生物制品批签发一览表

生 物 制 品 批 签 发 一 览 表
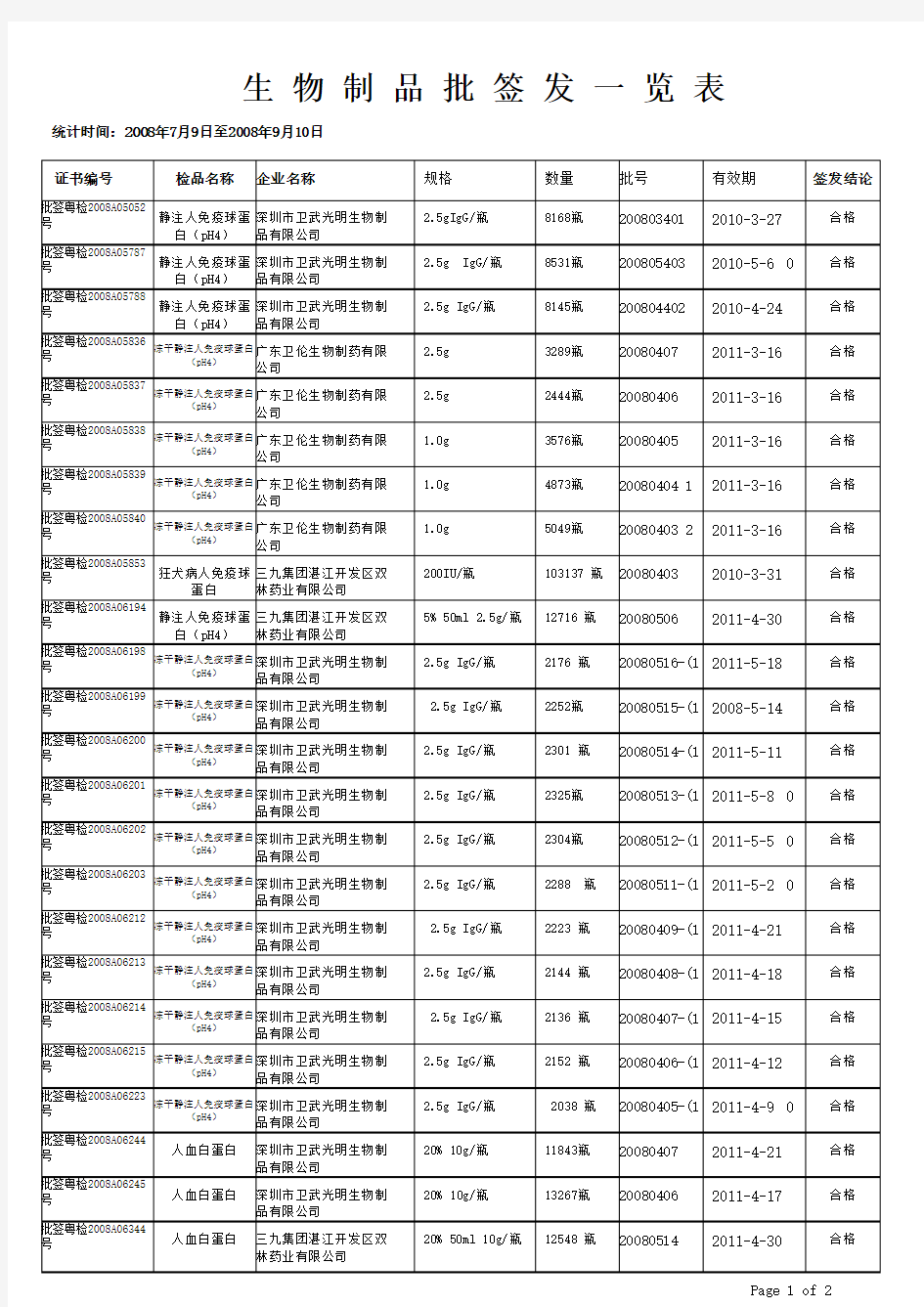
统计时间:2008年7月9日至2008年9月10日 证书编号
检品名称 签发结论
企业名称
规格
数量
有效期 批号 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司 2.5gIgG/瓶 8168瓶 2010-3-27 批签粤检2008A05052号
200803401 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司 2.5g IgG/瓶 8531瓶 2010-5-6 0批签粤检2008A05787号
200805403 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司
2.5g IgG/瓶 8145瓶 2010-4-24 批签粤检2008A05788号
200804402 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 2.5g 3289瓶 2011-3-16 批签粤检2008A05836
号 20080407
冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 2.5g 2444瓶 2011-3-16 批签粤检2008A05837
号 20080406 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 1.0g 3576瓶 2011-3-16
批签粤检2008A05838
号 20080405
冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 1.0g 4873瓶 2011-3-16 批签粤检2008A05839
号 20080404 1 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司
1.0g 5049瓶
2011-3-16
批签粤检2008A05840
号 20080403 2 狂犬病人免疫球蛋白 合格 三九集团湛江开发区双
林药业有限公司 200IU/瓶
103137 瓶 2010-3-31 批签粤检2008A05853号
20080403
静注人免疫球蛋白(pH4) 合格 三九集团湛江开发区双林药业有限公司
5% 50ml 2.5g/瓶 12716 瓶 2011-4-30
批签粤检2008A06194号
20080506 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2176 瓶 2011-5-18
批签粤检2008A06198
号 20080516-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2252瓶 2008-5-14
批签粤检2008A06199
号 20080515-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2301 瓶 2011-5-11 批签粤检2008A06200
号 20080514-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2325瓶 2011-5-8 0
批签粤检2008A06201
号 20080513-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2304瓶
2011-5-5 0
批签粤检2008A06202
号 20080512-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2288 瓶 2011-5-2 0
批签粤检2008A06203
号 20080511-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2223 瓶 2011-4-21
批签粤检2008A06212
号 20080409-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2144 瓶 2011-4-18
批签粤检2008A06213
号 20080408-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2136 瓶 2011-4-15
批签粤检2008A06214
号 20080407-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2152 瓶
2011-4-12
批签粤检2008A06215
号 20080406-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司
2.5g IgG/瓶 2038 瓶 2011-4-9 0
批签粤检2008A06223
号 20080405-(1
人血白蛋白 合格 深圳市卫武光明生物制
品有限公司 20% 10g/瓶 11843瓶 2011-4-21 批签粤检2008A06244号
20080407 人血白蛋白 合格 深圳市卫武光明生物制
品有限公司 20% 10g/瓶
13267瓶
2011-4-17 批签粤检2008A06245号
20080406
人血白蛋白 合格
三九集团湛江开发区双
林药业有限公司
20% 50ml 10g/瓶 12548 瓶 2011-4-30
批签粤检2008A06344号
20080514
生 物 制 品 批 签 发 一 览 表
统计时间:2008年7月9日至2008年9月10日 证书编号
检品名称 签发结论
企业名称
规格 数量 有效期 批号 人血白蛋白 合格 三九集团湛江开发区双
林药业有限公司 20% 50ml 10g/瓶 11229 瓶 2011-4-30 批签粤检2008A06581号
20080515 人血白蛋白 合格 深圳市卫武光明生物制
品有限公司
20 % 10g/瓶 11603瓶 2011-5-20
批签粤检2008A06587号
20080509
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 2098瓶 2011-5-21
批签粤检2008A06588
号 20080517-(1
冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司 2.5g IgG/瓶 1101瓶 2011-5-24 批签粤检2008A06589
号 20080518-1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制
