《中国药典》2020版—生物制品实验动物质量控制国家药品标准公示稿

生物制品实验动物质量控制
生物制品实验动物分为生物制品生产用实验动物和检定用实验动物。生产用实验动物是指用于生物制品生产的实验动物,检定用动物则是用于生物制品检定的实验动物。
本通则是对生物制品生产用和检定用实验动物微生物与寄生虫
学的质量控制要求。实验动物的管理应符合国家相关要求。
一、实验动物微生物学等级分类
按照实验动物携带微生物与寄生虫情况进行等级分类,分为普通级、清洁级、无特定病原体级和无菌级实验动物。
普通级实验动物[conventional (CV) animal]系指不携带所规定的重要人兽共患病和烈性传染病病原的实验动物。
清洁级实验动物[clean (CL) animal]系指不携带普通级实验动物的病原,并且不携带对动物危害大和对科学研究干扰大的病原体的实验动物。
无特定病原体级实验动物[specific pathogen free (SPF) animal]系指不携带普通级和清洁级实验动物的病原,并且不携带主要潜在感染或条件致病和对科学研究干扰大的病原体的实验动物。
无菌级实验动物[Germ Free (GF) animal]指无可检出的一切生命体的实验动物。
SPF 鸡胚是指由SPF 鸡所产的受精卵,在符合生物制品生产条件下,经孵化后所生成的鸡胚。
疫苗生产与检定应采用适宜级别的实验动物,具体应符合相关各
论的要求。
二、检测要求
1、外观要求实验动物应外观健康、无异常。
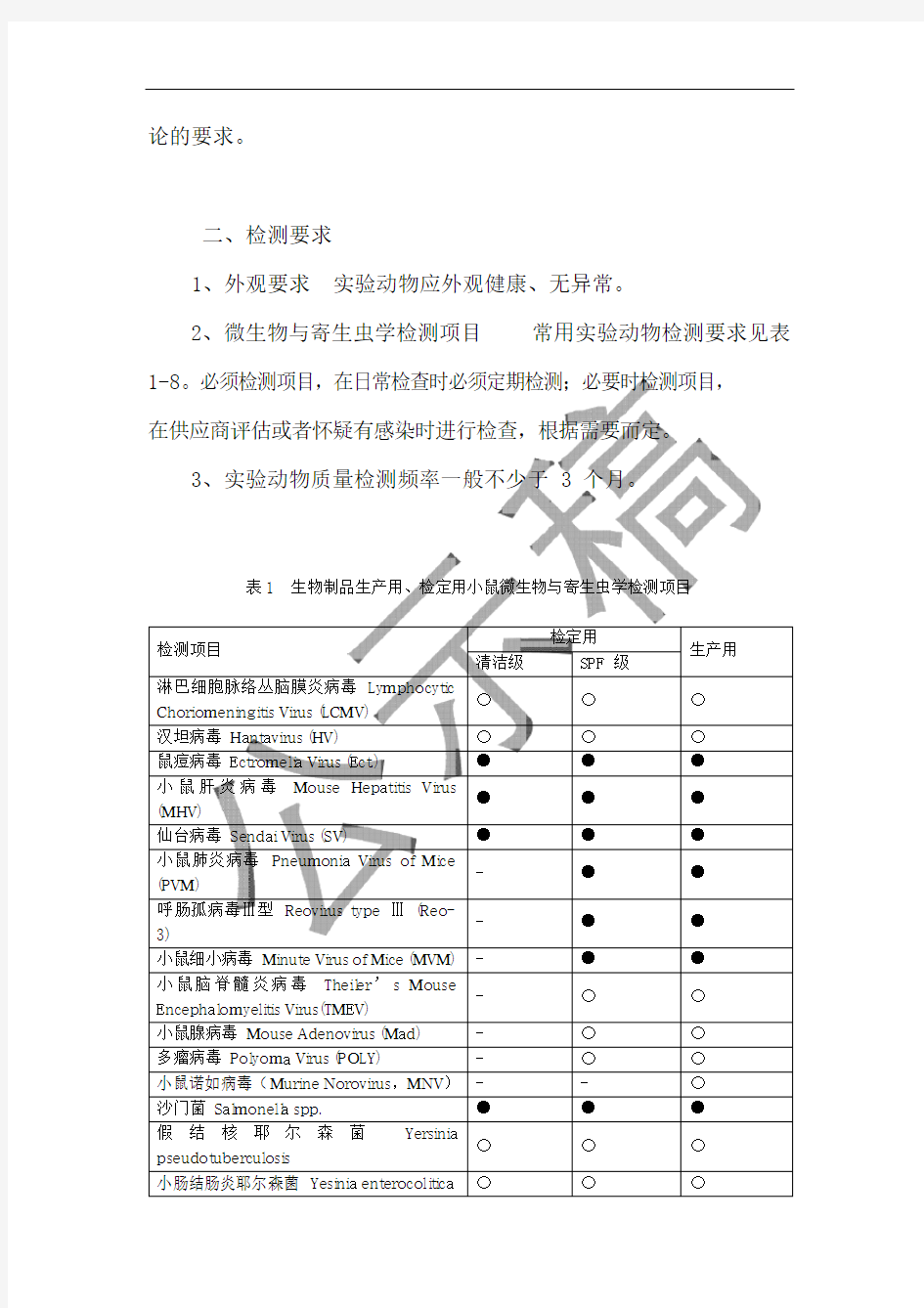
2、微生物与寄生虫学检测项目常用实验动物检测要求见表1-8。必须检测项目,在日常检查时必须定期检测;必要时检测项目,在供应商评估或者怀疑有感染时进行检查,根据需要而定。
3、实验动物质量检测频率一般不少于 3 个月。
表1 生物制品生产用、检定用小鼠微生物与寄生虫学检测项目
检测项目
检定用
生产用清洁级SPF 级
淋巴细胞脉络丛脑膜炎病毒L y m p h o c y t i c
C h o r i o m e n i n g i t i s V i r u s(L C M V)
〇〇〇汉坦病毒H a n t a v i r u s(H V)〇〇〇鼠痘病毒 E c t r o m e li a V i r u s(E c t.)●●●小鼠肝炎病毒M o u s e H e p a t i t i s V i r u s
(M H V)
●●●仙台病毒Se n d a i V i r u s(S V)●●●小鼠肺炎病毒Pn e u m o n i a V i r u s o f M i c e
(P V M)
- ●●呼肠孤病毒Ⅲ型R e o v i r u s t y p eⅢ(R e o-
3)
- ●●小鼠细小病毒M i nu t e V i r u s o f M i c e(M V M)- ●●小鼠脑脊髓炎病毒T h e il e r’s M o u s e
E n c e p h a l o m y e li t i s V i r u s(T M E V)
- 〇〇小鼠腺病毒M o u s e A d e n o v i r u s(M a d)- 〇〇多瘤病毒P o l y o m a V i r u s(P O L Y)- 〇〇小鼠诺如病毒(M u r i n e N o r o v i r u s,M N V)- - 〇沙门菌S a l m o n e ll a s pp.●●●假结核耶尔森菌Y e r s i n i a
p s e u do t u b e r c u l o s i s
〇〇〇小肠结肠炎耶尔森菌Y e s i n i a e n t e r o c o li t i c a 〇〇〇
2020年第1批国家药品标准批件公告(
2020年第1批国家药品标准批件公告(2020)2020年1月20日 修订批件 2 辛伐他汀分散片 片剂 XGB2020-001 WS1-(X-006)-2010Z-2020 各省、自治区、直辖市食品药品监督管理局 各省、自治区、直辖市食品药品监督管理局 2020年1月20日 修订颁布件 3 藿香正气胶囊 胶囊剂 ZGB2020-9 WS3-B-2266-96-20-2020 各省、自治区、直辖市食品药品监督管理局 各省、自治区、直辖市食品药品监督管理局 2020年1月21日 修订颁布件
4 妇科养血颗粒 颗粒剂 ZGB2020-15 WS-5001-(B-0001)-2014Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 5 xx补肾胶囊 胶囊剂 ZGB2020-13 WS-1046(ZD-0436)-2002-2012Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 6 复方蛤青胶囊 胶囊剂
ZGB2020-16 YBZ18112005-2009Z-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 7 xx养血糖浆 糖浆剂 ZGB2020-17 WS3-B-2339-97-2020 各省、自治区、直辖市食品药品监督管理局各省、自治区、直辖市食品药品监督管理局2020年1月21日 修订颁布件 8 xx 丸剂 ZGB2020-8 YBZ15012005-2014Z-2020 各省、自治区、直辖市食品药品监督管理局
9901 国家药品标准物质制备指导原则
