Bac-to-Bac杆状病毒表达系统

Bac-to-Bac杆状病毒表达系统
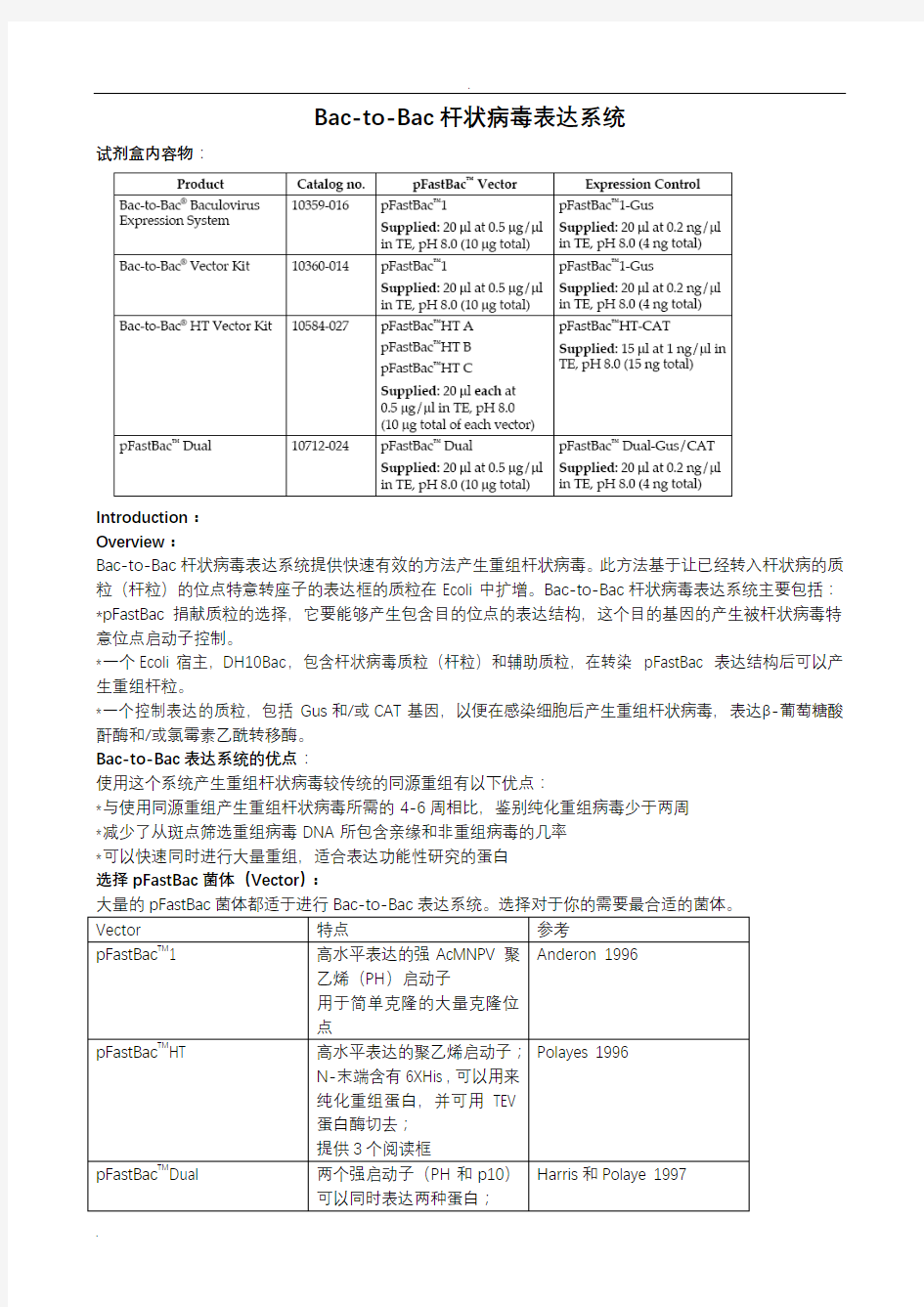
试剂盒内容物:
Introduction:
Overview:
Bac-to-Bac杆状病毒表达系统提供快速有效的方法产生重组杆状病毒。此方法基于让已经转入杆状病的质粒(杆粒)的位点特意转座子的表达框的质粒在Ecoli中扩增。Bac-to-Bac杆状病毒表达系统主要包括:*pFastBac捐献质粒的选择,它要能够产生包含目的位点的表达结构,这个目的基因的产生被杆状病毒特意位点启动子控制。
*一个Ecoli宿主,DH10Bac,包含杆状病毒质粒(杆粒)和辅助质粒,在转染pFastBac 表达结构后可以产生重组杆粒。
*一个控制表达的质粒,包括Gus和/或CAT基因,以便在感染细胞后产生重组杆状病毒,表达β-葡萄糖酸酐酶和/或氯霉素乙酰转移酶。
Bac-to-Bac表达系统的优点:
使用这个系统产生重组杆状病毒较传统的同源重组有以下优点:
*与使用同源重组产生重组杆状病毒所需的4-6周相比,鉴别纯化重组病毒少于两周
*减少了从斑点筛选重组病毒DNA所包含亲缘和非重组病毒的几率
*可以快速同时进行大量重组,适合表达功能性研究的蛋白
选择pFastBac菌体(Vector):
大量的pFastBac菌体都适于进行Bac-to-Bac表达系统。选择对于你的需要最合适的菌体。
指南提供了一个对于Bac-to-Bac表达系统的概述,并对以下提供指导:
1、克隆目的基因到pFastBac TM供体质粒的选择
2、转化pFastBac TM 结构到最高效的DH10Bac TM产生重组质粒
3、转染重组质粒DNA到昆虫细胞产生重组杆状病毒
4、扩增滴定(Amplify and titer)杆状病毒株,使用病毒株感染昆虫细胞表达目的重组蛋白
重要的:Bac-to-Bac杆状病毒表达系统是用来帮助你产生重组杆状病毒,在昆虫细胞中进行高水平表达目的基因的系统。虽然他可以帮助你很容易的产生杆状病毒表达你的重组蛋白,但是使用这系统更倾向于有杆状病毒生物学和昆虫表达背景的使用者。我们高度推荐使用者具有病毒和组织培养的技术和知识。
Bac-to-Bac表达系统
表达系统的成分:
表达系统简单高效的产生重组杆状不能给党。基于Luckow1993年的方法,此系统利用了位点特意的专座子Tn7区简化和提高重组质粒DNA
*系统的第一个成分是用来克隆目的基因的pFastBac TM菌株。基于pFastBac TM菌株的选择,表达基因被AcMNPV的PH或p10启动子控制,在昆虫细胞内高水平表达表达框被Tn7 的左右臂包围,并且包含一个庆大霉素抗性点和SV40多腺苷酸化信号,形成一个微型的Tn7
*第二个主要结构是Ecoli的DH10Bac TM品系,用来作为pFastBac TM菌株的宿主。DH10Bac TM细胞包含带有微型-attTn7靶位点的病毒质粒和辅助质粒(详细见下文)。一旦pFastBac TM表达质粒转入DH10Bac细胞,转化就会发生在pFastBac TM菌株的微型Tn7单位和微型-attTn7的靶位点之间产生重组质粒。这个转化反应发生在辅助质粒提供的转化蛋白处。
如果你已经完成了转化反应,你需要分离这个高分子量的重组质粒DNA并将质粒DNA转染如昆虫细胞趋产生重组杆状病毒,用来进行初步表达实验。在杆状病毒株扩增和滴定后,高效价的病毒株可以用来感染细胞,进行大规模的重组蛋白的表达。
杆状病毒菌株:
病毒菌株(杆粒),bMON14272(136KB),在DH10Bac Ecoli中包括:
*一个低拷贝的微型F复制子
*卡那霉素的抗性标记
*一个来自于pUC载体的编码LacZa肽的DNA片段,用来接触病毒转座子,Tn7(微型attTn7)已经被插入。插入的微型attTn7不会中断LacZa肽的阅读框。
杆粒在具有卡那抗性的大的质粒Ecoli DH10Bac中扩增,在显色物质如Bluo-gal 或X-gal和诱导剂IPTG 存在时,能补充染色体LacZ的缺失形成蓝斑(LacZ+)
辅助质粒:DH10Bac Ecoli也包含辅助质粒,pMON7124(13.2kb),他编码转移酶和四环素抗性基因。这个辅助质粒提供Tn7 转化功能。
图示Bac-toBac系统:下图刻画了重组病毒的产生和目的基因的表达
实验轮廓:
流程:下图揭示了表达目的基因的一般步骤
1、pFastBac donor 质粒(步骤:目的基因的克隆)得到
2、pFastBac重组体(转化至DN10Bac细胞(含有杆粒和helper))得到
3、含有重组杆粒的Ecoli细胞(重新划线)得到
4、验证过的含有重组杆粒的Ecoli细胞(过夜培养,分离重组杆粒DNA)得到
5、重组杆粒DNA (使用Cellfectin试剂感染细胞)得到
6、P1 重组杆状病毒株(>106pfu/ml)(感染昆虫细胞扩增病毒)得到
7、P2 重组杆状病毒株(>107pfu/ml)(滴定感染昆虫细胞)得到
8、蛋白的表达
培养昆虫细胞:
一般指导:
介绍:对于您的杆状病毒转移菌,我们推荐使用Sf9和Sf21昆虫细胞作为宿主。在开始你的转化试验和表达之前,确定你有收获的Sf9和Sf21,并将其冻存。
使用无血清的介质:昆虫细胞可能在无血清的条件下收获。我们推荐使用Sf900 Ⅱ SFM。Sf900 Ⅱ SFM 对于维持Sf9 和Sf21以及对于大规模生产重组蛋白,都是无蛋白的最优的介质。
昆虫细胞培养参考指导:维持和传代昆虫细胞在贴壁和悬浮条件下
冷冻细胞
使用无血清的介质
按比例增加细胞产量
一般指导:昆虫细胞对于环境很敏感,此外化学和营养因素,物理因素都可以影响昆虫细胞的成长。需要优化以得到最大产量。考虑以下培养条件:
*温度:细胞的感染和成长的最适合范围是27-28度
*PH:对于许多培养系统6.1-6.4时合适的范围。Sf900 Ⅱ SFM在此范围内支持一般的空气和开盖培养
*同渗重摩:鳞翅类细胞使用介质的最优同渗重摩是345-380 mOsm/kg
*通风:对于最优生长条件和蛋白的表达,昆虫细胞要求被动的通氧。积极的通氧系统要求溶氧饱和在10%-50%
*剪切力:悬浮培养产生机械剪切力。生长的昆虫细胞在含有血清的介质中(10%-20% FBS),对于细胞的剪切力一般会得到足够的保护。如果你的细胞在无血清条件下生长,加入剪切力保护剂例如PluronicF-68。注意:在Sf900 Ⅱ SFM中生长的细胞不需要加入剪切力保护剂。
转染的细胞:你需要的是对数期的细胞>95%发育能力,可以完成成功的转染。
产生重组的pFastBac TM菌株(Vector)
一般信息:
介绍:为了产生包含目的基因的重组质粒,使用Bac-to-Bac杆状病毒表达系统,你需要使用限制酶消化和连接,将你的目的基因克隆进入pFastBac 菌的其中一种。
一般分子生物学技术:为了帮助限制酶消化和连接DNA的序列,需要其他一般的分子生物学技术
扩增和保存质粒:pFastBac菌种和他的相应的表达对照质粒包含氨苄青霉素抗性基因,可以使用Ecoli 进行Amp筛选。为了扩增和保存pFastBac和pFastBac对照质粒,使用以下方法:
1、使用载体株系感染一个recA ,endA Ecoli株,例如Top10,DH10B或者DH5α
2、在含有100ug/ml Amp的LB琼脂糖平板选择转化株。
3、选择含有质粒的转化子制备甘油菌以便长期保存
克隆进入pFastBac TM1
介绍:为了帮助你设计策略克隆你的目的基因进入pFastBac TM1,参考以下建议和表格
克隆考虑事项:pFastBac TM1菌株是非融合菌株(无融合标签)。为了保证重组蛋白的表达,你的插入必须含有:
*一个ATG起始密码子用于转录起始
*一个终止密码子。注意:终止密码子包含在所有三个阅读框中的多克隆位点中
注意:重组蛋白的产生要求你的插入包含一个转录起始ATG。一般来说,转移载体包含完整的PH引导序列,可以提高表达的产量。蛋白翻译能够起始于多克隆位点上游突变的ATG(ATT),尽管如此,从这个位点起始的效率是低的,而且一般不干涉表达和重组蛋白的检测。
pFastBac TM1的多克隆位点:下图是pFastBac TM1的多克隆位点。限制位点标出来以便显示实际切刻位点。潜在的终止密码子下划线表示。pFastBac TM1的全序列可以从网站下载。pFastBac TM1的图谱和描述见后缀
克隆进入pFastBac TM HT A,B,C
介绍:pFastBac TM HT载体由三个读码框(A,B,C)提供的多克隆位点从而实现克隆目的基因,并且在N 端带有6XHis。
克隆:pFastBac TM HT菌株是融合菌株。为了保证表达重组蛋白,你必须:
*克隆你的基因要带有ATG,位于4050-4052碱基对间。这个将会产生融合表达,带有6XHis标签,可以用TEV切除
*你的插入要含有终止密码
注意:重组蛋白的产生要求你的插入包含一个转录起始ATG。一般来说,转移载体包含完整的PH引导序列,可以提高表达的产量。蛋白翻译能够起始于多克隆位点上游突变的ATG(ATT),尽管如此,从这个位点起始的效率是低的,而且一般不干涉表达和重组蛋白的检测
pFastBac TM HT A的多克隆位点:下图是pFastBac TM HT A的多克隆位点。其实ATG用黑体标出。限制性位点标出来以便显示实际切刻位点。
pFastBac TM HT B的多克隆位点:下图是pFastBac TM HT A的多克隆位点。其实ATG用黑体标出。限制性位点标出来以便显示实际切刻位点。框住的核苷酸显示出的是易变区域。
pFastBac TM HT C的多克隆位点:下图是pFastBac TM HT A的多克隆位点。其实ATG用黑体标出。限制性位点标出来以便显示实际切刻位点。框住的核苷酸显示出的是易变区域。注意pFastBac TM HT C在Xba Ⅰ位点内有一个终止密码子,他在N端标签框内。确定你的基因的5`端是在Xba Ⅰ位点上游开始。