品有限公司
2.5g IgG/瓶 942瓶 2011-5-24 批签粤检2008A06590
号 20080519-1 人血白蛋白 合格 深圳市卫武光明生物制
品有限公司 20% 10g/瓶
12235瓶
2011-5-3 0批签粤检2008A06591号
20080508
人血白蛋白 合格 三九集团湛江开发区双
林药业有限公司 20% 50ml 10g/瓶 11927 瓶 2011-5-31 批签粤检2008A06841号
20080616 人血白蛋白 合格 广东卫伦生物制药有限
公司
20%,10g/瓶 8717瓶 2013-4-30 批签粤检2008A06968号
20080507 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 2.5g 3350瓶 2011-3-22 批签粤检2008A06971
号 20080408
冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 2.5g 3864瓶 2011-3-22 批签粤检2008A06975
号 20080409
冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司 2.5g 3953瓶 2011-4-22 批签粤检2008A06976
号 20080511
冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限
公司
2.5g
3473瓶
2011-4-22 批签粤检2008A06977
号 20080512
人血白蛋白 合格 三九集团湛江开发区双
林药业有限公司 20% 50ml 10g/瓶 11149 瓶 2011-5-31 批签粤检2008A06988号
20080617 狂犬病人免疫球蛋白 合格 三九集团湛江开发区双
林药业有限公司 200IU/瓶 110529瓶 2010-4-28 批签粤检2008A07177号
20080504 人血白蛋白 合格 CSL Behring GmbH 10g/50ml/瓶 29,585瓶 2013-2-28 批签粤检2008A07282号
61644411B 人血白蛋白 合格 CSL Behring GmbH 10g/50ml/瓶 32,997瓶 2013-2-28 批签粤检2008A07283号
62244411B 人血白蛋白 合格 CSL Behring GmbH 10g/50ml/瓶
32,212瓶 2013-3-30 批签粤检2008A07284号
62844411B
人血白蛋白 合格 三九集团湛江开发区双
林药业有限公司 20% 50ml 10g/瓶 10809 瓶 2011-5-31 批签粤检2008A07297号
20080618 静注人免疫球蛋白(pH4) 合格 三九集团湛江开发区双林药业有限公司 5% 50ml 2.5g/瓶
14077 瓶 2011-5-31 批签粤检2008A07299号
20080607
人血白蛋白 合格 三九集团湛江开发区双
林药业有限公司 20% 50ml 10g/瓶 11630 瓶 2011-5-31 批签粤检2008A08107号
20080619 人血白蛋白 合格 深圳市卫武光明生物制
品有限公司 20% 10g/瓶 12931瓶 2011-6-10 批签粤检2008A08162号
20080614 人血白蛋白 合格 深圳市卫武光明生物制
品有限公司 20% 10g/瓶
13322瓶
2011-5-28 批签粤检2008A08163号
20080511 人血白蛋白 合格
广东卫伦生物制药有限
公司
20% 50ml 10g/瓶 8495瓶
2013-5-31
批签粤检2008A08471号
20080609
《生物制品批签发管理办法》
《生物制品批签发管理办法》(国家食品药品监督管理总局令第39号)2017年12月29日发布 国家食品药品监督管理总局令 第39号 《生物制品批签发管理办法》已于2017年12月20日经国家食品药品监督管理总局局务会议审议通过,现予公布,自2018年2月1日起施行。 局长:毕井泉 2017年12月29日 生物制品批签发管理办法 第一章总则 第一条为加强生物制品监督管理,规范生物制品批签发行为,保证生物制品安全、有效,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)有关规定,制定本办法。 第二条本办法所称生物制品批签发,是指国家食品药品监督管理总局(以下简称食品药品监管总局)对获得上市许可的疫苗类制品、血液制品、用于血源筛查的体外诊断试剂以及食品药品监管总局规定的其他生物制品,在每批产品上市销售前或者进口时,指定药品检验机构进行资料审核、现场核实、样品检验的监督管理行为。 未通过批签发的产品,不得上市销售或者进口。 第三条批签发申请人应当是持有药品批准证明文件的境内外制药企业。境外制药企业应当授权其驻我国境内办事机构或者我国境内企业法人作为代理人办理批签发。 批签发产品应当按照食品药品监管总局核准的工艺生产。企业对批签发产品生产、检验等过程中形成的资料、记录和数据的真实性负责。批签发资料应当经企业质量受权人审核并签发。 每批产品上市销售前或者进口时,批签发申请人应当主动提出批签发申请,依法履行批签发活动中的法定义务,保证申请批签发的产品质量可靠以及批签发申请资料、过程记录、试验数据和样品的真实性。 第四条食品药品监管总局主管全国生物制品批签发工作,负责规定批签发品种范围,指定批签发机构,指导批签发工作的实施。 省、自治区、直辖市食品药品监督管理部门负责本行政区域批签发申请人的日常监管,协助批签发机构开展现场核实,组织批签发产品的现场抽样及批签发不合格产品的处置,对批签发过程中发现的违法违规行为进行调查处理。
生物制品批签发管理办法》
国家食品药品监督管理局令 第11号 《生物制品批签发管理办法》于2004年6月4日经国家食品药品监督管理局局务会审议通过,现予公布。本管理办法自公布之日起施行。 二○○四年七月十三日 生物制品批签发管理办法 第一章总则 第一条为加强生物制品质量管理,保证生物制品安全、有效,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)及《中华人民共和国药品管理法实施条例》,制定本办法。
第二条生物制品批签发(以下简称批签发),是指国家对疫苗类制品、血液制品、用于血源筛查的体外生物诊断试剂以及国家食品药品监督管理局规定的其他生物制品,每批制品出厂上市或者进口时进行强制性检验、审核的制度。检验不合格或者审核不被批准者,不得上市或者进口。 第三条国家食品药品监督管理局主管全国生物制品批签发工作;承担生物制品批签发检验或者审核工作的药品检验机构由国家食品药品监督管理局指定。 第四条生物制品批签发检验或者审核的标准为现行的国家生物制品规程或者国家食品药品监督管理局批准的其他药品标准。 第二章申请 第五条按批签发管理的生物制品在生产、检验完成后,药品生产企业应当填写《生物制品批签发申请表》,向承担批签发检验或者审核的药品检验机构申请批签发。