9901 国家药品标准物质制备指导原则 本指导原则用于规范和指导国家药品标准物质的制备,保证国家药品标准的执行。 一、国家药品标准物质品种的确定 根据国家药品标准制定及修订的需要,确定药品标准物质的品种。 二、候选国家药品标准物质原料的选择 1.原料的选择应满足适用性、代表性及可获得性的原则。 2.原料的性质应符合使用要求。 3.原料的均匀性、稳定性及相应特性量值范围应适合该标准物质的用途。 三、候选国家药品标准物质的制备 1.根据候选药品标准物质的理化性质,选择合理的制备方法和工艺流程,防止相应特性量值的变化,并避免被污染。 2.对不易均匀的候选药品标准物质,在制备过程中除采取必要的均匀措施外,还应进行均匀性初检。 3.对相应特性量值不稳定的候选药品标准物质,在制备过程中应考察影响稳定性的因素,采取必要的措施保证其稳定性,并选择合适的储存条件。 4.当候选药品标准物质制备量大时,为便于保存可采取分级分装。 5.候选药品标准物质供应者须具备良好的实验条件和能力,并应提供以下资料。 (1)试验方法、量值、试验重复次数、必要的波谱及色谱等资料; (2)符合稳定性要求的储存条件(温度、湿度和光照等); (3)候选药品标准物质引湿性研究结果及说明; (4)加速稳定性研究结果; (5)有关物质的鉴别及百分比,国家药品标准中主组分的相对响应因子等具体资料; (6)涉及危害健康的最新的安全性资料。 四、候选国家药品标准物质的标定 候选药品标准物质按以下要求进行标定,必要时应与国际标准物质进行比对。 1.化学结构或组分的确证 (1)验证已知结构的化合物需要提供必要的理化参数及波谱数据,并提供相关文献及对比数据。如无文献记载,应提供完整的结构解析过程。 (2)对于不能用现代理化方法确定结构的药品标准物质,应选用适当的方法对其组分进行确证。 2.理化性质检查 应根据药品标准物质的特性和具体情况确定理化性质检验项目,如性状、熔点、比旋度、晶型以及干燥失重、引湿性等。 3.纯度及有关物质检查 应根据药品标准物质的使用要求确定纯度及有关物质的检查项,如反应中间体、副产物及相关杂质等。 4.均匀性检验 凡成批制备并分装成最小包装单元的候选药品标准物质,必须进行均匀性检验。对于分级分装的候选药品标准物质,凡由大包装分装成最小包装单元时,均应进行均匀性检验。
药品注册标准附件
附件4: 药品补充申请注册事项及申报资料要求 一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1.持有新药证书的药品生产企业申请该药品的批准文号。 2.使用药品商品名称。 3.增加中药的功能主治、天然药物适应症或者化学药品、生物制品国内已有批准的适应症。 4.变更用法用量或者变更适用人群范围但不改变给药途径。 5.变更药品规格。 6.变更药品处方中已有药用要求的辅料。 7.改变影响药品质量的生产工艺。 8.修改药品注册标准。 9.替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10.进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11.申请药品组合包装。 12.新药的技术转让。 13.修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14.改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15.改变进口药品的产地。 16.改变进口药品的国外包装厂。 17.进口药品在中国国内分包装。 18.其他。 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19.改变国内药品生产企业名称。
20.国内药品生产企业内部改变药品生产场地。 21.变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22.改变国内生产药品的有效期。 23.改变进口药品制剂所用原料药的产地。 24.变更进口药品外观,但不改变药品标准的。 25.根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。 26.补充完善进口药品说明书安全性内容。 27.按规定变更进口药品包装标签。 28.改变进口药品注册代理机构。 29.其他。 (三)省级食品药品监督管理部门备案的补充申请事项: 30.根据国家药品标准或者国家食品药品监督管理局的要求修改国内生产药品说明书。 31.补充完善国内生产药品说明书安全性内容。 32.按规定变更国内生产药品包装标签。 33.变更国内生产药品的包装规格。 34.改变国内生产药品制剂的原料药产地。 35.变更国内生产药品外观,但不改变药品标准的。 36.其他。 二、申报资料项目及其说明 1.药品批准证明文件及其附件的复印件: 包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名批准文件、药品标准颁布件、药品标准修订批件和统一换发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。附件包括上述批件的附件,如药品标准、说明书、标签样稿及其他附件。 2.证明性文件: (1)申请人是药品生产企业的,应当提供《药品生产许可证》及其变更记录页、营业执照、《药品生产质量管理规范》认证证书复印件。申请人不是药品生产企业的,应当提供其机构合法登记证明文件的复印件。
《中国药典》2020版—生物制品实验动物质量控制
生物制品实验动物质量控制 生物制品实验动物分为生物制品生产用实验动物和检定用实验动物。生产用实验动物是指用于生物制品生产的实验动物,检定用动物则是用于生物制品检定的实验动物。 本通则是对生物制品生产用和检定用实验动物微生物与寄生虫 学的质量控制要求。实验动物的管理应符合国家相关要求。 一、实验动物微生物学等级分类 按照实验动物携带微生物与寄生虫情况进行等级分类,分为普通级、清洁级、无特定病原体级和无菌级实验动物。 普通级实验动物[conventional (CV) animal]系指不携带所规定的重要人兽共患病和烈性传染病病原的实验动物。 清洁级实验动物[clean (CL) animal]系指不携带普通级实验动物的病原,并且不携带对动物危害大和对科学研究干扰大的病原体的实验动物。 无特定病原体级实验动物[specific pathogen free (SPF) animal]系指不携带普通级和清洁级实验动物的病原,并且不携带主要潜在感染或条件致病和对科学研究干扰大的病原体的实验动物。 无菌级实验动物[Germ Free (GF) animal]指无可检出的一切生命体的实验动物。 SPF 鸡胚是指由SPF 鸡所产的受精卵,在符合生物制品生产条件下,经孵化后所生成的鸡胚。 疫苗生产与检定应采用适宜级别的实验动物,具体应符合相关各