克隆进入pFastBac TM Dual
介绍:pFastBac TM Dual包含两个多克隆位点,可以同时表达两个异源基因,一个通过PH启动子控制,另一个通过P10启动子控制。参见下列建议以便有助于你的基因克隆
克隆事项:pFastBac TM Dual是一个非融合载体。为了保证重组蛋白的表达,你的插入必须含有以下两点
*一个ATG起始密码子
*如果你不使用多克隆位点中的终止密码子,就必须要有一个终止密码子
注意:重组蛋白的产生要求你的插入包含一个转录起始ATG。一般来说,转移载体包含完整的PH引导序列,可以提高表达的产量。对于插入克隆上游的聚乙烯启动子,注意到蛋白翻译能够起始于多克隆位点上游突变的ATG(ATT),尽管如此,从这个位点起始的效率是低的,而且一般不干涉表达和重组蛋白的检测。PH启动子下游的多克隆位点:下图是pFastBac TM Dual中PH启动子下游的多克隆位点的图示。限制性位点标记出来显示了实际的切刻位点。潜在的终止密码子有下划线标出。
P10启动子下游的多克隆位点:下图是pFastBac TM Dual的AcMNPV p10启动子下游的多克隆位点。限制性位点标记出来以便显示实际切刻位点。潜在的终止密码子标记下划线。
转化和分析
介绍:如果你已经完成了你的连接反应,你就要准备把你的pFastBac TM结构转化进Ecoli了。很多Ecoli 宿主菌和转化程序都是可以使用的。一般推荐转化Ecoli和分析转化在下面列出
Ecoli宿主:一旦你把你的插入序列克隆进入了pFastBac TM,你需要转化连接反应到Ecoli,并选择优Amp 抗性的转化株。你可以使用任何recA,end A Ecoli菌。包括Top10,DH10B或者DH5α进行转化。不要转化连接反应进入DH10Bac细胞。注意TOP10和DH10B感受态细胞可以从Invitrogen得到
转化方法:你可以选择使用任何方法对Ecoli进行转化。化学转化是最便利的方法,而电转对大质粒是最有效的方法。选择好转化株,使用含有100ug/ml Amp 的LB平板。
转化株分析:一旦你得到Amp抗性转化株,我们有以下推荐:
1、选出10个转化株,在含有100ug/ml的Amp的LB或S.O.B培养基过夜培养
2、使用你选的方法分离质粒DNA。我们推荐使用公司的试剂盒
3、使用限制性方法分析质粒,证明是重组质粒并证明插入起始的正确。使用限制性酶或者酶化合物对
Vector和插入端各进行一次酶切。
使用PCR对转化株进行分析:你也可以使用PCR方法对阳性转化株进行分析。使用合适的PCR引物和Amp 条件。如果你是第一次使用此方法,你最好使用限制性分析作平行试验。使用错误的引物或污染的平板可能会得到假象。以下方法可以给你提供便利。其他方法也是可用的。
需要的材料:高保真PCRSuperMix,合时的PCR前后引物
过程:1、对于每个样品,在0.5ml的小离心管加入48ul的高保真PCR SuperMix和各1ul的前后引物。
2、选出10个克隆,在上述离心管中进行重悬(记得做一个Patch板保护克隆以便进行更深的分析)
3、94度10分钟溶解细胞灭活核酸酶
4、20-30个循环进行扩增
5、延伸使用72度10分钟。4度储存
6、琼脂糖凝胶电泳检测
测序:对于你的结构需要进行测序证明目的基因是正确的起始以便表达。如果你是将基因克隆进入了pFastBac TM HT,证明的基因进入了N末端标签的读框中。
长期保存:一旦你证明了测序的正确,确定克隆的纯度并制作甘油菌长期保存。推荐储存质粒DNA在-20度
1、在含有100ug/ml的LB平板上对原始的克隆进行划线培养
2、挑单克隆在1-2ml含有Amp的LB抚育
3、培养至稳定期
4、在0.85ml的培养物中混合0.15ml的过滤甘油,转到冷冻小管
5、-80度储存
重组质粒的产生
转化到DH10Bac Ecoli
介绍:一旦你有了你的pFastBac TM结构,你就可以准备转化纯化的质粒DNA进入DH10Bac TM Ecoli,转变成为杆粒。你可以使用蓝/白斑选出含有重组杆粒的克隆。Bac-toBac表达系统提供高效DH10Bac TM感受态细胞。
阳性对照:每个pFastBac TM质粒都提供相应的阳性对照用来作为阳性转染和表达对照(下表)。根据pFastBac TM Vecctor的使用,我们推荐在你的DH10Bac转染试验中作相应的对照。
所需材料:开始之前你需要有以下材料
*纯化的pFastBac TM结构(200pg/ul储存于PH8.0的TE)
*阳性表达对照
*高效DH10Bac TM感受态细胞(表达系统提供,每个转化试验使用1管细胞)
*pUC19(与DH10Bac一起提供,用来做对照)
*LB平板,包含Kan,Gen(庆大),Tetracycline(四环素),Bluo-gal(卤代吲哚基-beta-D-半乳糖苷)和IPTG。
(每个转化3个板,使用新鲜平板,推荐见下文)
*LB板含有100ug/mlAmp(用于pUC19转化对照)
*S.O.C介质
*15ml圆底塑料管
*42度水域和37度摇床及37度培养箱。
你需要准备LB琼脂糖平板,包含50ug/ml Kan,7ug/ml Gentamicin,10ug/ml tetracycline,100ug/ml Biuo-gal,40ug/ml IPTG,用来进行DH10Bac TM转化株的筛选。可以从我公司预订抗生素,Bbluo-gal,IPTG,在后面有制备平板的指导。如果你正准备的LB平板使用的是事先混合好的,我们推荐使用Luria Broth Base 代替LB。使用LB板将会降低颜色敏感度,并可能降低克隆的数量。注意:在蓝/白斑筛选中使用Bluo-gal 代替X-Gal。Bluo-gal产生的蓝斑颜色要比X-Gal深。
准备转化:对于每个转化,你都需要一小瓶感受态细胞和三个平板。
*42度水域平衡 *37度烘板30分钟 *室温放置SOC介质 *对于每个转化试验预冷一个15ml管子
转化过程:按照以下过程用高效DH10Bac TM感受态细胞转化pFastBac TM结构。我们推荐做阳性对照的转化来帮助你证明你的结果。
1、整管高效DH10Bac TM感受态细胞冰上解冻。
2、每个转化试验,轻轻地混合,将100ul高效DH10Bac TM感受态细胞倒入预冷的15ml管子
3、向细胞中加入适量的质粒DNA,轻轻混合。千万不要上下吹吸混合
*pFastBac TM结构:1ng(5ul)
*pFastBac TM对照质粒:1ng
*pUC19对照:50pg(5ul)
4、冰上抚育30分钟
5、42度热激45秒
6、立即冰上2分钟
7、加入900ul室温的SOC培养基
8、pFastBac TM转化:37度225转震荡培养4小时。pUC19转化:37度225转震荡培养1小时
9、对于每个pFastBac TM的转化:准备10折连续(不知道什么意思)用SOC稀释的细胞(10-1,
10-2,10-3),每个稀释用100ul铺一个LB平板,平板包含50ug/ml Kan,7ug/ml Gentamicin,
10ug/ml tetracycline,100ug/ml Biuo-gal,40ug/ml IPTG。对于pUC19转化:用SOC培
养基1:100稀释细胞,铺100ul稀释物在含有100ug/mlAmp的LB平板。
10、37度抚育48小时。分析筛选的克隆(参见推荐)。注意:我们不推荐早于48小时挑单克隆,
因为这样可能会难于区分蓝白斑。
重要的:微型Tn7 插入到微型attTn7 附属位点会干扰LacZa肽的表达,所以包含重组质粒的克隆在蓝色背景下是白色的,可能隐藏在未改变的质粒中。选择白斑分析。真正的白斑克隆比较大;因此为了避免选择成假阳性,选择最大的,最独立的白斑。避免挑出现灰色的或者在中间的菌落,因为他们可能是包含空质粒的细胞核重组细胞的混合。
验证显性:1、挑10个白色的克隆,重新划线在新鲜LB平板(50ug/ml Kan,7ug/ml Gentamicin,10ug/ml tetracycline,100ug/ml Biuo-gal,40ug/ml IPTG)。37度过夜培养。
2、在包含Bluo-Gal和IPTG的重新划线的平板上选出单克隆证明是白色的显性,嫁接至有
50ug/ml Kan,7ug/ml Gentamicin,10ug/ml tetracycline的液体中
3、使用我公司的试剂盒抽提重组质粒DNA。选择性的,你可以使用附录提供的操作步骤。这个
过程源于抽提大的质粒DNA(>100kb),适用于分离质粒DNA。
4、分析重组质粒DNA证明成功转化到了杆粒中。我们推荐使用PCR分析杆粒DNA。
注意:适用琼脂糖凝胶电泳可以证明转化的成功与否,他可以分出高分子量的DNA。这个方
法要比PCR分析可信度低,因为高分子量DNA是很难见到的。
使用PCR分析重组质粒
介绍:重组杆粒DNA要大于135kb。既然限制性分析很难完成这么大的DNA分析,我们推荐使用PCR分析鉴定重组杆粒中的目的基因。杆粒包含M13 正负引物位点,位于LacZa互不区域的微型attTn7 位点两侧,
可以完成PCR检验。本节提供使用M13 引物的PCR检验指导。
使用M13引物进行PCR分析:使用PCR法证明你的目的基因在重组杆粒中,你可能会:
*使用M13Forward(-40)和M13 Reverse引物
*使用M13Forward(-40)或M13 Reverse引物于你的插入片段杂交成联合物
DNA聚合酶:你可能会使用任何DNA聚合酶在你的PCR试验中,包括Platinum Taq酶。如果希望PCR产物>4kb,我们推荐使用聚合酶混合物。例如高保真的Platinum Taq酶
得到PCR产物:使用下面过程扩增重组杆粒DNA,适用M13正反引物和Platinum Taq酶。如果你使用M13F 或M13 R与一个你的基因特意引物混合,你需要自己决定扩增的条件。如果你使用其它的聚合酶,依照供应商提供的建议。注意:如果你的插入片段>4kb扩增条件需要被优化.