第六条申请批签发的生物制品必须具有下列药品批准证明文件之一: ㈠药品批准文号; ㈡《进口药品注册证》或者《医药产品注册证》; ㈢体外生物诊断试剂批准注册证明。 第七条申请批签发的技术要求及相关资料的格式,由中国药品生物制品检定所负责组织制定,报国家食品药品监督管理局批准并发布。 第八条申请批签发时应当提交以下资料及样品:(一)生物制品批签发申请表; (二)药品生产企业质量保证部门负责人签字并加盖本部门印章的批制造及检验记录摘要; (三)检验所需的同批号样品; (四)与制品质量相关的其他资料; (五)进口预防用疫苗类生物制品应当同时提交生产国国家药品管理当局出具的批签发证明文件,并提供中文译本。
医药公司生物制品管理制度
医药公司生物制品 管理制度
***公司 生 物 制 品 管 理 制 度 签发日期:***年**月**日执行日期:***年**月**日
生物制品管理制度目录 1、生物制品购进管理制度 2、生物制品入库质量检查验收管理制度 3、生物制品仓储保管制度 4、生物制品养护管理制度 5、生物制品出库复核管理制度 6、进口生物制品管理制度 7、生物制品有效期管理制度 8、生物制品销售管理制度 9、生物制品运输管理制度 10、生物制品储存运输设施设备管理制度 11、生物制品不良反应报告制度 12、物流系统的装置及设备的配置情况
生物制品购进管理制度 1.0目的 明确本企业生物制品购进的管理。 2.0范围 适用于公司业务部。 3.0责任 公司业务部对本制度的实施负责。 4.0主要内容 1、认真学习《中华人民共和国药品管理法》、《药品经营质量管理规范》及《生物制品批 签发管理办法》等法律法规、行政规章,严格按照有关规定进行生物制品的采购工作,杜绝假、劣生物制品购进,确保生物制品质量安全有效。 2、严格执行公司采购药品的程序规定,坚持“按需进货,择优采购”的购进原则,编制 购进计划,可以是长期计划,也可以是临时计划。 3、负责生物制品购进的采购人员应熟悉生物制品的相关知识,以认真负责的态度、崇尚 科学和质量第一的精神,做好生物制品采购工作。 4、生物制品必须从生物制品生产企业或具有生物制品经营资格的批发企业购进,不得从 不具有生物制品经营资格的单位或个人购进。 5、购进时严格审查供货单位的《药品生产许可证》、GMP证书或《药品经营许可证》(注明 有“生物制品”经营范围)、GSP证书及《营业执照》,查验供货单位业务员委托书、身份证明,经核对无误后,方可进货,确保从合法资格的企业购进质量可靠的生物制品。 6、采购生物制品应当与生物制品生产企业或生物制品批发企业签订采购合同,约定生物制
生物制品批签发管理办法
一、填空题 1.批签发申请人应当是持有药品批准证明文件的境内外制药企业。 2.生物制品批签发审核、检验应当依据药品监管总局核准的药品注册标准,并应当同时符合中华人民共和国药典要求。 3. 食品药品监管总局委托中国食品药品检定研究院组织制定批签发技术要求和技术考核细则,对拟承担批签发工作或者扩大批签发品种范围的药品检验机构进行能力评估和考核,对其他批签发机构进行业务指导、技术培训和考核评估。 4. 疫苗类产品应当在60日内完成批签发,血液制品和用于血源筛查的体外诊断试剂应当在35日内完成批签发。需要复试的,批签发工作时限可延长该检验项目的两个检验周期,并告知批签发申请人。 5. 按照批签发管理的生物制品在销售时,应当出具该批产品的生物制品批签发证明复印件并加盖企业公章。 6. 批签发机构应当在本机构每一批产品批签发决定作出后7日内公开批签发结论等信息。 二.判断题 1.未通过批签发的产品,不得上市销售或者进口。(√) 2.食品药品监管总局组织制定批签发技术要求和技术考核细则,对拟承担批签发工作或者扩大批签发品种范围的药品检验机构进行能力评估和考核,对其他批签发机构进行业务指导、技术培训和考核评估。(×)中检院 3.批签发申请人召回产品的,可免除其依法应当承担的其他法律责任。(×) 单选题 A食品药品监管总局食品药品审核查验中心 B中国食品药品检定研究院 C食品药品监管总局指定的批签发机构 A食品药品监管总局食品药品审核查验中心 B中国食品药品检定研究院 C食品药品监管总局指定的批签发机构 D国家药品监督管理局 3.新批准上市的生物制品首次申请批签发前,批签发申请人应当在批签发信息管理系统内登记建档。登记时应当提交以下资料。(D) A生物制品批签发品种登记表; B药品批准证明文件; C合法生产的证明性文件; D以上都是 多选题 1.生物制品批签发,是指国家药品监督管理总局对获得上市许可的,在每批产品上市销售前或者进口时,指定药品检验机构进行资料审核、现场核实、样品检验的监
生物制品质量管理规定
生物制品质量管理规定 TYYGROUP system office room 【TYYUA16H-TYY-TYYYUA8Q8-
一、目的 确保生物制品的经营安全,加强经营过程中的监控管理措施,达到安全、合法经营的管理目标。 二、依据 《药品管理法》、《关于进一步加强生物制品管理的通知》(国食药监办[2008]613号)和《药品经营质量管理规范》及实施条例等相关法律法规。 三、适用范围 适用于公司生物制品的经营管理。 四、内容 生物制品是应用普通的或以基因工程、细胞工程、蛋白质工程、发酵工程等生物技术获得的微生物、细胞及各种动物和人源的组织和液体等生物材料制备的,用于人类疾病预防、治疗和诊断的药品。 1、生物制品的经营管理、药品质量以及安全管理中,企业法人是第一责任人。 2、凡购进生物制品,均应按规定配备专门的管理人员。
3、建立生物制品的专用账册及购进、入库验收、在库养护、出库复核、销售等各项记录,记录实行计算机管理。 4、生物制品购销业务中应票账货款相符。 5、生物制品账册及记录的保存期限应当自药品有效期期满之日起不少于5年。 6、生物制品的管理人员和直接业务人员应相对稳定,其管理人员和直接业务人员、储存和运输等人员每年接受相关业务培训,经考核合格后方可上岗。 7、严格执行药品电子监管码赋码和出入库“见码必扫”操作,确保正确核注核销,及时处理系统预警信息。加强对下游企业销售的管理,电子监管预警信息提示收货企业核注信息有误的必须立即暂停供货、进行调查,发现销售数量和流向等情况异常应及时报告。 一、目的 严格把好生物制品的购进业务质量关,确保依法经营并保证经营质量安全。 二、依据
生物制品批签发管理办法(修订稿)