论的要求。 二、检测要求 1、外观要求实验动物应外观健康、无异常。 2、微生物与寄生虫学检测项目常用实验动物检测要求见表1-8。必须检测项目,在日常检查时必须定期检测;必要时检测项目,在供应商评估或者怀疑有感染时进行检查,根据需要而定。 3、实验动物质量检测频率一般不少于 3 个月。 表 1 生物制品生产用、检定用小鼠微生物与寄生虫学检测项目 Hantavirus (HV) Ectromelia Virus (Ect.) 肝炎病毒Mouse Hepatitis 仙台病毒 Sendai Virus (SV) 小鼠肺炎病毒 Pneumonia Virus of Mice 呼肠孤病毒Ⅲ型Reovirus type Ⅲ (Reo 小鼠细小病毒 Minute Virus of Mice 脑脊髓病毒Theiler ’
国家药品标准物质协作标定实施细则
国家药品标准物质协作标定实施细则 第一条确定药品标准物质特性值的过程,是药品标准物质定值工作的一部分,为保证药品标准物质特性值的准确性,需要多个实验室协作研究。为建立规范的药品标准物质协作标定工作机制,制定本实施细则。 第二条本实施细则所称的药品标准物质协作标定,是指在多个具有同等能力的实验室间,使用一个或多个法定方法,各实验室所得试验数据按统计程序进行处理后,得到药品标准物质的特性值。 中检院标准物质管理处依照有关国家标准、国际准则制定有关协作标定工作的实施细则,统一管理和协调协作标定工作。 第四条中检院业务科室(以下简称组织者)负责组织与实施相关品种的协作标定工作,组织者主要的工作内容包括: (一)协作标定计划的起草; (二)协作标定作业指导书的编写; (三)协作标定数据的统计处理; (四)协作标定技术报告的起草。 第五条组织者应当于协作标定工作开展前向标准物质管理处提交《药品标准物质协作标定设计方案》(见附件
1)。包括:样品名称、申请科室、协作标定工作的必要性、拟参加实验室的要求和数量、日程安排、试验方法、数据的统计处理分析原理、协作标定费用预算等内容。 (一)协作标定的样品应通过分装前检查,并符合相关质量标准要求,分装后样品的均匀性与稳定性应符合要求。 (二)必要时协作标定的组织者可发放调查表(含拟标定实验方法的SOP),对拟参加的实验室进行该项检测的资质与能力进行调查。 (三)协作标定实验室的数目或独立定值组数应符合统计学的要求。 1. 参加实验室的数目 通常协作标定的实验室的数目为3-6个,随所需测定程序的复杂程度而变化。 对于化学对照品的协作标定,参加的实验室数目一般为3个。 对于生物标准物质的协作标定,参加的实验室数目不少于3个。 2.单元数及重复测量的次数 对于首批研制的化学对照品通常单个实验室可进行1-2个检测项的测定,在短时间内每个检测项需进行2-5次重复测量(即一份样品平行称样、测定2-5次)。对于原料量少且价格昂贵的品种每个单元的重复测量次数可减少为2次。
国家药品标准
药品管理法:国务院药品监督管理部门颁布的《中华人民共和国药典》和药品标准为国家药品标准。 第一百三十六条国家药品标准,是指国家食品药品监督管理局颁布的《中华人民共和国药典》、药品注册标准和其他药品标准,其内容包括质量指标、检验方法以及生产工艺等技术要求。 药品注册标准,是指国家食品药品监督管理局批准给申请人特定药品的标准,生产该药品的药品生产企业必须执行该注册标准。 药品注册标准不得低于中国药典的规定 1.药典标准 2.卫生部中药成方制剂一至二十一册 3.卫生部化学、生化、抗生素药品第一分册 4.卫生部药品标准(二部)一册至六册; 5.卫生部药品标准藏药第一册、蒙药分册、维吾尔药分册; 6.新药转正标准1至88册(正不断更新) 7.国家药品标准化学药品地标升国标一至十六册; 8.国家中成药标准汇编内科心系分册、内科肝胆分册、内科脾胃分册、内科气血津液分册、内科肺系(一耳鼻喉皮肤科分册、经络肢体脑系分册;)、(二)分册、内科肾系分册、外科妇科分册、骨伤科分册、口腔肿瘤儿科分册、眼科 9.国家注册标准(针对某一企业的标准,但同样是国家药品标准) 10.进口药品标准 1级标准:就是《中国药典》(中华人民共和国药典),是药典委员会制定,每5 年修订一次。2010年新版中国药典分3部,一部中药标准,二部化学药标准,三部生物制剂标准。另外2010年版第一增补本自2012年10月1日起施行。2级标准:是局颁标准(国家药监局)或部颁标准(卫生部),药品标准开头字母WS是卫生部批准的,药品标准开头字母 YB是国家药监局批准的。3级标准:基本已经废除,一般是指各省、自治区、直辖市制定的中药炮制或中药饮片标准。在药
药品补充申请注册事项及申报资料要求
附件4:药品补充申请注册事项及申报资料要求一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1、持有新药证书的药品生产企业申请该药品的批准文号。 2、使用药品商品名称。 3、增加中药的功能主治或者化学药品、生物制品国内已有批准的适应症。 4、变更用法用量或者变更适用人群范围但不改变给药途径。 5、变更药品规格。 6、变更药品处方中已有药用要求的辅料。 7、改变影响药品质量的生产工艺。 8、修改药品注册标准。 9、替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10、进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11、申请药品组合包装。 12、新药的技术转让。 