1、每个样品,在0.5ml微型管子中装入50ul的量进行PCR反映
重组杆粒DNA(100ng) 1ul
10XPCR Buffer 5ul
10mM dNTP Mix 5ul
50mM MgCl2 1.5ul
PCR引物(1.25ul/10uM) 2.5ul
蒸馏水 38.5
总体积 50ul
2、一滴矿物油覆盖
3、一下参量进行扩增
4、从反应产物吸取5-10ul进行琼脂糖电泳分析
你将会看到:如果转化发生了,而且你使用的是M13正负引物扩增,你将会在电泳上看到下列PCR产物大
小
如果你使用的是M13 和你的特异引物进行的扩增,你需要决定PCR产物是否是希望的。12页的图有助于你计算。
重组杆状病毒的产生
转染昆虫细胞:
介绍:一旦你证明了你的重组杆粒含有你的目的基因,就可以准备进行昆虫细胞的转染了,以产生重组杆状病毒。本章提供指导和建议以便完成昆虫细胞的转染
准备质粒:你可以使用任何方法准备纯化的重组杆粒DNA。杆粒DNA必须没有酚和NaCl的污染,他们会杀死细胞,盐类会干扰脂类复合物,降低转染效率。我们推荐使用本公司产品提质粒。
转染方法:推荐使用一个阳离子脂质体,例如Cellfectin试剂进行转染。此试剂用来提供给表达系统使用。
Cellfectin试剂:不做翻译
昆虫细胞系:推荐使用Sf9 和Sf21进行转染。High Five?and Mimic?Sf9不推荐因为他们转染效率低下。不过,如果你有了你的杆状病毒株,你可以进行High Five?and Mimic?Sf9表达试验。
转染介质:为了高效的转染,我们推荐在无Grace的昆虫细胞培养基中完成转染。注意Grace的昆虫细胞培养基应该不含有FBS(血糖),因为蛋白在血糖和其补充物中会与Cellfectin试剂互作,影响转染。
注意:如果你在Sf-900Ⅱ SFM中收获了Sf9或Sf21,你可以在不含有Grace的培养基中完成转染,转染后可以容易的将Sf-900Ⅱ SFM转变回去
阳性对照:如果你有了从pFastBac对照质粒中得到的重组杆粒,我们推荐,包括这个阳性对照在你的试验中和转化中能够评估你的结果。在这些杆粒中,包括β-葡糖甘酶(Gus)和/或氯霉素乙酰转移酶(CAT)将会在PH或者P10启动子控制下表达。在转染过后,表达的Gus和CAT可以适当进行验证。
所需材料:开始之前需要有下列材料:
*从pFastBac TM结构(500ng/ul TE溶解,PH8.0)中纯化的重组杆粒DNA
*从合适的pFastBac TM对照结构中纯化的重组杆粒DNA
*合适培养基收获的Sf9或者Sf21
*Cellfectin试剂(4度保存)
*Grace`s昆虫细胞培养基,未被补充的(培养基不含有补充剂,FBS或者抗生素)
*6孔培养板或者其它组织培养用具
*12X75mm灭菌管
*培养昆虫细胞的完全培养基
计算出你的转染试验所需的Sf9 细胞数量,进行扩增。转染前确定细胞健康且繁殖能力>97%。
转染条件:我们通常使用下列条件在Sf9中扩增杆状病毒株。对于你的转染试验这些条件应该作为一个起始点,你可以通过改变DNA和Cellfectin试剂的浓度以及细胞密度对条件进行优化。
转染步骤:适用下列步骤在6孔板进行Sf9 细胞的转染。如果你想在其它细胞培养用具中转染细胞,你需要对你的条件进行优化。记得使用无补充的Grace`s培养基,他不含有FBS或抗生素
1、在6孔板或35mm组织培养板,与每个孔2ml含有抗生素的培养基中(例如:2ml的SF900ⅡSFM
包含50单位/ml青霉素和50ug/ml链霉素)接种9X105Sf9细胞。
2、27度细胞最少接触1小时
3、对于每个转染的样品,准备杆粒DNA:Cellfectin试剂混合在12X75mm的灭菌管中
1)将1ug纯化的杆粒DNA稀释进入100ul未补充Grace培养基
2)完全混匀Cellfectin试剂,翻转混合5-10次。吸出6ulCellfectin试剂稀释进100ul 未补充Grace培养基。
3)使用稀释的Cellfectin试剂连接稀释的杆粒DNA(总体积约210ul)。轻混合,室温抚育15-45分钟。
4、在DNA/脂肪混合体抚育过程中,从细胞中除去培养基并用2ml未补充Grace培养基清洗。弃
去清洗培养基。
5、向每个含有混合体的管子中加入0.8ml未补充Grace培养基,清混匀,将混合体加入到含有细
胞的孔中。
6、27度培养5小时
7、弃去DNA/脂肪混合体,向细胞中加入2ml全培养基(例如SF900SFM含有抗生素)
8、27度潮湿条件抚育72小时或者指导你开始能够看到病毒感染的现象。提取P1病毒株。
分离P1病毒株
介绍:带有芽孢的病毒应该在转染以后释放入培养基72小时。但是,如果你的转染效率不好,细胞可能在转染后的4-5天也没有被病毒感染的迹象。转染过后的72小时,你需要亲自检查细胞是否有感染信号(下表)一旦细胞出现感染,使用下列步骤从培养基中收获细胞。
制备P1病毒株:1、一旦从第8步得到转染细胞,上述的晚期转染现象出现,就可以从每个孔中收集含有
病毒的细胞,并转入灭过菌的15ml带盖管子。500Xg离心5分钟除去细胞和大的碎片
2、将上清转到新的15ml离心管。这就是P1病毒株。4度避光储存。储存信息见下页
注意:如果你希望浓缩的病毒株,你可以在离心后,使用0.2um低蛋白吸附的滤器完
成。
病毒株的储存:按下列条件储存:
1、避光,4度
2、如果培养基不含血清,加入FBS至终浓度为2%,血清可以作为蛋白酶底物。
3、对于长期保存,重新扩增后保存整株病毒于-80度
4、不要在4度时储存经常使用的病毒。重复冻融会降低病毒的滴度。
下一步:得到你的纯化P1杆状病毒株之后,你可以:
1、扩增毒株(下节详细介绍)这个过程推荐得到最高滴度的毒株,而且在你的试验中优化
结果
2、决定你的毒株的滴定度(见病毒斑点试验)
3、如果你希望,纯化你的病毒结构
4、使用P1病毒株感染Sf9进行表达预试验。
注意:如果你想做一个小规模的或者是预试验,你可以直接使用P1毒株感染Sf9进行表达试验。由于毒株数量小,表达条件可能不能再生(如果滴度未知,MOI是不可知的)
扩增你的病毒株
介绍:P1毒株是小规模,低滴度的毒株,你需要使用这株去感染细胞,从而产生高滴度的P2株。转染Sf9细胞得到的首株的滴度一般为1X106到1X107pfu/ml。用以下步骤扩增得到的P2株滴度为1X107到1X108pfu/ml。本章提供P2株的制备和指导
所需材料:SF9或Sf21;P1病毒株;合适的培养用具;组织培养试剂;27度潮湿培养箱
重要的:扩增P1病毒株,需要感染Sf9或者Sf21 ,使用悬浮或单层培养。根据你的需要,你可以任何规模的扩增,但是你因你使用的P1菌株的量受到限制。一般扩增P1在10ml悬浮培养基中培养2X106细胞/ml 或在6孔板培养2X106细胞/ml。计算感染所需的Sf9细胞,随后Expand细胞。使用前确定你的细胞健康并且繁殖力大于97%
多样性感染(MOI):为了扩增毒株,在多样性感染细胞的范围是0.05-0.1。MOI就像每个细胞的病毒数量一样需要定义。使用下列公式计算获得特定的MOI需要多少病毒株:
注意:如果你没有滴定你的P1毒株,你需要假定滴度范围是1X106至107pfu/ml
例如:需要感染10ml细胞,2X106细胞/ml,使用MOI =0.1我们假定P1 毒株的滴度是5X106pfu/ml。
扩增步骤:依照下列步骤在6孔板扩增P1毒株
1、感染当天,准备Sf9和Sf21细胞上清和细胞板,2X106细胞/孔。接触培养细胞室温1小时
2、1小时后倒差显微镜检查细胞的吸附情况
3、每孔加入合适数量的P1毒株
4、27度潮湿培养箱抚育细胞48小时
5、感染后48小时,从每个孔收集2ml含有病毒的培养基,转到15ml的管子。500Xg离心5分
钟弃去细胞和碎片。注意:可以在感染晚期收获病毒(72小时)。每个杆状病毒的结构决定
了优化收获时间。过度培养将会由于细胞的调亡使生殖能力衰减
6、将上清转到15ml管子。这是P2毒株。4度避光保存。长期保存需要整株在-80度
7、检测你的P2毒株的滴度
梯度扩增步骤:一旦你有了高滴度的P2杆状病毒毒株,你可以梯度增加扩增病毒至任何体积。为了产生高滴度的P3,梯度扩增的细胞量和病毒体积要适当,遵照本章指导
从冰冻的主毒株获得高滴度毒株:如果你储存你的毒株在-80度,我们推荐使用此株产生另外的高滴度毒株进行表达试验。当病毒在-80度保存时,滴度会降低。下面为指导和扩增过程
病毒斑点试验(Performing a Viral Plaque Assy):
介绍:我们推荐使用斑点试验进行:决定你毒株的滴度;斑点纯化病毒(可选)
试验框架:决定病毒的滴度,你需要
1、在6孔板的Sf9细胞
2、准备10-flod连续稀释的病毒株
3、将不同稀释的杆状病毒加入到Sf9 和感染细胞中1小时
4、弃去病毒覆盖细胞层使用Plaquing培养基
5、抚育7-10天计算每个稀释物中的斑点
病毒滴度影响因素:影响滴度的因素很多:
1、目的基因的大小。滴度一般会随着目的基因的增大而减小
2、转染效率。为了高的转染效率,我们推荐使用Cellfectin试剂转染Sf9。准备DNA/脂质体混
合物在非补充Grace昆虫培养基
3、杆状病毒株的年龄。长期保存在4度或者-80度病毒的滴度都会减弱。如果你的病毒株已经
保存了6个月-1年我们推荐在使用前滴度或者重新滴度病毒
4、冻融次数。如果你储存在-80度,每次冻融都会降低病毒10%的滴度
5、错误的储存方式。为了经常使用,病毒株应该整株的保存在4度避光条件
所需材料:实验前所需材料:
1、你的澄清的杆状病毒株(使用前存在4度)
2、适当培养基中的Sf9或Sf21(每个被滴度的杆状病毒株为30ml对数期细胞,5X105细胞/ml)
3、SF900ⅡSFM或其它合适的完全生长培养基
4、Sf900培养基(1.3X)(100ml;或其它何时的培养基)
5、4%琼脂糖凝胶
6、灭菌的细胞培养级别的蒸馏水
7、100ml灭菌的玻璃瓶
8、6孔子之培养板(每个毒株2个板用来滴定)
9、无菌台 10、40度和70度水域 11、微波炉(可选) 12、27度潮湿培养箱注意:如果你在血清补充培养基培养Sf9 (完全TNM-FN)。你需要有以下试剂
1、Grace昆虫培养基,补充过的
2、Grace昆虫培养基(2X)
3、胎牛血清(FBS),高质量的热灭火的
准备斑点培养基(Plaquing Medium):斑点培养基由培养基和琼脂糖混合组成,对于斑点试验用来固定昆虫细胞。在开始下列步骤前立即准备斑点培养基。如果你培养Sf9在SF900ⅡSFM中,那准备Sf900斑点培养基。如果你培养是在TNM-FH,准备Grace斑点培养基。注意其它培养基也可以使用
1、在微波炉或者70度水域20-30分钟溶化4%的琼脂糖凝胶,溶化之后,下列物质放入40度水域
*空的,灭菌的100ml瓶子
*Sf-900培养基(1.3X)或者Grace昆虫细胞培养基(2X)
2、4%的琼脂糖凝胶液化后,将胶,培养基和空瓶子转到超净台
3、用下列方法快速制备斑点培养基:
Sf900 斑点培养基:混合30ml的Sf-900培养基(1.3X)和10ml的4%的琼脂糖凝胶在100ml