生物制品批签发管理办法 (修订稿)2015年12月10日 第一章总则 第一条为加强生物制品质量管理,保证生物制品安全、有效,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)、《中华人民共和国药品管理法实施条例》及有关规定,制定本办法。 第二条生物制品批签发(以下简称批签发),是指国家食品药品监督管理总局(以下称食品药品监管总局)指定的药品检定机构(以下称批签发机构)对疫苗类制品、血液制品、用于血源筛查的体外生物诊断试剂以及食品药品监管总局规定的其他生物制品,每批产品在销售前或者进口时进行检验、审核的质量评估过程。批签发不合格的生物制品不得销售或进口。 第三条食品药品监管总局负责全国生物制品批签发管理工作,负责指定批签发机构。 中国食品药品检定研究院(以下称中检院)负责制定批签发机构评估指南,指导、培训和考核其他批签发机构的批签发工作。 批签发机构负责承担批签发的具体工作,包括受理、资料审核、样品检验、现场核查、签发等。 省、自治区、直辖市药品监督管理部门负责组织批签发的现场抽样及批签发不合格产品的处置和调查等工作。 第四条生物制品生产企业应当按照《药品生产质量管理规范》组织生产。生物制品批签发审核检验的标准为《中华人民共和国药典》、食品药品监管总局批准发布的其他药品标准和技术规范性文件。 第五条批签发申请人对提供的证明性文件、资料及样品的真实性负责。 第六条批签发机构应当向公众公开生物制品批签发合格和不合格信息。 批签发机构应当在本机构网站或者申请受理场所公开批签发申请程序、收费标准和依据、时限等信息。 第七条食品药品监管总局建立统一的批签发申请系统,向申请人提供可查询的批签发进度、检验结果和签发结论等信息。 中检院负责批签发申请系统的日常运行和维护。 第二章批签发机构 第八条批签发机构应当具有实施批签发工作的质量保证体系,具备以下条件:(一)配备与批签发工作相适应的设施、设备和仪器; (二)检验人员应具备相应品种的审核和检验能力,数量应能满足批签发工作需求; (三)已建立符合要求的批签发工作文件; (四)其他与批签发工作相关的能力和条件。 第九条申请批签发工作的药品检定机构可根据具备的批签发条件,向省级药品监督管理部门提出申请,经省级药品监督管理部门初步审查,符合条件的,向食品药品监管总局申请相应品种批签发。
新版GSP质量管理工作程序2
新版GSP质量管理工作程序2 1、药品采购操纵程序 (2) 2、药品验收检查程序 (7) 3、药品入库储存操纵程序 (12) 4、药品在库养护程序 (14) 5、药品出库复核程序 (16) 6、销后退回药品处理程序 (18) 7、购进退出药品处理程序 (19) 8、不合格药品的确认及处理程序 (21) 9、药品抽样检查程序 (23) 10、质量记录和凭证操纵程序 (25) 11、质量治理体系内部评审程序 (29) 12、质量治理体系文件编制、审批、修订、撤销程序 (32) 13、直调药品工作程序 (38) 14、药品召回治理程序 (40) 15、药品电子监管治理程序 (43) 16、阴凉、冷藏条件储存药品治理治理程序(含储存、运输过程、运输车治理) (45) 17、药品收货程序 (43)
1目的:依法采购药品,防止假劣药品进入本企业,保证经营药品的质量。 2引用标准及制定依据: 2.1《中华人民共和国药典》; 2.2《中华人民共和国药品治理法》及事实上施条例。 2.3《药品经营质量治理规范》(90号令)。 2.4《药品流通治理方法》(局令第26号) 3适用范畴:本程序适用于公司药品采购全过程的操纵治理。 4定义: 4.1药品:是指用于预防、治疗、诊断人的疾病,有目的地调剂人的生理机能并规定有适应症或者功能主治、用法和用量的物质,包括中药材、中药饮片、中成药、化学原料药及其制剂、抗生素、生化药品、放射性药品、血清、疫苗、血液制品和诊断药品等。 4.2供方:提供产品的组织。 4.3顾客:同意产品的组织。 4.4合格(符合):满足要求。 4.5不合格(不符合):未满足要求。 4.6验证:通过提供客观证据对规定要求已得到满足的认定。 4.7确认:通过提供客观证据对特定的预期用途或应用要求已得到满足的认定。
生物制品批签发申请表
生物制品批签发申请表
“生物制品批签发申请表”填写说明 本表仅适用于申请人(单位)向中国食品药品检定研究院提出的生物制品批签发申请事项。每张申请表要求填写一个批号的内容,张贴申请制品的标签,并加盖申报单位公章。具体填写要求如下: 制品名称填写申请批签发制品注册的通用名称及英文名称,必须与送检样品标签名称和相关批件内容一致。 商品名填写申请批签发制品注册的商品名称,必须与样品标签名称一致。 申报单位填写申请批签发的单位名称及部门名称。 生产单位填写生产批签发制品的单位名称及部门名称,必须与送检样品标签和相关批件内容一致。 产地填写生产批签发制品的企业所在的国家或地区。 药品批准文号/进口药品注册证号/医药产品注册证号填写批签发制品的药品批准文号,或进口药品注册证号,或医药产品注册证号。 送审项目按实际情况分别在相应栏目前划√。供选择项目包括:“记录摘要”、“全套制检记录”、“检品及相应制检记录摘要”、“检品及相应全套制检记录摘要”。 批号填写申请批签发制品的批号,并应与样品标签一致。 批量/进口量填写国内批签发填写生产批签发制品的批量,进口批签发填写批签发制品的进口量。 生产日期填写生产批签发制品的日期,并应与样品标签一致。 有效期至填写批签发制品的有效期截止日期,并应与样品标签一致。要去按“ 年月日”的格式填写。 检品量填写抽取的样品数量,数量单位应与包装规格相对应。 检验项目一般进口产品填写“全检”;国内产品填写“部分检验”。其他情况按主管业务处、科室确认的项目填写。 规格填写申请批签发制品注册的药品规格。必须与送检样品标签和相关批件内容一致。如10ml/支,5mg/1ml/支、0.2mg/片,等等。 剂型填写申请批签发制品注册的剂型,必须与送检样品标签和相关批件内容一致。。
新版gsp4:药品收货与验收
附录4 药品收货与验收 第一条企业应当按照国家有关法律法规及《药品经营质量管理规范》(以下简称《规范》),制定药品收货与验收标准。对药品收货与验收过程中出现的不符合质量标准或疑似假、劣药的情况,应当交由质量管理部门按照有关规定进行处理,必要时上报药品监督管理部门。 第二条药品到货时,收货人员应当对运输工具和运输状况进行检查。 (一)检查运输工具是否密闭,如发现运输工具内有雨淋、腐蚀、污染等可能影响药品质量的现象,及时通知采购部门并报质量管理部门处理。 (二)根据运输单据所载明的启运日期,检查是否符合协议约定的在途时限,对不符合约定时限的,报质量管理部门处理。 (三)供货方委托运输药品的,企业采购部门要提前向供货单位索要委托的承运方式、承运单位、启运时间等信息,并将上述情况提前通知收货人员;收货人员在药品到货后,要逐一核对上述内容,内容不一致的,通知采购部门并报质量管理部门处理。
