入10g酵母膏,分装后121℃高压灭菌15min。 3.麦芽汁-蛋白胨培养基 1000mL麦芽汁中加入1g蛋白胨,分装后121℃高压灭菌15min。 4.葡萄糖天门冬素培养基 葡萄糖10.0 g 天门冬素0.5 g K2HPO4 0.5 g 蒸馏水1000.0 mL 调pH 7.2-7.4,分装后121℃高压灭菌15min。 5.高氏合成1号培养基 可溶性淀粉20.0 g KNO3 1.0 g K2HPO40.5 g MgSO4·7H2O 0.5 g NaCl 0.5 g FeSO4·7H2O 0.01 g 蒸馏水1000.0 mL 调pH 7.2-7.4,分装后121℃高压灭菌15min。 6.麦芽汁培养基 200 mL麦芽汁分装后,121℃高压灭菌15min。 7.0.5%蛋白胨培养基 取2.0g葡萄糖,0.6g酵母膏,1.0g蛋白胨,4.0g琼脂,加蒸馏水至200mL,分装后121℃高压灭菌15min。 二、培养物制备 将送检菌种或转种的纯培养物(适宜的培养基斜面,28±1℃培养5-7d),确证为纯培养物后,分别接种于适宜的产毒培养基中,一般菌种接种于麦芽汁-酵母膏、麦芽汁-蛋白胨及马铃薯-酵母膏-蔗糖三种产毒培养基中,放线菌接种于葡萄糖天门冬素培养基和高氏合成1号培养基中,红发夫酵母接种于麦芽汁培养基和0.5%蛋白胨培养基中,置28±1℃培养14d。