瓶子里,轻摇。
Grace`s斑点培养基:20ml热灭活的FBS加入到100ml Grace昆虫细胞培养基(2X)的瓶子。
将25ml含有血清的Grace昆虫细胞培养基(2X)和12.5ml细胞培养级别的灭菌蒸馏水以及
12.5ml溶化的琼脂糖胶混合在100ml空瓶子,轻轻混匀
4、使用之前将斑点培养基的瓶子放回40度水域
斑点试验的过程:使用以下步骤在6孔板完成斑点试验以决定拟的杆状病毒株的滴度。如果你有了杆状病毒表达对照,我们推荐你对这个也作滴度。记得在试验中做一个阴性对照(无病毒)注意:下列过程提供的量适合滴度一个病毒株(每个病毒株2个6孔板)。如果你想滴度更多,,相应的增加试剂量
1、转染当天,收获Sf9 ,在Sf900 ⅡSMF中制备30ml细胞上清,5X105细胞/ml。(或其它完全培
养基)。如果你有阴性对照,你需要另外的一个板子
2、是细胞在板底定居,盖盖室温抚育1小时
3、1小时培养之后,相差显微镜观察细胞。Sf9应该是已经吸附而且50%已经融合。
4、准备8个梯度稀释(10-1到10-8)的纯化病毒株,在Sf900 ⅡSMF或者未补充的Grace培养基,
无FBS。为了做这些,你需要在12ml管子顺序稀释0.5ml的病毒株或前面稀释过的4.5ml培养
基。需要完成8个管子。
5、将装有Sf9的6孔板和稀释过病毒的管子转移到超净台。标记上板子,需要2体积(一个样品一
个复制品)按如下方法:10-3,10-4,10-5,10-6,10-7,10-8。
6、从孔中除去培养基,立即用1ml适合的稀释的病毒代替。
7、室温抚育1小时
8、抚育1小时后,从40度水域中除去细胞和培养基瓶。
9、从最高的稀释到最低的稀释(10-8至10-3)顺序,从孔中除去含有病毒的培养基,使用2ml斑点
培养基代替。尽快操作避免细胞单层干燥
10、移动板子之前使用琼脂糖培养基覆盖10-20分钟
11、27度潮湿培养箱抚育7-10天之道斑点可见并可以计数。如果你想着色斑点计数,见下面,
计算滴度,见前面
注意:为了改进斑点的可见度,用中性红染色平板。其它平板染料比如水晶蓝不推荐,因为他们包含的有机溶剂能杀死宿主细胞。你可以使用下列之一:1、准备琼脂糖溶液包含有中性红,将溶液覆盖感染后的板子子上4天。感染后7-10天计算板子数量。2、准备中性红溶液加入到计数前1-2小时的平板(感染后7-10天)。重要的:如果你想斑点纯化你的病毒,不要染色斑点使用中性红,它可以诱导重组病毒的突变所需材料:开始试验前你需要有下列物质:
1、中性红
2、细胞培养级别的蒸馏水
3、Sf900 ⅡSMF或其它培养基(如果制备琼脂糖溶液)
4、4%琼脂糖凝胶(如果准备琼脂糖溶液)
5、40水域(如果制备琼脂糖溶液)
中性红染色过程:
准备琼脂糖覆盖的中性红(第4天使用)
1、在 Sf900 ⅡSMF(或其它全培养基)中制备1mg/ml的中性红。过滤除菌。
2、40度水域中50ml的管子混合下列试剂:1mg/ml的中性红(1.5ml);Sf900 ⅡSMF(16.5ml)
3、微波炉溶化4%的琼脂糖凝胶,40度水域5分钟
4、将含有中性红溶液的50ml管子和凝胶转到超净台。加入6ml琼脂糖凝胶到中性红溶液
5、每个孔用1ml中性红覆盖。琼脂糖凝胶凝固后,将平板放回27度潮湿培养箱知道斑点可以计数。
红色单层中将会出现清楚地斑点
准备中性红染色(第7-10天计数前使用)
1、准备1mg/ml的中性红加入到细胞培养级别的蒸馏水
2、每个孔加入0.5ml中性红溶液,室温抚育1-2小时
3、用滤纸或吸耳球轻轻除去过量的燃料,计算斑点。在红的背景下,透明胶上的斑点会清楚可见计算滴度:是用下列公式计算你的毒株的滴度。注意优化范围是6孔板上每个孔计算3-20个斑滴度(pfu/ml)=斑的数量X稀释因子X(1/接种体积ml/孔)
例如:我们在每孔含有10-6病毒稀释的孔中加入了1ml接种体积,得到了20个斑。使用公式得到滴度:滴度=20(斑)X106X(1/1ml/孔)=2X107pfu/ml
你将会看到:当滴度杆状病毒株时,一般得到的滴度范围为:P1为1X106-1X107pfu/ml;P2为1X107-1X108 pfu/ml。
注意:如果你的P1毒株滴度小于为1X106pfu/ml或P2小于1X107 pfu/ml,推荐生产新的毒株。
斑点纯化:你可以通过斑点纯化从单个的病毒克隆得到毒株。使用下列步骤
所需材料:Well-Space的病毒斑点的平板(来自斑点试验);活性大于95%的对数期的Sf9或Sf21;
灭菌的巴斯德吸管和曲径瓶
过程:1、培养Sf9或者Sf21参照斑点试验的1-3步
2、使用巴斯德管和曲径瓶,小心的选择一个清澈斑点,将琼脂糖盖子(含有病毒)转到1.5ml离
心管,其中包含有500ul全培养基。使用漩涡震荡混合均匀
3、每个孔加入100ul琼脂糖盖子溶液
4、27度潮湿培养箱抚育72小时
5、从每个孔收集含有毒株的培养基,转到15ml灭菌管。500Xg离心5分钟除去细胞和残片
6、将上清转到新鲜的15ml管子。这个是斑点纯化的毒株
7、以后过程为扩增杆状病毒株
表达重组蛋白
介绍:一旦你有了合适滴度的pFastBac 杆状病毒株,你可以感染昆虫细胞进行重组蛋白试验
阳性对照:如果你有高滴度的pFastBac 杆状病毒对照结构,你可能希望做一个你的试验对照。一旦你的对照病毒感染了你的细胞,基因编码的Gus和/或CAT将会限制表达,以便进行简单的试验。
表达指导:下面提供了一个使用重组病毒感染昆虫细胞表达目的蛋白的指导
*细胞系:取决于你的需要和目的基因,你可以使用任何昆虫细胞包括Sf9和Sf21或Mimic TM Sf9进行表达。细胞可以进行悬浮和贴壁培养。注意:你如果表达分泌蛋白,需要使用High
Five细胞改进表达
*培养条件:一般在无血清条件下使用Sf900 ⅡSMF或表达FiveSFM培养细胞。依据你的需要和目的蛋白,注意可能在感染后期需要添加0.1%-0.5%的FBS或BSA以便保护重组蛋白不
被水解。Protien-Based蛋白酶抑制剂一般比人工合成的蛋白酶抑制剂便宜而且高效
*感染条件:推荐在细胞处于中对数期时(密度为1X106-2X106细胞/ml)感染细胞。
确定在感染时培养不受营养或环境限制
*MOI:理想的MOI因细胞系间,相关感染动力学毒株或使用的克隆不同而不同。对于蛋白表达试验,应该为每个毒株,介质,反映和细胞系建立一个剂量反映,来证明感染参量的
最优。最为试验的起始,感染细胞使用的MOI为1-5
*时间进程:推荐实施一个时间进程决定表达蛋白的表达动力学,因为许多蛋白可能会在培养之中被细胞蛋白酶降解。注意:分泌蛋白的最大表达一般在感染30-72小时看到,非
分泌表达蛋白48-96小时。
最优的表达:大量因素影响着最优表达,包括细胞系,MOI,你的目的,基因种类等。你可以使用下列优化的条件表达你的重组蛋白
*细胞系:一定量MOI的感染的Sf9,Sf21,HighFive或Mimic TM Sf9。重组蛋白表达的试验在不同的感染后时间(24,48,72,96小时)。选择细胞系提供重组蛋白的最好表达条件
*MOI:不同的MOIs的感染的细胞量和蛋白试验。使用重组蛋白的最优水平提供的MOI
*时间进程:固定的MOI感染的细胞和试验在感染后不同时间表达重组蛋白(24,48,96)选择
最优条件
分析重组蛋白表达:是用下列过程分析你的重组蛋白。下列过程使用与24孔形式的感染后24-96小时收获的细胞进行分析。其它程序也可以参考
1、24孔板接种Sf96X105/孔.细胞接触至少30分钟
2、弃去培养基用新鲜的培养基清洗一次。换上300ul新鲜培养基
3、在希望的MOI值时每孔加入毒株。包括合适的对照
4、27度潮湿培养
5、合适的时间收获细胞或培养基(分泌表达)(感染后24,48,72,96)。如果收获细胞,弃去培养基,使用无血清培养漂洗一次。1XSDS-PAGE Buffer 400ul溶解细胞
6、-20度冷冻或煮沸样品最少3分钟,使用SDS-PAGE分离蛋白
重组蛋白的检测:可以使用任何方法,包括目的蛋白功能性检验和Western印记。如果你做WB,需要有你目的蛋白的抗体。
注意:如果你使用的是pFastBac TM HT表达蛋白,N末端的6XHis标签和TEV识别位点将会增加你的目的蛋白约3KD
β-葡糖甘酸酶试验:如果你的试验中包括了pFastBac TM1Gus或者pFastBac TM Dual-Gus/CAT结构,你需要使用下列方法进行β-葡糖甘酸酶试验。其他方法也可用:
*使用X-葡糖甘酸在琼脂糖板上检验篮斑
*定性的快速鉴定β-葡糖甘酸酶,使用X-葡糖甘酸混合小量的细胞观察它的蓝色。简单的,混合5ul 20mg/ml的X-葡糖甘酸(溶于DMSO或二甲基甲酰胺)到50ul细胞培养基。2小时内观察蓝色的变化
CAT蛋白试验:如果你的试验中包括了pFastBac TM HT-CAT或者pFastBac TM Dual-Gus/CAT结构,你需要使用你选择的方法进行CAT表达试验。CAT试验有显影试剂盒,如果做WB,可以从公司买抗血清
纯化重组蛋白:你可以使用任何方法纯化蛋白。注意:涂过你的目的基因在pFastBac TM HT的6XHis标签筐内,你可以使用亲和层析介质例如ProBond或Ni-NTA。
使用TEV蛋白酶除去N末端融合标签:如果你使用pFastBac TM HT表达融合蛋白,你可以使用TEV蛋白酶除去N末端融合的标签。注意:由于你可隆重使用的限制酶,增加得AA可能会出现在你的蛋白N末端
问题解决
TM
产生重组杆粒DNA:当转化到DH10Bac时你可能会遇到下列情况,及解决方法
过表达慢病毒载体构建和包装手册 version1
过表达慢病毒载体构建和包装手册 Version1.0 吉凯基因 二零一一年五月
目录 简介 (3) 第一部分过表达慢病毒载体的制备 实验流程 (4) 实验材料 (5) 过表达克隆制备 (6) 第二部分慢病毒包装与滴度检测 实验流程 (17) 实验材料 (18) L e n t i v i r u s病毒包装 (21) 病毒的收获及浓缩 (22) L e n t i v i r u s滴度测定 (24) 参考文献 (33)
简介 慢病毒(Lentivirus)载体是以人类免疫缺陷型病毒(HIV)为基础发展起来的基因治疗载体,它对分裂细胞和非分裂细胞均具有感染能力,并可以在体内较长期的表达且安全性高。吉凯基因提供的慢病毒为“自杀”性病毒,即病毒感染目的细胞后不会再感染其他细胞,也不会利用宿主细胞产生新的病毒颗粒。慢病毒中的毒性基因已经被剔除并被外源性目的基因所取代,属于假型病毒。但该病毒仍然具有可能的潜在的生物学危险,吉凯基因建议不要使用编码已知或可能会致癌的基因的假型病毒,除非已经完全公认某个基因肯定没有致癌性,否则均不建议采用假型病毒进行生物学实验。 吉凯基因慢病毒载体系统由GV慢病毒载体系列、pHelper 1.0载体和pHelper 2.0载体三质粒组成。GV慢载体中含有HIV的基本元件5’LTR和3’LTR以及其他辅助元件,例如WRE (woodchuck hepatitis virus posttranscriptional regulatory element)。通常根据不同的实验目的针对GV载体改造以进行基因功能研究。pHelper 1.0载体中含有HIV病毒的gag基因,编码病毒主要的结构蛋白;pol基因,编码病毒特异性的酶;rev基因,编码调节gag和pol基因表达的调节因子。pHelper 2.0载体中含有单纯疱疹病毒来源的VSV-G基因,提供病毒包装所需要的包膜蛋白。 吉凯基因过表达慢病毒产品可通过对GV慢病毒载体的改造和病毒包装,获得带有特定基因序列的慢病毒颗粒,以满足不同的实验需求。 本手册为吉凯基因RNAi慢病毒载体的构建和病毒包装的通用操作流程,目的是为了方便大家交流使用,部分细节内容未能做到一一详述,敬请谅解。同时希望大家能够针对手册中的错误和问题,提出宝贵的意见。