(四)冷藏、冷冻药品到货时,查验冷藏车、车载冷藏箱或保温箱的温度状况,核查并留存运输过程和到货时的温度记录;对未采用规定的冷藏设备运输或温度不符合要求的,应当拒收,同时对药品进行控制管理,做好记录并报质量管理部门处理。 第三条药品到货时,收货人员应当查验随货同行单(票)以及相关的药品采购记录。无随货同行单(票)或无采购记录的应当拒收;随货同行单(票)记载的供货单位、生产厂商、药品的通用名称、剂型、规格、批号、数量、收货单位、收货地址、发货日期等内容,与采购记录以及本企业实际情况不符的,应当拒收,并通知采购部门处理。 第四条应当依据随货同行单(票)核对药品实物。随货同行单(票)中记载的药品的通用名称、剂型、规格、批号、数量、生产厂商等内容,与药品实物不符的,应当拒收,并通知采购部门进行处理。 第五条收货过程中,对于随货同行单(票)或到货药品与采购记录的有关内容不相符的,由采购部门负责与供货单位核实和处理。 (一)对于随货同行单(票)内容中,除数量以外的其他内容与采购记录、药品实物不符的,经供货单位确认并提供正确的随货同行单(票)后,方可收货。 (二)对于随货同行单(票)与采购记录、药品实物数
2018年生物制品批签发管理办法培训试题及答案
生物制品批签发管理办法 培训试题2018.1 岗位:姓名:成绩: 一、单选题(每题4分,共20分) 1、《生物制品批签发管理办法》(原国家食品药品监督管理局令第39号)自起 施行。() A、2017年12月29日 B、2018年2月1日 C、2017年12月20日 D、2018年1月1日 2、批签发机构应当在本机构每一批产品批签发决定作出后内公开批签发结论等信息。() A、1 日 B、3日 C、5日 D、7日 3、批签发资料应当经企业审核并签发。() A、QA负责人 B、生产负责人 C、QC负责人 D、质量受权人 4、食品药品监管总局委托组织制定批签发技术要求和技术考核细则。() A、总局药品审评认证中心 B、中国食品药品检定研究院 C、国家药典委员会 D、北京市食品药品监督管理局 5、批签发机构及其所负责的批签发品种由确定。()
A、食品药品监管总局 B、中国食品药品检定研究院 C、省级食品药品监督管理局 D、县级食品药品监督管理局 二、多选题(每题4分,共20分) 1、生物制品批签发申请表、、生物制品批签发复审结果通知书的格式由中检院统一制定并公布。() A、生物制品批签发登记表 B、生物制品批签发证明 C、生物制品不予批签发通知书 D、生物制品批签发复审申请表 2、食品药品监管总局建立统一的批签发信息管理系统,该系统可用于:。() A、公布批签发机构确定及调整情况 B、向批签发申请人提供可查询的批签发进度、批签发结论 C、汇总公开已完成批签发的产品批签发结论以及重大问题处理决定等信息 D、办理《药品生产许可证》变更 3、食品药品监管总局指定的批签发机构负责批签发的等工作,并依法作出批签发决定。() A、受理 B、资料审核 C、现场核实 D、样品检验 4、新批准上市的生物制品首次申请批签发前,批签发申请人应当在批签发信息管理系统内登记建档。登记时应当提交以下资料:() A、生物制品批签发品种登记表 B、药品批准证明文件 C、合法生产的证明性文件 D、上市后变更的批准证明性文件
药品购进验收管理制度
药品购进验收管理制度 1、药品进货必须严格执行《药品管理法》、《医疗机构药品监督管理办法(试行)》、《合同法》等法律、法规。 2、购进药品以质量为前提,所需任何药品必须由资质合法的药品经营企业或药品生产企业组织配送。药品购进前应认真审核供货方资质,签订《药品质量保证协议》,质量协议中有明确的质量条款内容,并将加盖供货方原印章的证、照复印件存档备查。 3、采购药品时应索取合法票据(增值税发票和电脑清单),验收员按照清单,在药品货柜上对药品的品名、规格、批准文号、注册商标、有效期、数量、生产企业、生产批号、供货单位及药品合格证等逐一进行验收,对其进行外观质量、包装质量进行感观检查。发现质量不合格,应履行拒收职能,及时退回。 4、验收外用药品,其包装的标签或说明书上有规定的标识和警示说明。处方药和非处方药按分类管理要求,标签、说明书上有相应的警示语或忠告语;非处方药的包装有国家规定的专有标识。 5、验收进口药品要索取加盖了供货单位质管科原印章的《进口药品注册证》或《医药产品注册证》和《进口药品检验报告书》或《进口药品通关单》复印件。 6、验收实行批签发管理的生物制品(血液制品、疫苗)要索取加盖了供货单位质管科原印章的《生物制品批签发合格证》。 7、药品验收后,验收员要及时做好药品购进验收记录,记录药品的品名、规格、批准文号、有效期、数量、生产企业、生产批号、供货单位、进货日期、验收日期、验收结论等。
8、对进货情况应每年年终进行认真总结、分析,对进货过程中出现的相关问题,加以改进。 药品陈列、储存和养护管理制度 1、药房、库应地面光洁、墙面平整,门窗结构紧密,配备必要的防尘、防虫、防鼠设施;陈列和储存药品的货柜、货架等应保持清洁卫生。 2、药品储存应实行色标管理、分开存放并标示,其中待验库(区)、退货库(区)为黄色、合格品库(区)为绿色、不合格品库(区)为红色。 3、药品陈列和储存区域应配备检测和调节温湿度的设施设备,阴凉库控制在2-20℃,常温库控制在2-30℃。需冷藏的药品,应配备有相应的冷藏设施,温度控制在2-10℃。 4、对药品与非药品、内服药与外用药分开陈列和储存,并按药品的用途或剂型分类摆放,标签使用恰当,放置准确,字迹清晰。拆零药品存放于拆零专柜,并保留药品的原包装或标签。 5、养护员应做好药品陈列、储存区域的温、湿度管理工作,每日上午9:30分至10:30分间、下午2:30至3:30分间各记录一次温湿度。如温、湿度超标,及时采取相应的通风、降温、增温、除湿、加湿等措施,确保药品存放环境达到要求。 6、对药品要按月进行质量检查,并做好质量检查记录,3个月内到期失效的药品为近效期药品,对近效期药品应及时在效期
检验证明书格式
检验证明书格式 篇一:检验证明书格式 中华人民共和国出入境检验检疫出境货物报检单报检单位 (加盖公章):报检单位登记号: * 编号联系人: 电话: 报检日期: 年月 日注:有“*”号栏由出入境检验检疫机关填写◆国家出入境检验检疫局制篇二:质量证明书(格式)压力容器产品质量证明书产品名称 产品编号 质量保证工程师(签章)单位法定代表人(签章) 质量检验专用(公章)产品合格证制造单位制造许可证编号 产品名称类别设计单位设计批准书编号图号订货单位产品编号制造编号制造完成日期年月日 本压力容器产品经质量检验符合《压力容器安全技术监察规程》、设计图样和技术条件的 要求。 产品技术特性