13、修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14、改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15、改变进口药品的产地。 16、改变进口药品的国外包装厂。 17、进口药品在中国国内分包装。 18、其它 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19、改变国内药品生产企业名称。 20、国内药品生产企业内部改变药品生产场地。 21、变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22、改变国内生产药品的有效期。 23、改变进口药品制剂所用原料药的产地。 24、变更进口药品外观,但不改变药品标准的。 25、根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。 26、补充完善进口药品说明书安全性内容。 27、按规定变更进口药品包装标签。
生物制品生产用原材料及辅料的质量控制规程推荐WORD范文
生物制品生产用原材料及辅料的质量控制规程 生物制品是采用生物技术制备而成的具有活性的药品。生物制品的生产工艺复杂且易受多种因素影响,生产过程中使用的各种材料来源复杂,可能引入外源因子或毒性化学材料;产品组成成分复杂且一般不能进行终端灭菌,产品的质量控制仅靠成品检定难以保证其安全性和有效性。因此,对生物制品生产用原材料和辅料进行严格的质量控制,是降低制品中外源因子或有毒杂质污染风险,保证生物制品安全有效的必要措施。 本规程是对生物制品生产企业在生物制品生产过程中使用的原材料和辅料质量控制的通用性要求。 一、生物制品生产用原材料 生物制品生产用原材料系指生物制品生产过程中使用的所有生物材料和化学材料。 本规程所述原材料不包括用于生物制品生产的起始原材料(如细胞基质、 菌毒种、生产用人血浆和动物免疫血清等) 1.分类 按照来源可将生物制品生产用原材料分为两大类,一类为生物原材料,主要包括来源于微生物,人和动物细胞、组织、体液成分,以及采用重组技术或生物合成技术生产的生物原材料等;另一类为化学原材料,包括无机和有机化学材料。 2.风险等级分级及用于生产的质量控制要求 根据原材料的来源、生产以及对生物制品潜在的毒性和外源因子污染风险等,将生物制品生产用原材料按风险级别从低到高分为以下四级,各级生物制品原 材料至少应进行的质量控制要求见附表1;对于不同风险级别原材料的质量控制,应充分考虑来源于动物(或人)的生物原材料可能带来的外源因子污染的安全性风险。 生产过程中应避免使用毒性较大的化学原材料,有机溶剂的使用应符合本版药 典附录“残留溶剂检测”的相关要求。 第1级为较低风险的原材料,为已获得上市许可的生物制品或药品无菌制剂。如人血白蛋白、各种氨基酸、抗生素注射剂等。 第2级为低风险原材料,这类原材料为已有国家药品标准、取得国家药品批准文号并按照我国现行药品GMP生产的用于生物制品培养基成分以及提取、纯化、灭活等过程的化学原料药和药用级非动物来源的蛋白水解酶等。 第3级为中等风险等级原材料,这类原材料为非药用,包括生物制品生产用培养基成分、非动物来源蛋白水解酶、用于靶向纯化的单克隆抗体,以及用于 生物制品提取、纯化、灭活的化学试剂等。这类生物制品原材料的质量控制要求应
国家药品标准物质管理办法
国家药品标准物质管理办法 第一条为进一步规范国家药品标准物质的管理,根据《中华人民共和国药品管理法》、《药品注册管理办法》及《体外诊断试剂注册管理办法(试行)》的相关规定,制定本办法。 第二条国家药品标准物质的标定、审核、批准以及国家药品标准物质的对外供应,应遵守本办法。 第三条国家药品标准物质是指供国家药品标准中物理和化学测试及生物方法试验用,具有确定特性量值,用于校准设备、评价测量方法或者给供试药品赋值的材料或物质。 国家药品标准物质分为生物标准品、生物参考品、化学对照品、对照药材/对照提取物、医疗器械对照材料和标准品、体外诊断试剂标准品、药用辅料对照品及药包材对照物质等。 第四条中国食品药品检定研究院(以下简称中检院)负责对标定的药品标准物质从原材料选择、制备方法、标定方法、标定结果、定值准确性、量值溯源、稳定性、分装与包装条件、成本等进行全面审核,并作出可否作为国家药品标准物质的结论。 第五条中检院设立标准物质管理处,负责国家药品标准物质工作的综合、组织、协调和管理,中检院设立国家药品标准物质委员会,负责国家药品标准物质全面技术审核,并负责相关技术指导规范的审定。
第六条标准物质管理处负责组织药品注册申请与药品标准修订中新增的药品标准物质原料的申报与受理,申报的药品标准物质原料及其研究资料,应符合《国家药品标准物质申报、备案办法》及相关类别药品标准物质原料的要求。 第七条国家药品标准物质的原材料应满足国家药品标准的要求,由标准物质管理处负责组织相关品种原料的制备或采购。对于特殊来源要求的药品标准物质,可通过采取自行制备、向有关机构或特定人群收集的方式,获得符合国家药品标准要求的原材料。对于药品标准物质原材料供应不足或难以获得的品种,应由标准物质管理处汇总后报送相关单位,并提请国家食品药品监督管理局协调解决。 第八条国家药品标准物质的标定及定值方法,应遵照国家药品标准、中检院发布的《国家药品标准物质技术规范》以及具体类别标准物质定值方法的要求。 第九条标准物质管理处可以组织有能力的药品检验所、研究机构和生产企业等单位协作标定国家药品标准物质。 第十条参照国际惯例,国家药品标准物质不规定有效期,但应在规定的储存和使用条件下,定期进行特性量值的稳定性核查。 第十一条国家药品标准物质更换批号、停止使用及撤销的品种,需经国家药品标准物质委员会批准后,标准物质管理处及时向社会公布。
注册变更国家局