慢病毒系统简介及应用
慢病毒包装系统简介及应用 一、慢病毒包装简介及其用途 慢病毒(Lentivirus)载体是以HIV-1(人类免疫缺陷I 型病毒)为基础发展起来的基因治疗载体。区别一般的逆转录病毒载体,它对分裂细胞和非分裂细胞均具有感染能力。慢病毒载体的研究发展得很快,研究的也非常深入。该载体可以将外源基因有效地整合到宿主染色体上,从而达到持久性表达。在感染能力方面可有效地感染神经元细胞、肝细胞、心肌细胞、肿瘤细胞、内皮细胞、干细胞等多种类型的细胞,从而达到良好的基因治疗效果,在美国已经开展了临床研究,效果非常理想,因此具有广阔的应用前景。 目前慢病毒也被广泛地应用于表达RNAi的研究中。由于有些类型细胞脂质体转染效果差,转移到细胞内的siRNA半衰期短,体外合成siRNA对基因表达的抑制作用通常是短暂的,因而使其应用受到较大的限制。采用事先在体外构建能够表达siRNA的载体,然后转移到细胞内转录siRNA的策略,不但使脂质体有效转染的细胞种类增加,而且对基因表达抑制效果也不逊色于体外合成siRNA,在长期稳定表达载体的细胞中,甚至可以发挥长期阻断基因表达的作用。在所构建的siRNA表达载体中,是由RNA聚合酶Ⅲ启动子来指导RNA合成的,这是因为RNA聚合酶Ⅲ有明确的起始和终止序列,而且合成的RNA不会带poly A尾。当RNA聚合酶Ⅲ遇到连续4个或5个T时,它指导的转录就会停止,在转录产物3' 端形成1~4个U。U6和H1 RNA启动子是两种RNA聚合酶Ⅲ依赖的启动子,其特点是启动子自身元素均位于转录区的上游,适合于表达~21ntRNA和~50ntRNA茎环结构(stem loop)。在siRNA表达载体中,构成siRNA 的正义与反义链,可由各自的启动子分别转录,然后两条链互补结合形成siRNA;也可由载体直接表达小发卡状RNA(small hairpin RNA, shRNA),载体包含位于RNA聚合酶Ⅲ启动子和4~5T转录终止位点之间的茎环结构序列,转录后即可折叠成具有1~4个U 3 '突出端的茎环结构,在细胞内进一步加工成siRNA。构建载体前通常要通过合成siRNA的方法,寻找高效的siRNA,然后从中挑选符合载体要求的序列,将其引入siRNA表达载体。 慢病毒载体(Lentiviral vector)较逆转录病毒载体有更广的宿主范围,慢病毒能够有效感染非周期性和有丝分裂后的细胞。慢病毒载体能够产生表达shRNA的高滴度的慢病毒,在周期性和非周期性细胞、干细胞、受精卵以及分化的后代细胞中表达shRNA,实现在多种类型的细胞和转基因小鼠中特异而稳定的基因表达的功能性沉默,为在原代的人和动物细胞组织中快速而高效地研究基因功能,以及产生特定基因表达降低的动物提供了可能性。 慢病毒表达载体包含了包装、转染、稳定整合所需要的遗传信息。慢病毒包装质粒可提供所有的转录并包装RNA到重组的假病毒载体所需要的所有辅助蛋白。为产生高滴度的病毒颗粒,需要利用表达载体和包装质粒同时共转染细胞,在细胞中进行病毒的包装,包装好的假病毒颗粒分泌到细胞外的培养基中,离心取得上清液后,可以直接用于宿主细胞的感染,目的基因进入到宿主细胞之后,经过反转录,整合到基因组,从而高水平的表达效应分子。 二、这一系统的目的,主要是为了解决以下问题: 1. 对于一些较难转染的细胞,如原代细胞、干细胞、不分化的细胞等,能大大提高目的基因转导效率,而且目的基因整合到宿主细胞基因组的几率大大增加,这就为RNAi,cDNA克隆以及报告基因的研究提供了一个有利的途径。 2. 进行稳转细胞株的筛选;
杆状病毒表达系统简介
体外基因表达系统包括原核细胞系统和真核细胞系统。原核细胞系统主要是大肠杆菌细胞,它操作简便、周期短收益大及表达产物稳定,但是表达基因的相对分子质量有限,不宜过大,且不能对表达产物进行一些翻译后加工、修饰。真核细胞系统包括 CHO等哺乳动物细胞、酵母细胞和昆虫细胞等。昆虫细胞表达系统(即杆状病毒表达系统)具有独特的生物学特性,日益受到人们的重视。 1、杆状病毒的生物学特性 杆状病毒只来源于无脊椎动物,虽然已发现600多种杆状病毒,但进行分子生物学研究的不到20种。杆状病毒的基因组为单一闭合环状双链DNA分子,大小为80~160 kb,其基因组可在昆虫细胞核复制和转录。DNA复制后组装在杆状病毒的核衣内,后者具有较大的柔韧性,可容纳较大片段的外源DNA插入,因此是表达大片段DNA的理想载体。其中,用作外源基因表达载体的杆状病毒,目前仅限于核型多角体病毒(nuclear polyhedrosis virus,NPV)。该病毒颗粒在细胞内可由多角体蛋白包裹形成长度约1~5 m的包含体病毒,呈多角体形状。核型多角体病毒有两种形式:一种为包含体病毒(occluded virus,OV),另一种则为细胞外芽生病毒(budded virus,BV)。它们在病毒感染中扮演的角色不同,包含体病毒是昆虫间水平感染的病毒形式,昆虫往往是食入污染OV的食物后引起感染。包含体病毒外层裹了一层蛋白晶体,即为29 000的多角体蛋白,它对病毒的水平感染起以下作用: ①保护病毒颗粒在外界传播过程中免遭环境因素的破坏而失活。 ②保证病毒颗粒在适当的位置释放,引起感染。 昆虫中肠上皮局部的强碱性环境(pH=10.5),可使病毒颗粒释放蛋白酶溶解多角体。BV病毒是个体内细胞间的感染形式,由细胞芽生出BV,进入血淋巴系统中感染其它部位的细胞或直接在临近细胞内感染。 近几十年,有关杆状病毒基因结构、功能和表达调节的研究进展迅速,其中研究最深入的是苜蓿银蚊夜蛾(autogra—phacalifornica)多核型多角体病毒(multiple nuclear polyhedro-sis virus,MNPV),简称AcMNPV或AcNPV。该病毒是杆状病毒科 Baculoviridae 的原型,是一种大的、带外壳的双链DNA病毒,能感染30多种鳞翅目昆虫,被广泛用作基因表达系统载体。其它作为表达载体的杆状病毒,主要是来自家蚕的NP~(bombyx moil,BmNP~)。由于家蚕幼虫体内系统适合大规模地制备生产外源蛋白,且成本低,显示出良好的应用前景。本文主要介绍 AcNPV病毒,BmNPV在许多方面与其具有共同的特征。 AcNPV的基因表达分为4个阶段:立即早期基因表达、早期基因表达、晚期基因表达和极晚期基因表达。前两个阶段的基因表达早于DNA复制,而后两个阶段的基因表达则伴随着一系列的病毒DNA合成。其中在极晚期基因表达过程中,有两种高效表达的蛋白,它们是多角体蛋白和P10蛋白:多角体蛋白是形成包含体的主要成分,感染后期在细胞中的积累可高达30%~50%,是病毒复制非必需成分,但对病毒粒子却有保护作用,可使之保持稳定和感染能力另一类高效表达的极晚期蛋白为P10蛋白,也是一类病毒复制非必需成分,可在细胞中形成纤维状物质,可能与细胞溶解有关。多角体基因和P10基因现在都已被定位和克隆这两个基因的启动子具有较强的启动能力,因此这两个基因位点成为杆状病毒表达载体系统理想的外源基因插入位点。 杆状病毒基因组的结构和功能研究 杆状病毒基因组为双链环状 DNA分子。DNA以超螺旋方式被压缩包装在杆状核衣壳(rod.shaped nueleocapsid)内,核衣壳包被脂质蛋白囊膜(envelope)后形成病毒粒子。核衣壳包括衣壳(capsid)蛋白和髓核(COle)。其中衣壳蛋白是杆状病毒粒子的主要结构蛋白;髓核由病毒DNA分子和与其密切相关的碱性蛋白构成。碱性蛋白同DNA紧密结合以维持其复杂有序的超螺旋结构。 目前已知基因组全序列的杆状病毒有苜蓿丫纹夜蛾核型多角体病毒(AcMNPV)b]、家蚕核多角
Bac-to-bac表达系统中文版说明书
Bac-to-bac表达系统中文版说明书
Bac-to-Bac杆状病毒表达系统 试剂盒内容物: Introduction: Overview: Bac-to-Bac杆状病毒表达系统提供快速有效的方法产生重组杆状病毒。此方法基于让已经转入杆状病的质粒(杆粒)的位点特意转座子的表达框的质粒在Ecoli中扩增。Bac-to-Bac杆状病毒表达系统主要包括: *pFastBac捐献质粒的选择,它要能够产生包含目的位点的表达结构,这个目的基因的产生被杆状病毒特意位点启动子控制。 *一个Ecoli宿主,DH10Bac,包含杆状病毒质粒(杆粒)和辅助质粒,在转染pFastBac 表达结构后可以产生重组杆粒。*一个控制表达的质粒,包括Gus和/或CAT基因,以便在感
染细胞后产生重组杆状病毒,表达β-葡萄糖酸酐酶和/或氯霉素乙酰转移酶。 Bac-to-Bac表达系统的优点: 使用这个系统产生重组杆状病毒较传统的同源重组有以下优点: *与使用同源重组产生重组杆状病毒所需的4-6周相比,鉴别纯化重组病毒少于两周 *减少了从斑点筛选重组病毒DNA所包含亲缘和非重组病毒的几率 *可以快速同时进行大量重组,适合表达功能性研究的蛋白 选择pFastBac菌体(Vector): 大量的pFastBac菌体都适于进行Bac-to-Bac表达系统。选择对于你的需要最合适的菌体。
指南用途: 指南提供了一个对于Bac-to-Bac表达系统的概述,并对以下提供指导: 1、克隆目的基因到pFastBac TM供体质粒的选择 2、转化pFastBac TM 结构到最高效的DH10Bac TM产生重组质粒
慢病毒Cas9表达系统使用说明
慢病毒Cas9表达系统使用说明 本说明书用于: ?构建Cas9蛋白表达细胞系 ?在Cas9蛋白表达细胞系中敲除目的基因 适用于以下产品 货号货号 pLV‐Cas9载体系列 CR2001,CR2002 pLV‐Cas9‐Nick载体系列 CR2003,CR2004 pGR载体系列 CR2011~CR2013 pGR‐EGFP载体系列 CR2014~CR2016 北京英茂盛业生物科技有限公司 北京市昌平区沙河镇青年创业大厦B‐916 Tel:010‐62495135 Emai:order@https://www.360docs.net/doc/7f12356310.html, Web site:https://www.360docs.net/doc/7f12356310.html,