产品编号产品主要受压元件使用材料一览表审核人:填表人: 年月日 共页第页产品焊接试板力学和弯曲性能检验报告产品编号理化责任师:填表人:年月日共页第页篇三:检验检疫证明格式(英文)(公司英文名称抬头)transmitting animal spongiform encephalopathy (tse)/bovine spongiform encephalopathy (bse) declaration we declare and certify that the product:xxx (产品名称) manufactured at xxx (工厂名称) contains no ingredients of animal origin and no material derived from, or exposed to animals affected by or under quarantine for transmitting animal spongiform encephalopathy(tse)/bovine spongiform encephalopathy(bse). the product is produced in facility where no animal, animal products, animal by -products, veterinary vaccines or animal pathogens are maintained and are packed in a packaging that can be identified as to the nature of the products and the country of origin. date:
生物制品批签发管理办法
生物制品批签发管 理办法
生物制品批签发管理办法 (试行) 第一章总则 第一条为加强生物制品管理,保证生物制品安全、有效,根据《中华人民共和国药品管理法》及《中华人民共和国药品管理法实施条例》,制定本办法。 第二条生物制品批签发(下简称批签发)是指国家对疫苗类制品、血液制品、用于血源筛查的体外生物诊断试剂以及国家药品监督管理局规定的其它生物制品,每批制品出厂销售前或者进口时实行强制性审查、检验和批准的制度。 依据本办法规定实行批签发的生物制品未经批签发的,不得销售或者进口,禁止使用。 第三条国家药品监督管理局授权其设置或者确定的药品检验机构承担生物制品批签发工作。 第四条实行批签发管理的生物制品品种由国家药品监督管理局确定并公布。 生物制品批签发审查、检验标准为现行的国家生物制品规程和国家药品监督管理局批准的其它标准。
第二章申请 第五条药品生产企业在完成生物制品的生产、检验后,填写《生物制品批签发申请表》,向承担批签发的药品检验机构申请批签发。 第六条申请批签发的生物制品必须具有下列药品批准证明文件之一: (一)药品批准文号。 (二)《进口药品注册证》或者《医药产品注册证》。(三)体外生物诊断试剂批准注册证明。 第七条申请批签发的技术要求及相关资料的格式,由中国药品生物制品检定所负责制定。 第八条申请批签发时须提交以下资料及样品: (一)生物制品批签发申请表。 (二)药品生产企业质量保证部门负责人签字并加盖本部门印章的批制造及检验记录摘要。 (三)检验所需的同批号样品。 (四)与制品质量相关的其它资料。 (五)进口生物制品应同时提交生产国国家药品管理当局出具的批签发证明文件,并提供中文译本。 第三章审查、检验与签发
生物制品批签发一览表
生 物 制 品 批 签 发 一 览 表 统计时间:2008年7月9日至2008年9月10日 证书编号 检品名称 签发结论 企业名称 规格 数量 有效期 批号 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司 2.5gIgG/瓶 8168瓶 2010-3-27 批签粤检2008A05052号 200803401 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司 2.5g IgG/瓶 8531瓶 2010-5-6 0批签粤检2008A05787号 200805403 静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制品有限公司 2.5g IgG/瓶 8145瓶 2010-4-24 批签粤检2008A05788号 200804402 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限 公司 2.5g 3289瓶 2011-3-16 批签粤检2008A05836 号 20080407 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限 公司 2.5g 2444瓶 2011-3-16 批签粤检2008A05837 号 20080406 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限 公司 1.0g 3576瓶 2011-3-16 批签粤检2008A05838 号 20080405 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限 公司 1.0g 4873瓶 2011-3-16 批签粤检2008A05839 号 20080404 1 冻干静注人免疫球蛋白(pH4) 合格 广东卫伦生物制药有限 公司 1.0g 5049瓶 2011-3-16 批签粤检2008A05840 号 20080403 2 狂犬病人免疫球蛋白 合格 三九集团湛江开发区双 林药业有限公司 200IU/瓶 103137 瓶 2010-3-31 批签粤检2008A05853号 20080403 静注人免疫球蛋白(pH4) 合格 三九集团湛江开发区双林药业有限公司 5% 50ml 2.5g/瓶 12716 瓶 2011-4-30 批签粤检2008A06194号 20080506 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2176 瓶 2011-5-18 批签粤检2008A06198 号 20080516-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2252瓶 2008-5-14 批签粤检2008A06199 号 20080515-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2301 瓶 2011-5-11 批签粤检2008A06200 号 20080514-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2325瓶 2011-5-8 0 批签粤检2008A06201 号 20080513-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2304瓶 2011-5-5 0 批签粤检2008A06202 号 20080512-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2288 瓶 2011-5-2 0 批签粤检2008A06203 号 