药品补充申请注册事项及申报资料要求 一、注册事项 (一)国家食品药品监督管理局审批的补充申请事项: 1.持有新药证书的药品生产企业申请该药品的批准文号。 2.使用药品商品名称。 3.增加中药的功能主治、天然药物适应症或者化学药品、生物制品国内已有批准的适应症。 4.变更用法用量或者变更适用人群范围但不改变给药途径。 5.变更药品规格。 6.变更药品处方中已有药用要求的辅料。 7.改变影响药品质量的生产工艺。 8.修改药品注册标准。 9.替代或减去国家药品标准处方中的毒性药材或处于濒危状态的药材。 10.进口药品、国内生产的注射剂、眼用制剂、气雾剂、粉雾剂、喷雾剂变更直接接触药品的包装材料或者容器;使用新型直接接触药品的包装材料或者容器。 11.申请药品组合包装。 12.新药的技术转让。 13.修订或增加中药、天然药物说明书中药理毒理、临床试验、药代动力学等项目。 14.改变进口药品注册证的登记项目,如药品名称、制药厂商名称、注册地址、药品有效期、包装规格等。 15.改变进口药品的产地。 16.改变进口药品的国外包装厂。 17.进口药品在中国国内分包装。 18.其他。 (二)省级食品药品监督管理部门批准国家食品药品监督管理局备案或国家食品药品监督管理局直接备案的进口药品补充申请事项: 19.改变国内药品生产企业名称。 20.国内药品生产企业内部改变药品生产场地。 21.变更直接接触药品的包装材料或者容器(除上述第10事项外)。 22.改变国内生产药品的有效期。 23.改变进口药品制剂所用原料药的产地。 24.变更进口药品外观,但不改变药品标准的。 25.根据国家药品标准或者国家食品药品监督管理局的要求修改进口药品说明书。
生物制品质量控制
生物制品质量控制 生物制品是以微生物、细胞、动物或人源组织和体液等为原料,应用传统技术或现代生物技术制成,用于人类疾病的预防、治疗和诊断。人用生物制品包括:细菌类疫苗(含类毒素)、病毒类疫苗、抗毒素及抗血清、血液制品、细胞因子、生长因子、酶、体内及体外诊断制品,以及其他生物活性制剂,如毒素、抗原、变态反应原、单克隆抗体、抗原抗体复合物、免疫调节及微生态制剂等。根据生物制品的用途可分为预防用生物制品、治疗用生物制品和诊断用生物制品三大类。 其起始材料均为生物活性物质; 生物制品生产加工全过程是生物学过程,是无菌操作过程; 有些生物制品的生产过程是有毒或有菌的过程; 生物制品多为蛋白质或多肽类物质,分子量较大,并具有复杂的分子结构, 较不稳定,易失活,易被微生物污染,易被酶解破坏。 其质量控制和质量检定是采用生物学分析方法,其效价或生物活性检定有其变异性; 生物制品原材料、中间品、成品、运输、贮存、甚至使用保持在“冷链”系统中; 特别是预防制品使用对象不是病人,而是健康人群; 生物制品的质量控制实行生产全过程监控; 特点: 1、起步晚、发展速度快,前景美好; 2、有巨大的科研价值、重大的经济效益和巨大的社会效益:因为生物制品多涉及国民的健康、长寿,社会稳定和经济的快速发展。 3、研发的产品面广,具有高速的成长性和广阔的发展空间。 2、人源性生物制品的特点: 1.效价高、医疗可靠2安全性好、不易产生副作用3稳定性好4资源有限、研究意义重 “GMP”是英文Good Manufacturing Practice 的缩写,中文的意思是“良好作业规范”,或是“优良制造标准”,是一种特别注重在生产过程中实施对产品质量与卫生安全的自主性管理制度。它是一套适用于制药、食品等行业的强制性标准,要求企业从原料、人员、设施设备、生产过程、包装运输、质量控制等方面按国家有关法规达到卫生质量要求,形成一套可操作的作业规范帮助企业改善企业卫生环境,及时发现生产过程中存在的问题,加以改善。第一条为规范药品生产质量管理,根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》,制定本规范。第二条企业应当建立药品质量管理体系。该体系应当涵盖影响药品质量的所有因素,包括确保药品质量符合预定用途的有组织、有计划的全部活动。第三条本规范作为质量管理体系的一部分,是药品生产管理和质量控制的基本要求,旨在最大限度地降低药品生产过程中污染、交叉污染以及混淆、差错等风险,确保持续稳定地生产出符合预定用途和注册要求的药品。第四条企业应当严格执行本规范,坚持诚实守信,禁止任何虚假、欺骗行为。 GLP(Good laboratory practice of drug) 药品非临床研究质量管理规范。GLP是英文Good Laboratory Practice 的缩写,中文直译为优良实验室规范。GLP是就实验室实验研究从计划、实验、监督、记录到实验报告等一系列管理而制定的法规性文件,涉及到实验室工作的所有方面。它主要是针对医药、农药、食品添加剂、化妆品、兽药等进行的安全性评价实验而制定的规范。制定GLP的主要目的是严格控制化学品安全性评价试验的各个环节,即严格控制可能影响实验结果准确性的各种主客观因素,降低试验误差,确保实验结果的真实性。1999年11月1日起国家食品药品监督管理局发布施行《药品非临床研究质量管理规范》。系指对从事实验研究的规划设计、执行实施、管理监督和记录报告的实验室的组织管理、工
药品-试剂-标准物质管理制度
实验室药品试剂标准物质管理制度 1目的 1.1规范实验室药品试剂的管理,遵循既有利于使用,又保证安全的原则,管好用好化学品,加强安全教育。 1.2为确保试验室所需的参考标准和标准物质,始终处于受控状态的规范管理,确保检验结果的准确性,可靠性,可比性。 2范围 2.1实验室内所用药品试剂及标准物质。 3职责 3.1 药品试剂保管人员严格执行药品保管制度,做到药品试剂的安全保管和使用领取。 3.2标准物质的购置由化学分析责任人提出申请,经试验室主任审批后,材料员到国家标准物质管理中心购买。 3.3实验室管理人员负责实验室操作人员药品试剂的安全使用。 3.4实验室操作人员准确掌握试剂药品的安全使用,防止出现误操作。 技术负责人负责对标准物质购置计划的审核与标准物质报废的批准。
