原子转移自由基聚合及可控自由基聚合

活性可控自由基聚合
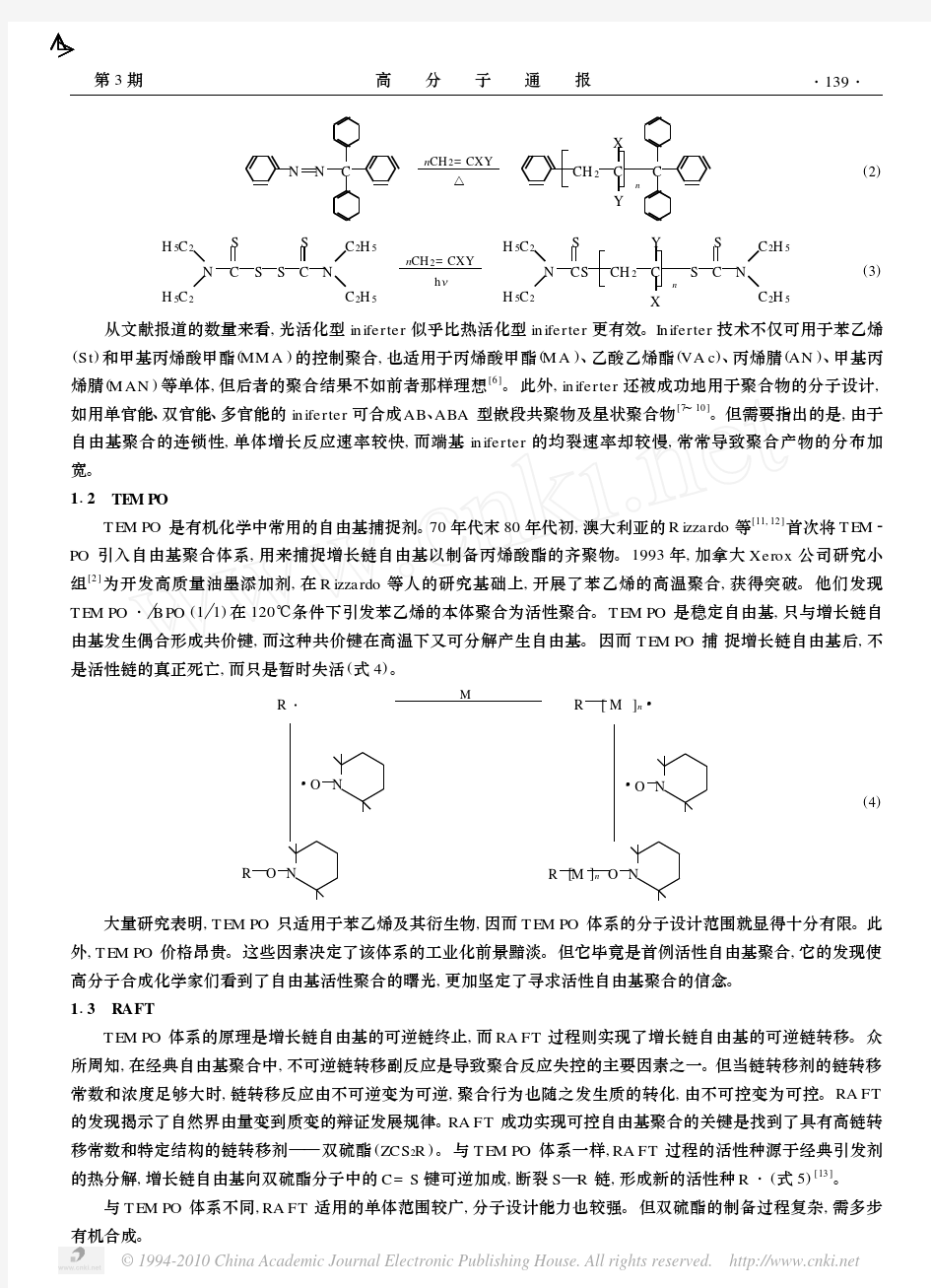
活性/可控自由基聚合 在20世纪50、60年代,自由基聚合达到了它的鼎盛时期。但由于存在链转移和链终止反应,传统自由基聚合不能较好地控制分子量及大分子结构[1]。1956年美国科学家Szwarc等提出了活性聚合的概念[2],活性聚合具有无终止、无转移、引发速率远远大于链增长速率等特点,与传统自由基聚合相比能更好地实现对分子结构的控制,是实现分子设计、合成具有特定结构和性能聚合物的重要手段。但离子型活性聚合反应条件比较苛刻、适用单体较少,且只能在非水介质中进行,导致工业化成本居高不下,较难广泛实现工业化。鉴于活性聚合和自由基聚合各自的优缺点,高分子合成化学家们联想到将二者结合,即可控活性自由基聚合(CRP)或活性可控自由基聚合。CRP可以合成具有新型拓扑结构的聚合物、不同成分的聚合物以及在高分子或各种化合物的不同部分链接官能团,适用单体较多,产物的应用较广,工业化成本较低。目前实现“活性”/可控自由基聚合可分以下几种途径: (1) 稳定“活性”自由基聚合(SFRP);(2) 原子转移自由基聚合(ATRP);(3)可逆加成-断裂链转移聚合(RAFT)。 一、稳定“活性”自由基聚合(SFRP) SFRP属于非催化性体系,是利用稳定自由基来控制自由基聚合。其机理是按照下面的可逆反应进行:外加的稳定自由基X·可与活性自由基P·迅速进行失活反应,生成“休眠种”P-X,P-X能可逆分解,又形成X·及活性种自由基P·而链增长。有研究表明,使用烷氧胺作引发剂效果好[3]。
反应体系中的自由基活性种P·可抑制在较低的浓度,这样就可以减少自由基活性种之间的不可逆终止作用,从而聚合反应得到控制。稳定自由基X·,主要有TEMPO(2,2,6,6-四甲基-1-哌啶氮氧自由基)和CoⅡ·,TEMPO属于稳定的有机自由基;CoⅡ·属于稳定的有机金属自由基。氮氧稳定自由基这类体系聚合的一大特点是聚合工艺较简单,可合成一些具有特殊结构的大分子,如树枝-线状杂化结构、聚苯乙烯嵌段共聚物等[4,5],其缺点是氮氧自由基的价格较贵,合成困难,只适用于苯乙烯及其衍生物,并且聚合慢,温度需在110℃~140℃之间,在聚合过程中增长链自由基和氮氧自由基可发生歧化终止的副反应而影响控制程度。不过,Moad、Thang等[6]认为,这些缺点是可以避免的,他们采用新的一类氮氧自由基2,2,5,5-(tetraalkylimida zolidin-4-one-1-oxyl)或其衍生物替代TEMPO组成的聚合体系,得到了分子量可控和窄分子量分布的均聚物、无规共聚物和嵌段共聚物,同时这类聚合反应具有比TEMPO聚合体系更好的活性聚合特征,并且具有较易合成、无挥发性和副反应较少等优点。另外一种方法是利用电子效应作用于氮氧自由基[7]。