20080511-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2223 瓶 2011-4-21 批签粤检2008A06212 号 20080409-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2144 瓶 2011-4-18 批签粤检2008A06213 号 20080408-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2136 瓶 2011-4-15 批签粤检2008A06214 号 20080407-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2152 瓶 2011-4-12 批签粤检2008A06215 号 20080406-(1 冻干静注人免疫球蛋白(pH4) 合格 深圳市卫武光明生物制 品有限公司 2.5g IgG/瓶 2038 瓶 2011-4-9 0 批签粤检2008A06223 号 20080405-(1 人血白蛋白 合格 深圳市卫武光明生物制 品有限公司 20% 10g/瓶 11843瓶 2011-4-21 批签粤检2008A06244号 20080407 人血白蛋白 合格 深圳市卫武光明生物制 品有限公司 20% 10g/瓶 13267瓶 2011-4-17 批签粤检2008A06245号 20080406 人血白蛋白 合格 三九集团湛江开发区双 林药业有限公司 20% 50ml 10g/瓶 12548 瓶 2011-4-30 批签粤检2008A06344号 20080514
(完整版)某市药品经营企业GSP认证跟踪检查方案
某市药品经营企业GSP认证跟踪检查方案 根据《药品GSP认证管理办法》规定,决定对某市辖区内药品经营企业实施跟踪现场检查。检查方案如下: 一、跟踪检查范围:辖区内所有药品经营企业 二、检查时间:某月某日—某月某日 三、检查内容: (一)药品批发企业检查重点 1、企业管理机构及质量负责人是否有变动,是否符合要求;技术人员队伍是否符合要求,是否稳定; 2、企业员工的培训情况。培训内容是否包括兴奋剂知识、蛋肽类有关规定,有无要求掌握蛋肽类相关品种与含兴奋剂的单方制剂品种; 3、企业质量管理制度是否按照现行法规进行了修订,是否增加了“生物制品批签发的管理规定”“销售人员网上备案制度及对与本公司发生业务来往的销售人员进行网上核查”“首营企业、首营品种网上核查的管理”等内容; 4、企业《药品经营许可证》是否存在未履行手续私自变更许可事项的情况(变更经营地址、增加或减少仓库面积、增加或者注销经营范围、调整质量负责人等); 5、仓储、冷藏运输、检测仪器、调节温湿度等设施设备运行、记录、维护情况;
6、冷藏药品的存储条件是否符合规定,冷库有无安装自动温湿度记录仪; 7、检查企业是否取得经营蛋白同化制剂、肽类激素、二类精神药品资格,有无违规购进、销售情况;有无按规定实行三专; 8、检查重点品种的购进、销售是否符合规定,是否按规定索取相关资料。如进口药品是否索要《进口药品检验报告书》和《进口药品注册证》、生物制品是否索要《生物制品批签发合格证》、终止妊娠药品是否有违规销售给零售企业、个体诊所和未取得《母婴保健专项技术服务合格证》的医疗机构等;随机抽查5个品种检查合法票据(对提供不出合法票据的品种进行记录); 9、检查中药饮片的购进渠道和包装,要求包装必须印有生产企业的相关信息,不得为经营企业分装的包装。抽查3-5个中药饮片品种,检查购进渠道和合法票据情况(重点为省局培训过的20种); 10、检查该企业销售人员是否在省局网上备案,核查供货企业及其销售人员的资质证明文件是否齐全,供货方销售人员是否在网上进行了核查; 11、认证以来所经营药品的购进、验收、储存、养护、销售各环节手续、记录是否合法和齐全; 12、检查现场管理情况:仓库内五区是否齐全,色标管理是否到位,各区是否正常使用;药品是否按规定储存在相应的仓库中,储存状况是否符合要求,有无混品种、混批号存放、有无外包装破损、挤压
2018年生物制品批签发管理办法
生物制品批签发管理办法 第一章总则 第一条为加强生物制品监督管理,规范生物制品批签发行为,保证生物制品安全、有效,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)有关规定,制定本办法。 第二条本办法所称生物制品批签发,是指国家食品药品监督管理总局(以下简称食品药品监管总局)对获得上市许可的疫苗类制品、血液制品、用于血源筛查的体外诊断试剂以及食品药品监管总局规定的其他生物制品,在每批产品上市销售前或者进口时,指定药品检验机构进行资料审核、现场核实、样品检验的监督管理行为。 未通过批签发的产品,不得上市销售或者进口。 第三条批签发申请人应当是持有药品批准证明文件的 境内外制药企业。境外制药企业应当授权其驻我国境内办事机构或者我国境内企业法人作为代理人办理批签发。 批签发产品应当按照食品药品监管总局核准的工艺生产。企业对批签发产品生产、检验等过程中形成的资料、记录和数据的真实性负责。批签发资料应当经企业质量受权人审核并签发。 每批产品上市销售前或者进口时,批签发申请人应当主 动提出批签发申请,依法履行批签发活动中的法定义务,保
证申请批签发的产品质量可靠以及批签发申请资料、过程记录、试验数据和样品的真实性。 第四条食品药品监管总局主管全国生物制品批签发工作,负责规定批签发品种范围,指定批签发机构,指导批签发工作的实施。 省、自治区、直辖市食品药品监督管理部门负责本行政 区域批签发申请人的日常监管,协助批签发机构开展现场核实,组织批签发产品的现场抽样及批签发不合格产品的处置,对批签发过程中发现的违法违规行为进行调查处理。 食品药品监管总局指定的批签发机构负责批签发的受理、资料审核、现场核实、样品检验等工作,并依法作出批签发决定。 食品药品监管总局委托中国食品药品检定研究院(以下 简称中检院)组织制定批签发技术要求和技术考核细则,对拟承担批签发工作或者扩大批签发品种范围的药品检验机 构进行能力评估和考核,对其他批签发机构进行业务指导、技术培训和考核评估。 食品药品监管总局食品药品审核查验中心(以下简称核 查中心)负责批签发过程中的现场检查工作。 第五条食品药品监管总局对批签发产品建立基于风险 的监督管理体系。必要时,可以通过现场核实验证批签发申请资料的真实性、可靠性。
国家食品药品监督管理局关于人用狂犬病疫苗实施批签发管理的通知