3.5检验组负责监督对标准物质管理的执行情况,及报废的审核。 4药品试剂及标准物质的保管 4.1化学试剂应指定专人保管,并有账目。 4.2标准物质购入后,有理化检验工程师与化学分析室负责人共同检查验收。化学分析责任人负责上账登记、标识并设专人保管,填写标准物质管理一览表。 4.3在固体试剂和液体试剂及化学性质不同或灭火方法相抵触的化学试剂应分柜存放。 4.4受光照易变质、易燃、易爆、易产生有毒气体的化学试剂应存放在阴凉通风处;易燃、易爆物应远离火源。 4.5易挥发试剂应贮存在有通风设备的房间内。 4.6剧毒试剂应专柜存放,双人双锁保管。 4.7试剂使用应有记录,剧毒试剂的领用需实验室负责人签字。 4.8配制的试剂应贴上标识,注明试剂名称、浓度、配制时间、有效期及配制人。 4.9定期检查试剂是否过期,过期试剂应及时妥善处理。 4.10配制的试剂除有特殊规定外,存放期不应超过3个月。 4.11应定期检查试剂是否齐全,所缺试剂应及时申请购买。
关于“生物制品质量控制分析方法验证技术审评一般原则”中几个验证参数的说明
发布日期20050718 栏目生物制品评价>>生物制品质量控制 关于“生物制品质量控制分析方法验证技术审评一般原则”中几个验证参数 标题 的说明 作者常卫红 部门 正文内容 审评五部生物制品室常卫红 摘要:本文对《生物制品质量控制分析方法验证技术审评一般原则》中有关验证参数的问题进行了阐述 质控分析方法的验证就是根据方法的需要测定该方法的专属性、准确性、精密度、线性、范围、检测限度、定量限度、耐用性等几个指标中的一个或 几个,所以上述参数的验证是本技术审评一般原则中的核心内容。本文拟就 该部分内容进行补充说明。 1、精密度 由于各种生物活性测定方法的变异均较大,所以精密度往往不太理想。 对于一些新建立的活性测定方法而言,测定结果不稳定、难以进行定量及无 法准确标示等问题已成为质控中的主要问题。针对上述情况,本技术审评一 般原则拟强调,作为测定有效成分含量水平的生物活性指标,需采用定量的 测定方法,并尽可能减少方法的变异。参照国外的有关标准,对各种测定方 法,提出了一般的可接受标准。 经课题研究组讨论认为,本原则中所提出的可接受标准,一般情况下均可以做到。对于个别难以做到的,可结合制品特性及其临床效应等进行综合 评估,如基本不影响到产品的安全、有效和质量可控,也可考虑适当放宽。 鉴于生物学测定方法的多样性和复杂性,本原则的正文中未对精密度测
定的重复次数作明确要求。从理论上考虑,测定次数主要取决于方法学的误差,变异大的方法应增加测定次数;从可操作性角度考虑,对于复杂、成本高及周期长的方法,重复次数太多,试验难度将很大。所以关于验证次数的设计,需综合考虑,应以基本达到验证的目的为准则。验证设计方案中的重复次数供参考。 2、线性 线性关系一般是指检测结果与样品含量的直线相关性,而且一般情况下线性关系是定量测定的基础,所以应尽可能摸索出存在较好线性关系的测定方法并进行线性验证。但对于某些生物活性测定方法而言,其线性范围较小(如细胞测定中呈S曲线),这时采用曲线拟合的方法应更合理。鉴于目前曲线拟合的方法在生物活性测定中被广泛采用,所以本原则中的线性验证亦包括了有关曲线拟合的内容。 3、专属性和准确性 专属性和准确性不是每个生物学测定方法都需要测定的参数,但同样重要,只是对于不同品种的不同方法应有不同的考虑,所以对于这两个参数进行了较详细的说明,并列举了一些具体情况,提请研制单位在建立方法和进行方法验证过程中能有所考虑。 4、耐用性 由于生物学测定结果对分析条件往往比较敏感,所以方法的耐用性验证是非常重要的。为保证测定结果稳定,研究者应对此项工作给予充分重视。但是考虑到目前国内的相关研究现状,在此项研究工作的起步阶段,为不致使验证工作量过大,暂提出:根据具体情况,可针对一个或几个关键的参数进行验证。今后将视国内相关研究的进展,逐步提高此项验证要求。 5、检测限度和定量限度 这两项是杂质检查中需测定的参数。根据目前国内的实际状况,部分生物制品尚未建立杂质检查项目,但是对于已建立的杂质检测方法,应参照本原则进行验证。 对于生物活性测定而言,精密度、线性和范围是非常重要的验证参数,所以本原则不但提出了一般性的验证要求,而且附上了推荐的验证设计方案(参照WHO的建议),供研制单位参考。 最后需要强调的是,分析方法的验证工作最终是为了保证产品的质量可控,以此为目的,各种质控分析方法之间存在着一定的内在联系,所以在评
国家标准物质技术规范审批稿
国家标准物质技术规范 YKK standardization office【 YKK5AB- YKK08- YKK2C- YKK18】
国家药品标准物质技术规范 第一章总则 第一条为保证国家药品标准物质的质量,规范药品标准物质的研制工作,根据《国家药品标准物质管理办法》,制定本技术规范。 第二条本技术规范适用于中国食品药品检定研究院(以下简称中检院)研究、制备、标定、审核、供应的国家药品标准物质。 第三条国家药品标准物质系指供药品质量标准中理化测试及生物方法试验用,具有确定特性,用以校准设备、评价测量方法或给供试药品定性或赋值的物质。 (一)理化检测用国家药品标准物质系指用于药品质量标准中物理和化学测试用,具有确定特性,用以鉴别、检查、含量测定、校准设备的对照品,按用途分为下列四类: 1. 含量测定用化学对照品:系指具有确定的量值,用于测定药品中特定成分含量的标准物质。 2. 鉴别或杂质检查用化学对照品:系指具有特定化学性质,用于鉴别或确定药品某些特定成分的标准物质。 3. 对照药材/对照提取物:系指用于鉴别中药材或中成药中某一类成分或组分的对照物质。
4. 校正仪器/系统适用性试验用对照品:系指具有特 定化学性质用于校正检测仪器或供系统适用性实验用的标 准物质。 (二)生物检测用国家药品标准物质系指用于生物制 品效价、活性、含量测定或其特性鉴别、检查的生物标准 品或生物参考物质,可分为生物标准品和生物参考品。 1.生物标准品系指用国际生物标准品标定的,或由我 国自行研制的(尚无国际生物标准品者)用于定量测定某一 制品效价或毒性的标准物质,其生物学活性以国际单位(IU)或以单位(U)表示。 2.生物参考品系指用国际生物参考品标定的,或由我 国自行研制的(尚无国际生物参考品者)用于微生物(或其产物)的定性鉴定或疾病诊断的生物试剂、生物材料或特异性抗血清;或指用于定量检测某些制品的生物效价的参考物质,如用于麻疹活疫苗滴度或类毒素絮状单位测定的参考品,其效价以特定活性单位表示,不以国际单位(IU)表 示。 第二章国家药品标准物质的制备 第四条在建立新的国家药品标准物质时,研制部门 应提交研制申请,标准物质管理处负责评估研制的必要