目录 1、产品简介 (2) 1.1CRISPR/gRNA基因敲除原理 (2) 1.2慢病毒Cas9表达系统特点 (2) 2、Cas9表达慢病毒制备 (3) 2.1试剂准备 (3) 2.2简要实验流程 (4) 2.3实验前准备 (4) 2.4病毒制备步骤 (5) 2.4 PEG纯化慢病毒 (6) 3、筛选Cas9表达稳定细胞株 (7) 3.1 试剂 (7) 3.2 实验前准备 (7) 3.3 筛选细胞系实验步骤 (8) 3.4 Cas9表达细胞系检测 (9) 4、用pTYNE载体对Cas9表达细胞系进行验证 (10) 4.1验证Cas9蛋白表达细胞系 (10) 4.2验证Cas9Nicknase蛋白表达细胞系 (11) 5、pGR和pGR‐EGFP载体构建 (13) 5.1 pGR和pGR‐EGFP载体图谱 (13) 5.2 靶点设计 (13) 5.3 pGR载体构建步骤 (14) 附录1 用到的产品 (17) 附录2 引物列表 (17) 1
慢病毒载体,稳定表达
慢病毒载体,稳定表达 一、慢病毒 逆转录病毒(Retrovirus):是一种RNA病毒,在复制时需在逆转录酶的作用下首先将RNA 转变为cDNA,再在DNA复制、转录、翻译等蛋白酶作用下扩增。主要包括RNA肿瘤病毒、慢病毒及泡沫病毒等三种亚科。 慢病毒(Lentivirus):属于逆转录病毒科,名称源自该种病毒长达数年的潜伏期。 最经典的慢病毒是由HIV病毒改造而来,而且HIV-1/HIV-2系统也得到了广泛的应用,除了HIV病毒系统以外,后续还有猿类免疫缺陷病毒(simian immunodeficiency virus, SIV)载体系统、猫免疫缺陷病毒(felines immunodeficiency virus, FIV)载体系统、绵羊梅迪-维斯纳病毒(MMV)载体系统和马传染性贫血(EIA)载体系统等。 慢病毒结构: 2个调节基因: (1)tat基因:反式激活因子,对HIV基因起正调控作用。 (2)rev基因:病毒蛋白表达调节因子,增加gag和env基因对结构蛋白的表达。 4个辅助蛋白(附属)基因: (1)vif和vpu调节感染性病毒颗粒的产生; (2)vpr和nef参与疾病的表现。 慢病毒的优势: 1.慢病毒携带的基因组可整合到宿主基因组,使宿主细胞长时间稳定表达外源基因; 2.可感染分裂和非分裂细胞; 3.低免疫原性,直接注射活体组织不易造成免疫反应,适用于动物实验; 4.可以更换特异性启动子; 5.野生型的HIV大小约为9.8 kb,插入片段可长达5-6 kb;
二、慢病毒载体 慢病毒载体(Lentivirus)是一类改造自人免疫缺陷病毒(HIV)的病毒载体,是逆转录病毒的一种,基因组是RNA,其毒性基因已经被剔除并被外源性目的基因所取代,属于假型病毒。可利用逆转录酶将外源基因整合到基因组中实现稳定表达,具有感染分裂期与非分裂期细胞的特性。 慢病毒包装过程: 慢病毒基因组进入细胞后,在细胞浆中反转录为DNA,形成DNA整合前复合体,进入细胞核后,DNA整合到细胞基因组中。整合后的DNA转录成mRNA,回到细胞浆中,表达目的蛋白;或产生小RNA。慢病毒介导的基因表达或小RNA干扰作用持续且稳定,并随细胞基因组的分裂而分裂。 慢病毒包装和侵染细胞的过程(元和生物) 三、慢病毒的使用和优势 慢病毒的使用量的取决因素:滴度,感染体积,MOI ,细胞密度 滴度(Titer):单位体积液体中有感染能力的病毒或噬菌体数目。单位:TU/mL (活性滴度单位)、copies/mL (物理滴度单位) 检测方法:定量PCR检测干扰后细胞基因组中外源DNA拷贝数。 实验原理:慢病毒介导外源基因以逆转录方式整合进目的细胞基因组。 图3 MOI(multiplicity of infection):感染复数或者复感染指数。指感染时病毒和细胞数量的比值。在实验中也将某个细胞达到80%感染时所需的MOI 值定义为这个细胞的MOI值。加的病毒量(μl)=细胞数×MOI/滴度(…/ml) ×1000。 最后,818 一些有关慢病毒方面的产品: 1.关于慢病毒载体构建方面: ORF表达克隆产品【LPP-货号-载体-100,ORF/Promoter/lncRNA慢病毒】 shRNA克隆产品【LPP-货号-载体-050,shRNA慢病毒】 miRNA克隆产品【LPP-货号-载体-050,miRNA/inhibitor慢病毒】
昆虫杆状病毒诱导宿主行为变化及其分子机制
Science of Sericulture 蚕业科学 收稿日期:2013-06-14接受日期:2013-06-30资助项目:国家自然科学基金项目(No.31272506)。第一作者信息:王国宝(1987-),男,博士研究生。 E-mail :gbwang0216@163.com 通信作者信息:吴小锋,教授,博士生导师。 E-mail :wuxiaofeng@zju.edu.cn * Corresponding author.E-mail :wuxiaofeng@zju.edu.cn 2013,39(5):1005-1010 ISSN 0257-4799;CN 32-1115/S E-mail :CYKE@chinajournal.net.cn 昆虫杆状病毒诱导宿主行为变化及其分子机制 王国宝吴小锋 (浙江大学动物科学学院,杭州310058) 摘要最近的研究发现杆状病毒感染能够诱导宿主昆虫产生行为变化,典型的表现为寄主运动能力的增强。从生物学的 角度分析,这是杆状病毒有利于自身传播的操控策略。本文综述了3种典型杆状病毒诱导宿主昆虫行为发生变化的现象以及其可能的分子机制。其中,舞毒蛾核型多角体病毒(LdMNPV )的egt 基因能够引起吉普赛舞毒蛾(Lymantria dispar )出现异常活跃的攀爬行为;而家蚕(Bombyx mori )与甜菜夜蛾(Spodoptera exigua )幼虫分别被家蚕核型多角体病毒(BmNPV )和苜蓿银纹夜蛾核型多角体病毒(AcMNPV )感染后,出现爬行异常活跃的行为则是由于病毒中ptp 基因的存在。不同种类的杆状病毒对宿主昆虫行为的操控策略不同,对杆状病毒操控宿主行为的分子机制的探索是一个新的研究领域,其研究成果不仅有助于揭示杆状病毒与寄主的互作关系,而且将为农林病虫害的生物防治提供新的参考策略。关键词 杆状病毒;egt 基因;ptp 基因;昆虫宿主;行为 中图分类号 S884.5+1 文献标识码 A 文章编号0257-4799(2013)05-1005-06 Host Behavior Alteration and Its Underlying Molecular Mechanism upon Infection of Insect Baculovirus WANG Guo-Bao WU Xiao-Feng * (College of Animal Sciences ,Zhejiang University ,Hangzhou 310058,China )Abstract Recent studies discovered that baculovirus infection could induce behavior alteration on their host insects.A typ- ical consequence of it is the enhanced locomotor activity.In view of biology ,it is a manipulatory strategy of baculovirus that favors its own transmission.This paper describes behavior alterations in host insect caused by three types of baculov-irus and possible molecular mechanisms underlying the alterations.Among them ,the egt gene of LdMNPV is essential for the hyperactive climbing behavior of Lymantria dispar ,while the ptp gene of BmNPV and AcMNPV is the cause of abnor-mal wandering behavior of Bombyx mori and Spodoptera exigua lavrae.Different types of baculovirus have various mecha-nisms in manipulating host insect behavior.Studies on the underlying molecular mechanisms of such behavior manipula-tions constitute a new and fascinating research field.It will not only uncover the interactive relationship between baculovirus and their host insects but also provide new strategy for biological control of agricultural and forestry pests /diseases.Key words Baculovirus ;egt gene ;ptp gene ;Insect host ;Behavior 杆状病毒是一个庞大的病毒家族,其基因组大 小在80 180kb 之间,为共价闭合环状双链DNA ,包含100多个基因。有趣的是,其中超过10%的基因是从原始宿主中通过水平转移获得的,这些基因 被利用后更加有利于病毒的复制和传播[1] 。已发现的几百种昆虫杆状病毒分为杆状病毒属(NPV )和颗粒体病毒属(GV )2个属。杆状病毒在感染循环过程中会产生遗传物质完全一致但表型具有差
慢病毒转染手册
慢病毒(Lentivirus)载体是以HIV-1(人类免疫缺陷I型病毒)为基础发展起来的基因治疗载体。区别一般的逆转录病毒载体,它对分裂细胞和非分裂细胞均具有感染能力。 基本概述 慢病毒载体的研究发展得很快,研究的也非常深入。该载体可以将外源基因有效地整合到宿主染色体上,从而达到持久性表达。在感染能力方面可有效地感染神经元细胞、肝细胞、心肌细胞、肿瘤细胞、内皮细胞、干细胞等多种类型的细胞,从而达到良好的的基因治疗效果,在美国已经开展了临床研究,效果非常理想,因此具有广阔的应用前景。 慢病毒的应用 目前慢病毒也被广泛地应用于表达RNAi的研究中。由于有些类型细胞脂质体转染效果差,转移到细胞内的siRNA半衰期短,体外合成siRNA对基因表达的抑制作用通常是短暂的,因而使其应用受到较大的限制。采用事先在体外构建能够表达siRNA的载体, 然后转移到细胞内转录siRNA的策略,不但使脂质体有效转染的细胞种类增加,而且对基因表达抑制效果也不逊色于体外合成siRNA,在长期稳定表达载体的细胞中,甚至可以发挥长期阻断基因表达的作用。在所构建的siRNA表达载体中,是由RNA聚合酶Ⅲ启动子来指导RNA合成的,这是因为RNA聚合酶Ⅲ有明确的起始和终止序列,而且合成的RNA不会带poly A尾。当RNA聚合酶Ⅲ遇到连续4个或5个T时,它指导的转录就会停止,在转录产物3’端形成1~4个U。U6和H1 RNA启动子是两种RNA聚合酶Ⅲ依赖的启动子,其特点是启动子自身元素均位于转录区的上游,适合于表达~21ntRNA和~50ntRNA茎环结构(stem loop)。在siRNA表达载体中,构成siRNA的正义与反义链,可由各自的启动子分别转录,然后两条链互补结合形成siRNA;也可由载体直接表达小发卡状RNA(small hairpin RNA, shRNA), 载体包含位于RNA聚合酶Ⅲ启动子和4~5T转录终止位点之间的茎环结构序列,转录后即可折叠成具有1~4 个U 3 ’ 突出端的茎环结构,在细胞内进一步加工成siRNA。构建载体前通常要通过合成siRNA的方法,寻找高效的siRNA,然后从中挑选符合载体要求的序列,将其引入siRNA表达载体。 慢病毒载体