国家食品药品监督管理局关于人用狂犬病疫苗实施批签发管 理的通知 【法规类别】药品管理 【发文字号】国食药监注[2005]327号 【失效依据】本篇法规已被《国家食品药品监督管理局关于清理规章和规范性文件的公告》(发布日期:2007年3月22日实施日期:2007年3月22日)宣布失效或废止(原因:已被《关于进一步实施生物制品批签发工作的通知》(国食药监注〔2005〕424号)代替)【发布部门】国家食品药品监督管理局(原国家药品监督管理局)(已撤销) 【发布日期】2005.06.30 【实施日期】2005.06.30 【时效性】失效 【效力级别】部门规范性文件 国家食品药品监督管理局关于人用狂犬病疫苗实施批签发管理的通知 (国食药监注[2005]327号) 各省、自治区、直辖市食品药品监督管理局(药品监督管理局): 为了进一步加强人用狂犬病疫苗的管理,根据《药品管理法实施条例》及《生物制品批签发管理办法》(局令第11号)的相关规定,经研究,决定将人用狂犬病疫苗纳入生物制品批签发管理,现将有关事项通知如下: 一、自2005年7月1日起人用狂犬病疫苗纳入生物制品批签发管理。2005年7月1日以
后出厂的人用狂犬病疫苗需获得生物制品批签发合格证书后方可上市销售,2005年7月1日以前已上市的人用狂犬病疫苗可在效期内继续流通、使用。相关表格及要求由中国药品生物制品检定所负责制订并发送。 二、授权中国药品生物制品检定所承担人用狂犬病疫苗的批签发工作,负责批记录摘要的审核、项目检验及人用狂犬病疫苗批签发文件的签发。北京、吉林、上海、湖北、广东、四川、甘肃等授权生物制品批签发任务的药品检验机构负责管理辖区内人用狂犬病疫苗的现场抽样和其中部分项目的检验工作。具体分工由中国药品生物制品检定所负责组织确定,并予以公布。 三、各授权开展生物制品批签发工作的药品检验机构须严格执行《生物制品批签发管理办法》的相关规定,认真做好人用狂犬病疫苗的批签发工作。如在具体实施过程中发现的问题,应及时向我局报告。 国家食品药品监督管理局 二00五年六月三十日 附件:
生物制品批签发管理办法
生物制品批签发管理办法 (试行) 第一章总则 第一条为加强生物制品管理,保证生物制品安全、有效,根据《中华人民共和国药品管理法》及《中华人民共和国药品管理法实施条例》,制定本办法。 第二条生物制品批签发(下简称批签发)是指国家对疫苗类制品、血液制品、用于血源筛查的体外生物诊断试剂以及国家药品监督管理局规定的其他生物制品,每批制品出厂销售前或者进口时实行强制性审查、检验和批准的制度。 依据本办法规定实行批签发的生物制品未经批签发的,不得销售或者进口,禁止使用。 第三条国家药品监督管理局授权其设置或者确定的药品检验机构承担生物制品批签发工作。 第四条实行批签发管理的生物制品品种由国家药品监督管理局确定并公布。 生物制品批签发审查、检验标准为现行的国家生物制品规程和国家药品监督管理局批准的其他标准。 第二章申请
第五条药品生产企业在完成生物制品的生产、检验后,填写《生物制品批签发申请表》,向承担批签发的药品检验机构申请批签发。 第六条申请批签发的生物制品必须具有下列药品批准证明文件之一: (一)药品批准文号。 (二)《进口药品注册证》或者《医药产品注册证》。 (三)体外生物诊断试剂批准注册证明。 第七条申请批签发的技术要求及相关资料的格式,由中国药品生物制品检定所负责制定。 第八条申请批签发时须提交以下资料及样品: (一)生物制品批签发申请表。 (二)药品生产企业质量保证部门负责人签字并加盖本部门印章的批制造及检验记录摘要。 (三)检验所需的同批号样品。 (四)与制品质量相关的其它资料。 (五)进口生物制品应同时提交生产国国家药品管理当局出具的批签发证明文件,并提供中文译本。 第三章审查、检验与签发
医院药品入库验收标准操作程序【最新版】
医院药品入库验收标准操作程序 目的:制定药品入库验收标准操作程序,确保药品质量和数量。 范围:药库 责任人:药库管理人员 程序: 1、药库保管员在待验区按公司的随货同行联认真验收:逐一检查药品的品名、规格、数量、生产厂家、批号、效期及外观质量包装等,验收合格后在随货联签字。 2、特殊药品的验收:麻醉药品、一类精神药品验收除以上要求外,必须严格按随货联药品片剂每盒查看,注射剂每支查看,验收合格后双人签字。准确无误才能入库,并放入保险柜上锁。麻醉药品、一类精神药品需在麻醉药品印鉴卡登记盖章。 3、验收进口药品,应有《进口药品注册证》和同批号的《进口药品检验报告书》复印件。进口生物制品应有《生物制品进口批件》,以上批准文件应加盖供货单位质量管理机构原印章,以上材料经查验
无误后由药库管理员保存归档。 4、验收生物制品应有《生物制品批签发合格证》(《生物制品检验报告单》),以上材料经查验无误后由药库管理员保存归档。 5、药库管理员应及时做好验收台帐,务必要准确无误,记录:供货单位、药品名称、规格、剂型、数量、生产厂家、批号、有效期、进口药品注册证号等,附验收结论。 6、在验收药品时,零星拼装成件的药品以及容易变质得药品应全部逐件点验,整件包装药品按件数开箱抽查。 7、在验收中发现质量有疑问,如药品数量不符、错发等,应暂缓进库,与供货企业业务员联系,查明原因,对外观不合格的药品,填写拒绝签收。 8、药品验收后,保管员在验收单上签字。合格药品分类入库,上架存放,部分品种即入登货位卡,随货发票交药品入账责任人登记入账。 9、药品验收记录保存至超过药品有效期一年,但不得少于三年。
生物制品批签发管理办法-试题
质量技能大赛试题 (生物制品批签发管理办法) 一、填空题 1.批签发申请人应当是持有的境内外制药企业。 2.生物制品批签发审核、检验应当依据药品监管总局核准的,并应当同时符合要求。 3. 食品药品监管总局委托组织制定批签发技术要求和技术考核细则,对拟承担批签发工作或者扩大批签发品种范围的药品检验机构进行能力评估和考核,对其他批签发机构进行业务指导、技术培训和考核评估。 4. 疫苗类产品应当在内完成批签发,血液制品和用于血源筛查的体外诊断试剂应当在35日内完成批签发。需要复试的,批签发工作时限可延长该检验项目的检验周期,并告知批签发申请人。 5. 按照批签发管理的生物制品在销售时,应当出具该批产品 的。 6. 批签发机构应当在本机构每一批产品批签发决定作出后内公开批签发结论等信息。 二.判断题 1.未通过批签发的产品,不得上市销售或者进口。() 2.食品药品监管总局组织制定批签发技术要求和技术考核细则,对拟承担批
签发工作或者扩大批签发品种范围的药品检验机构进行能力评估和考核,对其他批签发机构进行业务指导、技术培训和考核评估。() 3.批签发申请人召回产品的,可免除其依法应当承担的其他法律责任。() 单选题 1.负责批签发过程中的现场检查工作。() A食品药品监管总局食品药品审核查验中心 B中国食品药品检定研究院 C食品药品监管总局指定的批签发机构 D国家药品监督管理局 2.批签发机构及其所负责的批签发品种由确定。() A食品药品监管总局食品药品审核查验中心 B中国食品药品检定研究院 C食品药品监管总局指定的批签发机构 D国家药品监督管理局 3.新批准上市的生物制品首次申请批签发前,批签发申请人应当在批签发信息管理系统内登记建档。登记时应当提交以下资料。() A生物制品批签发品种登记表; B药品批准证明文件; C合法生产的证明性文件; D以上都是