药品标准
第三节药品标准 一、药品标准概述 1.药品标准是指对药品的质量指标、生产工艺和检验方法所作的技术要求和规定,内容包括药品的名称、成分或处方的组成;含量及其检查、检验方法;制剂的辅料;允许的杂质及其限量要求以及药品的作用、用途、用法、用量;注意事项;贮藏方法等。中药材、中成药、化学原料药及其制剂,生物制品等应根据各自的特点设置不同的项目。 2.国家药品标准《药品管理法》规定,国务院药品监督管理部门颁布的《中国药典》和药品标准为国家药品标准。 国家药品标准包括《中国药典》及增补本,经国家食品药品监督管理局批准的药品注册标准和颁布的其他药品标准,以及与药品质量指标、生产工艺和检验方法相关的技术指导原则和规范。 二、药品标准的分类 依据《药品管理法》规定,我国的药品标准分为国家药品标准和炮制规范。 1.国家药品标准分类《中国药典》、国家食品药品监督管理局颁布的药品标准和药品注册标准。 (1)《中国药典》由国家药典委员会编纂,国家食品药品监督管理局颁布。《中国药典》是国家药品标准的核心,是国家为保证药品质量、保护人民用药安全有效而制定的法典。 《中国药典》于1953年编纂出版第一版以后,相继于1963年、1977年分别编纂出版。从1985年起每5年修订颁布新版药典,现行版为2010年版《中国药典》。 2010年版《中国药典》是新中国成立以来第九版药典,本版药典收载品种总计4567个,与2005年版《中国药典》相比新增品种1386个;一部收载药材及饮片、植物油脂和提取物、成方和单味制剂共2165个,其中新增1019个,修订634个;二部收载化学药品、抗生素、生化药品、放射性药品及药用辅料共2271个,其中新增330个,修订1500个;三部收载生物制品131个品种,其中新增37个,修订94个;药典附录新增47个,修订154个。药用辅料标准新增130多种。 (2)国家食品药品监督管理局颁布的药品标准这类药品标准是指未列入《中国药典》而由国家食品药品监督管理局颁布的药品标准,以及与药品质量指标、生产工艺和检验方法相关的技术指导原则和规范。 (3)药品注册标准是指国家食品药品监督管理局批准经申请人特定药品的标准,生产该药品的生产企业必须执行该注册标准。 根据《标准化法》规定和国际惯例,国家标准是市场准入的最低标准,原则上行业标准高于国家标准,企业标准应高于行业标准。所以,药品注册标准不得
关于在药品包装标签上印制说明书二维码的指导意见
关于在药品包装标签上印制说明书二维码的指导意见 我局为进一步落实国家药品监督管理局《药品说明书和标签管理规定》的有关要求,根据国家相关法律法规,结合我省实际,制定此指导意见。 一、适用范围 (一)药品说明书文字内容多,因客观原因导致纸张尺寸小,说明书字号小于5号标准字号时,企业应以药品说明书二维码方式在药品包装上印刷,并以药品补充申请形式向省局提出备案; (二)药品说明书字号大于等于5号标准字号,企业可根据需要在药品包装上印制药品说明书二维码,并以药品补充申请形式向省局提出备案。 二、申报形式及资料 (一)根据国家药品标准或国家药监局的要求修改国内生产药品说明书和按规定变更国内生产药品包装标签的备案事项,申报资料如下: 1、药品批准证明文件及其附件:包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名称批准文件、药品标准颁布件、药品标准修订批准和统一制发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。
附件包括上述批件的附件,如药品质量标准、说明书、标签样稿及其他附件。 2、修订的药品说明书样稿,并附详细修订说明。 3、修订药品标签样稿,在左下角预留二维码印制位置,并附详细修订说明。 4、提供药品处方、生产工艺、质量标准。 5、生产药品制剂所用原料药的来源。改变原料药来源的,应当提供批准证明文件。 6、提供新的国家药品标准或者国家药品监督管理局要求修改说明书的文件。 7、提供新修订说明书的w o r d版,字号不得小于5号。 (二)补充完善国内生产药品说明书安全性内容和按规定变更国内生产药品包装标签的备案事项,提供如下资料: 1、药品批准证明文件及其附件:包括与申请事项有关的本品各种批准文件,如药品注册批件、补充申请批件、商品名称批准文件、药品标准颁布件、药品标准修订批准和统一制发药品批准文号的文件、《新药证书》、《进口药品注册证》、《医药产品注册证》等。附件包括上述批件的附件,如药品质量标准、说明书、标签样稿及其他附件。 2、修订的药品说明书样稿,并附详细修订说明。 3、修订药品标签样稿,在左下角预留二维码印制位置,并附详细修订说明。
国家药品标准物质制备指导原则-en
Guideline for Preparation of National Pharmaceutical Reference Standards This guideline is indicated for normalizing and guiding preparation of national pharmaceutical reference standards and ensuring implementation of national drug standards. I. Establishment of varieties of national pharmaceutical reference standards Varieties of reference standards to be prepared should be established according to use requirements (varieties, purpose) put forward in setup and revision of national drug standards. II. Selection of candidate materials for reference standards 1. For selection of materials, the principles of suitability, representation and availability should be followed, and the quantity needed should be satisfied. 