杆状病毒——昆虫细胞表达系统
实验材料: 1. 重组杆状病毒质粒:Bacmid/nsp-6及阳性对照Bacmid/CAT,已构建成功。 2. 昆虫细胞Sf9、High Five及其相关培养基、转染试剂均购自Invitrogen公司。抗His单克隆抗体购自Oncogene公司,CAT-ELISA试剂盒购自Roche。 实验步骤: 一、昆虫细胞转染: 1. Sf9细胞计数,取6孔板中的两孔,每孔加入9×10 5个细胞(其中一孔设为正常对照),并以全培培养至少1小时,使细胞贴壁。 2.准备重组质粒和细胞转染试剂的混合物: a. 溶解1μg纯化重组杆状病毒重组质粒于100μl 无添加成分的Grace’s Medium。 b. 转染试剂充分摇匀后取6μl加入100μl 无添加成分的Grace’s Medium,混匀。 c. 将上述稀释好的质粒及稀释好的转染剂混匀,室温孵育20min。 3.重组质粒与转染剂混合液孵育的同时,以2ml无添加成分的Grace’s Medium洗涤待转染的一孔细胞并弃去洗液。 4.取0.8ml无添加成分的Grace’s Medium加入质粒与转染剂的混合液中,轻轻混匀后,总体积约为1ml。加入上步洗涤后的细胞孔中,27℃继续培养5h。 5.移除质粒、转染剂混合物,加入2ml全培。27℃湿盒孵育,直到病变现象产生。 二、病毒贮液的制备: 1. 病毒感染晚期(正常24-72h)可见细胞停止生长、黏附,呈颗粒状外观。即收集含病毒的培养上清,500g离心5min,去除细胞和碎片。 2. 上清即为P1病毒贮液,移入新的离心管中4℃避光保存。长期保存分装冻存于-80℃。 3. 病毒贮液的扩增,按以下公式进行所需病毒P1贮液的量: 感染所需病毒贮液量(ml)= [MOI(pfu/cell) ×细胞数÷病毒贮液效价(pfu/ml)] 注:若不进行病毒空斑测定,P1贮液效价按照1×10 6到1×10 7计。 4. 扩增P1液制备P2病毒贮液方法如下: a. 转染当天,取2×106个细胞/孔加入六孔板中,贴壁生长至少1h。 b. 每孔加入适量的P1贮液,27℃湿盒孵育48h。 c. 根均细胞病变情况(约48h后)收集各孔中的病毒上清液,500g离心5min取上清,即为P2病毒贮液。4℃避光保存。长期保存分装冻存于-80℃。 d. 制备了高效价的P2液后,按上述方法扩增P3贮液,用于高效表达。 三、病毒贮液初步鉴定 1. SDS-PAGE蛋白电泳:取P2或P3病毒贮液浓缩后上样(或直接上样)进行SDS-PAGE 蛋白电泳,初步根据分子量对表达蛋白进行鉴定。CAT-his融合蛋白分子量约为28KDa,nsp6-his融合蛋白分子量约为34KDa。 2. western blot:以小鼠抗his单克隆抗体鉴定在相应条带出是否有his标记。 3. 以CAT-ELISA检测试剂盒检测Bacmid/CAT对照的蛋白表达情况。方法详见CAT-ELISA 试剂和说明书。 结果 1. CAT-ELISA检测试剂盒成功检测出对照CAT质粒在细胞中的表达,同时可测于上清和细
杆状病毒表达蛋白
杆状病毒表达蛋白 1 donor vector的构建 1.1 外源基因的连接 1.2 转化 1.2.1 取10μL连接产物放入100μL感受态细胞中。 1.2.2 冰浴30min。 1.2.3 42℃,90s。 1.2.4 马上放入冰浴中,2min。 1.2.5 加入800μL LB培养基,37℃,150rpm摇1hr。(LB 37℃预热) 1.2.6 取200μL涂Amp+LB平板。 1.2.7 37℃,恒温培养箱,倒置培养12-16hr。 1.3 鉴定 1.3.1 挑取10个单菌落于Amp+LB培养基中,37℃,225rpm,摇过夜。 1.3.2 小量提取质粒,操作步骤见附录2。 1.3.3 用SalI及XbaI双酶切鉴定连接结果。 1.3.4 取1ml菌液测序。 1.4 菌种的保存 1.4.1挑取单菌落于1-2ml Amp+LB培养基中。 1.4.2 37℃,225rpm摇至平稳期。 1.4.3 0.85ml细菌培养物+0.15ml灭菌甘油,混匀。 1.4.4 -80℃保存。 2 转座 2.1 转座反应 2.1.1 将DH10Bac放入冰中备用。 2.1.2 轻轻混匀,取100μL DH10Bac细胞于遇冷的15ml圆底灭菌管中。 2.1.3 加入下列质粒DNA并轻混匀。 pFastBac TM construct 1ng(5μL) pFastBac TM control 1ng PUC19 control 50pg 2.1.4于冰中放置30min。 2.1.5 42℃,热激45s,不要摇动。 2.1.6 马上放入冰浴中,2min。 2.1.7 加入900μL室温LB培养基。 2.1.8 37℃,225rpm摇4hr。 2.1.9准备一系列10倍稀释的细菌培养物(10-1、10-2、10-3),每板加入100μL的稀释菌液。(平板为:50μg/ml kana、7μg/ml的genta、10μg/ml tet、100μg/ml Bluo-gal、40μg/ml IPTG)。 2.1.10 37℃,倒置培养48hr,挑取白斑进行分析。
杆状病毒表达系统(The Baculovirus Expression System)
Baculovirus Facility in Cambridge The Baculovirus Expression System The Baculovirus Expression Vector System (BEVS) has been widely used in research and scientific industrial communities for the production of high levels (up to 1000mg/mL) of properly post-translationally modified (folding, disulfide bond formation, oligomerization, glycosylation, acylation, proteolytic cleavage), biologically active and functional recombinant proteins. The Baculovirus Expression Vector System is based on the introduction of a foreign gene into nonessential for viral replication genome regio n via of homologous recombination with a transfer vector containing target gene. The resulting recombinant Baculovirus lacks one of nonessential gene (polh, v-cath, chiA etc.) replaced with foreign gene encoding heterologous protein which can be expressed in cultured insect cells and insect larvae. Baculovirus Facility in Cambridge offers services on protein expression and production - from recombinant baculovirus production to large scale protein expression. Several features make the Baculovirus Expression Vector System attractive for researchers: - High levels of heterologous gene expression are often achieved compared to other eukaryotic expression systems, particularly for intracellular proteins. In many cases, the recombinant proteins are soluble, post-translationally modified and easily recovered from infected cells late in infection when host protein synthesis is diminished. -The cell lines used for AcMNPV propagation grow well in suspension cultures, permitting the production of recombinant proteins in large-scale bioreactors. - Expression of hetero-oligomeric protein complexes can be achieved by simultaneously infecting cells with two or more viruses or by infecting cells with recombinant viruses containing two or more expression cassettes. - Baculoviruses have a restricted host range, limited to specific invertebrate species. They are safer to work with than most mammalian viruses since they are noninfectious to vertebrates.
杆状病毒介绍
杆状病毒 关键词:昆虫病毒,杆状病毒,核型多角体病毒,颗粒体病毒,质型多角体病毒 杆状病毒是一类在自然界中专一性感染节肢动物的DNA病毒,病毒粒子呈杆状,基因组为双链环状DNA分子,DNA以超螺旋形式压缩包装在杆状衣壳内,大小在90~180 Kb之间。目前杆状病毒作为高效、安全的无公害生物虫剂广泛应用于害虫防治。杆状病毒只来源于无脊椎动物,虽然已发现600多种杆状病毒,但进行分子生物学研究的不到20种。杆状病毒的基因组为单一闭合环状双链DNA 分子,大小为80~160 kb,其基因组可在昆虫细胞核复制和转录。DNA复制后组装在杆状病毒的核衣内,后者具有较大的柔韧性,可容纳较大片段的外源DNA 插入,因此是表达大片段DNA的理想载体。其中,用作外源基因表达载体的杆状病毒,目前仅限于核型多角体病毒(nuclear polyhedrosis virus,NPV)。该病毒颗粒在细胞内可由多角体蛋白包裹形成长度约1~5 m的包含体病毒,呈多角体形状。核型多角体病毒有两种形式: 一种为包含体病毒(occluded virus,OV), 另一种则为细胞外芽生病毒(budded virus,BV)。 它们在病毒感染中扮演的角色不同,包含体病毒是昆虫间水平感染的病毒形式,昆虫往往是食入污染OV的食物后引起感染。包含体病毒外层裹了一层蛋白晶体,即为29 000的多角体蛋白,它对病毒的水平感染起以下作用:①保护病毒颗粒在外界传播过程中免遭环境因素的破坏而失活。②保证病毒颗粒在适当的位置释放,引起感染。昆虫中肠上皮局部的强碱性环境(pH=10.5),可使病毒颗粒释放蛋白酶溶解多角体。BV病毒是个体内细胞间的感染形式,由细胞芽生出BV,进入血淋巴系统中感染其它部位的细胞或直接在临近细胞内感染。近几十年,有关杆状病毒基因结构、功能和表达调节的研究进展迅速,其中研究最深入的是mùxu苜蓿银蚊夜蛾(autogra—phacalifornica)多核型多角体病毒(multiple nuclear polyhedro-sis virus,MNPV),简称AcMNPV或AcNPV。该病毒是杆状病毒科 Baculoviridae的原型,是一种大的、带外壳的双链DNA病毒,能感染30多种鳞翅目昆虫,被广泛用作基因表达系统载体。其它作为表达载体的杆状病毒,主要是来自家蚕的NP~(bombyx moil,BmNP~)。由于家蚕幼虫体内系统适合大规模地制备生产外源蛋白,且成本低,显示出良好的应用前景。本文主要介绍 AcNPV病毒,BmNPV在许多方面与其具有共同的特征。 AcNPV的基因表达分为4个阶段:立即早期基因表达、早期基因表达、晚期基因表达和极晚期基因表达。前两个阶段的基因表达早于DNA复制,而后两个阶段的基因表达则伴随着一系列的病毒DNA合成。其中在极晚期基因表达过程中,有两种高效表达的蛋白,它们是多角体蛋白和P10蛋白:多角体蛋白是形成包含体的主要成分,感染后期在细胞中的积累可高达30%~50%,是病毒复制非必需成分,但对病毒粒子却有保护作用,可使之保持稳定和感染能力另一类高效表达的极晚期蛋白为P10蛋白,也是一类病毒复制非必需成分,可在细胞中形成纤维状物质,可能与细胞溶解有关。多角体基因和P10基因现在都已被定位和克隆这两个基因的启动子具有较强的启动能力,因此这两个基因位点成为杆状病毒表达载体系统理想的外源基因插入位点。
杆状病毒对昆虫有什么危害
杆状病毒对昆虫有什么危害 杆状病毒感染会让昆虫患病,目前发现的有: 1.颗粒病体 根据39蜂疗网调查目前仅见于鳞翅目昆虫。其自然侵染过程与细胞病变均类似于核型多角体病,但病虫症状与核型多角体病不同,病虫皮色变灰或乳黄,虫尸以腹部前端1~2对腹足握持植物枝条,虫尸以“∧”型倒挂。幼虫被感染后至少至4日龄才发病,死于化蛹前,病虫生存期常大于21天。 2.核型多角体病 现已发现280余种核型多角体病,约占昆虫病毒病总数的40%,大多侵染鳞翅目昆虫。 核型多角体病的自然感染过程为:昆虫吞食了被病毒污染的食物,病毒即进入中肠,在昆虫中肠碱性消化液的作用下,多角体被溶解,释放出病毒粒子,游离病毒粒子的囊膜与中肠上皮细胞绒毛的膜融合,核衣壳侵入细胞中,脱壳后,病毒的DNA经细胞核膜的核孔侵入细胞核内,开始其增殖过程。 随病毒的增殖,细胞表现的病理变化为:细胞核内染色质凝集成块,核仁增大,数目增多,RNA合成旺盛,合成出的RNA不断转移到细胞质中;凝集的染色质块集中于细胞核中部形成网状结构的病毒发生基质,在病毒发生基质中病毒的DNA大量合成。随后,在病毒发生基质表面核衣壳开始装配,并不断移到细胞核周围,大部分包入新形成的囊膜内,成为成熟的病毒粒子。最后在病毒发生基质周围形成一个环状带,在带上开始多角体的结晶,病毒粒子随机地包入多角体中,多角体约到一定大小后停止生长,在其表面形成了一层难溶的多角体膜。从多角体开始形成时起,病毒发生基质开始缩小,待多角体充满细胞核后,病毒发生基质消失,核膨大,破裂,细胞随之崩解。 小部分未被包入多角体的病毒粒子,可随细胞崩解进入昆虫血体腔。血体胶中病毒粒子的靶细胞为:气管皮膜细胞,脂肪细胞,肌肉细胞,真皮细胞,血细胞及神经、生殖腺、丝腺等几乎所有组织的细胞。 最后新形成的大量多用体充满了昆虫整个血体腔。 蛀虫幼虫被感染后,4~5天体液是乳白色,厌食,不喜运动,多数移到植物枝条顶部后死亡,虫体软化,脚失去握持力,仅以1~2对臀足附着在植物枝条上,最后松弛倒挂死之。体内组织完全溶解,变成黑褐色,表皮完整但脆弱易破裂。从感染到处亡约1~2周。
Bac-to-Bac杆状病毒表达系统
Bac-to-Bac杆状病毒表达系统 试剂盒内容物: Introduction: Overview: Bac-to-Bac杆状病毒表达系统提供快速有效的方法产生重组杆状病毒。此方法基于让已经转入杆状病的质粒(杆粒)的位点特意转座子的表达框的质粒在Ecoli中扩增。Bac-to-Bac杆状病毒表达系统主要包括:*pFastBac捐献质粒的选择,它要能够产生包含目的位点的表达结构,这个目的基因的产生被杆状病毒特意位点启动子控制。 *一个Ecoli宿主,DH10Bac,包含杆状病毒质粒(杆粒)和辅助质粒,在转染pFastBac 表达结构后可以产生重组杆粒。 *一个控制表达的质粒,包括Gus和/或CA T基因,以便在感染细胞后产生重组杆状病毒,表达β-葡萄糖酸酐酶和/或氯霉素乙酰转移酶。 Bac-to-Bac表达系统的优点: 使用这个系统产生重组杆状病毒较传统的同源重组有以下优点: *与使用同源重组产生重组杆状病毒所需的4-6周相比,鉴别纯化重组病毒少于两周 *减少了从斑点筛选重组病毒DNA所包含亲缘和非重组病毒的几率 *可以快速同时进行大量重组,适合表达功能性研究的蛋白 选择pFastBac菌体(Vector): 大量的pFastBac菌体都适于进行Bac-to-Bac表达系统。选择对于你的需要最合适的菌体。
指南用途: 指南提供了一个对于Bac-to-Bac表达系统的概述,并对以下提供指导: 1、克隆目的基因到pFastBac TM供体质粒的选择 2、转化pFastBac TM 结构到最高效的DH10Bac TM产生重组质粒 3、转染重组质粒DNA到昆虫细胞产生重组杆状病毒 4、扩增滴定(Amplify and titer)杆状病毒株,使用病毒株感染昆虫细胞表达目的重组蛋白 重要的:Bac-to-Bac杆状病毒表达系统是用来帮助你产生重组杆状病毒,在昆虫细胞中进行高水平表达目的基因的系统。虽然他可以帮助你很容易的产生杆状病毒表达你的重组蛋白,但是使用这系统更倾向于有杆状病毒生物学和昆虫表达背景的使用者。我们高度推荐使用者具有病毒和组织培养的技术和知识。 Bac-to-Bac表达系统 表达系统的成分: 表达系统简单高效的产生重组杆状不能给党。基于Luckow1993年的方法,此系统利用了位点特意的专座子Tn7区简化和提高重组质粒DNA *系统的第一个成分是用来克隆目的基因的pFastBac TM菌株。基于pFastBac TM菌株的选择,表达基因被AcMNPV的PH或p10启动子控制,在昆虫细胞内高水平表达表达框被Tn7 的左右臂包围,并且包含一个庆大霉素抗性点和SV40多腺苷酸化信号,形成一个微型的Tn7 *第二个主要结构是Ecoli的DH10Bac TM品系,用来作为pFastBac TM菌株的宿主。DH10Bac TM细胞包含带有微型-attTn7靶位点的病毒质粒和辅助质粒(详细见下文)。一旦pFastBac TM表达质粒转入DH10Bac细胞,转化就会发生在pFastBac TM菌株的微型Tn7单位和微型-attTn7的靶位点之间产生重组质粒。这个转化反应发生在辅助质粒提供的转化蛋白处。 如果你已经完成了转化反应,你需要分离这个高分子量的重组质粒DNA并将质粒DNA转染如昆虫细胞趋产生重组杆状病毒,用来进行初步表达实验。在杆状病毒株扩增和滴定后,高效价的病毒株可以用来感染细胞,进行大规模的重组蛋白的表达。 杆状病毒菌株: 病毒菌株(杆粒),bMON14272(136KB),在DH10Bac Ecoli中包括: *一个低拷贝的微型F复制子 *卡那霉素的抗性标记 *一个来自于pUC载体的编码LacZa肽的DNA片段,用来接触病毒转座子,Tn7(微型attTn7)已经被插入。插入的微型attTn7不会中断LacZa肽的阅读框。 杆粒在具有卡那抗性的大的质粒Ecoli DH10Bac中扩增,在显色物质如Bluo-gal 或X-gal和诱导剂IPTG 存在时,能补充染色体LacZ的缺失形成蓝斑(LacZ+) 辅助质粒:DH10Bac Ecoli也包含辅助质粒,pMON7124(13.2kb),他编码转移酶和四环素抗性基因。这个辅助质粒提供Tn7 转化功能。 图示Bac-toBac系统:下图刻画了重组病毒的产生和目的基因的表达