2. Properties of materials should comply with the use requirements. 3. Quantity value spectra of homogeneity, stability and other relevant properties of material should be suitable for purposes of the corresponding reference standards. III. Preparation of candidate reference standards 1. Select rational preparation procedures and crafts according to physicochemical properties of candidate reference standards and prevent contamination and value changes of corresponding properties. 2. For candidate reference standards the corresponding property values of which cannot be homogenized easily, not only necessary homogenizing measures should be taken in the process of preparation, but also preliminary homogeneity test should be carried out. 3. Where values of corresponding properties of a candidate reference standard show the trend to be unstable, factors affecting its stability should be paid attention to in the process of preparation and necessary measures should be taken to improve its stability, and a suitable environment should be selected for storage. 4. When the prepared amount of a candidate reference standard is large, graded subpackaging may be applied for convenient storage. 5. Suppliers of candidate reference standards should have favorable experimental conditions and capacity and provide the following data: 1) Completed data including test methods, values, number of repeated times of tests, necessary spectra and chromatograms; (2) Storage conditions (temperature, humidity and light irradiation) complying with stability requirements; (3) Hygroscopicity study results or hydroscopicity description of candidate reference standards; (4) Results of accelerated stability study; (5) Specific data about identification and percentages of related substances, relative response factors of principal ingredients in national drug standards; (5) Latest safety data related to health risks. IV. Standardization of candidate national drug reference standards Candidate reference standards are standardized as per the following requirements. Conditions permitting, comparison with international reference standards or pharmacopoeial reference standards of advanced countries and regions such as Europe and America should be performed: 1. Characterization of chemical structures or components