用CoⅡ·类稳定自由基体系聚合得到的聚合物分子量不高,分子量分布较宽[8]。可以相信,通过使用新型氮氧自由基,此体系完全可以扩展到(甲基)丙烯酸和其它单体。 二、原子转移自由基聚合(ATRP) [9] 自由基是一种十分活泼的活性种,在自由基聚合中极易发生链转移和链终止,所以要抑制副反应,聚合体系中必须具有低而恒定的自由基浓度;但又要维持可观的反应速度(自由基浓度不能太低);为解决这一矛盾,高分子化学家们受活性正离子聚合体系的启发,将可逆链转移和链终止的概念引入自由基聚合,通过在活性种和休眠种之间建立一个快速交换反应,成功的实现了矛盾的对立统一。
原子转移自由基聚合及其应用新进展(精)
原子转移自由基聚合及其应用新进展 原子转移自由基聚合(ATRP),是近几年迅速发展并有着重要应用价值的一种活性聚合技术。自从1956 年Szwarc[1]等报道了一种没有链转移和链终止的负离子聚合技术以来,活性聚合的研究性得到了巨大的发展,并一直是高分子学术界高度重视的领域。1983年Webster等[2]成功地实现了适用于丙烯酸酯类单体的基团转移聚合。随后又成功的实现了开环聚合[3]、活性正离子聚合[4,5]、络合负离子聚合[6] 以及无金属离子的活性负离子聚合[7]。1993年Xerox公司在苯乙烯的普通自由基聚合体系中加入有机自由基捕捉剂(Tempo体系)[8],使反应体系在聚合过程中自由基保持较低的浓度,从而抑制了自由基的副反应。第一次实现了" 活性"自由基聚合。与此同时,1995年《美国化学会志》报道了CarnegieMellon大学Matyjaszewski教授和王锦山博士共同开发的原子转移自由基聚合(ATRP)[9],成功地实现了真正意义上的"活性"/可控自由基聚合,取得了活性自由基聚合领域的历史性突破。 1. ATRP基本原理 ATRP的基本原理如Figure 1.1所示: Figure 1.1 Mechanism of atom transfer radical polymerization
式中,R-X是引发剂卤代烃(X-般为Cl或Br),M t n为过渡金属络合物,它由过渡金属离子和配位剂构成。在引发阶段,处于低氧化态的过渡金属络合物(盐)M t n从一有机卤化物-X中夺取卤原子X,生成引发自由基R·及处于高氧化态的金属络合物(盐) M t n + 1 -X。R·引发可给出卤原子X,即M t n + 1-X 与R·/R-M·发生减活反应生成R-X/R-M-X。如果R-Mn-X (n = 1, 2, ...)与R-X-样可与M t n发生促活反应生成相应的R-Mn及M t n + 1-X,同时若R-Mn·与M t n + 1-X又可反过来发生减活反应生成R-Mn-X及M t n,在自由基聚合反应进行的同时,就会始终伴随着一个自由基活性种Mn·与有机大分子卤化物休眠种Mn-X的可逆转换平衡反应。卤原子的可逆转移控制着[Mn·],而一个快速的卤原子转换速率将控制着分子量及分子量分布。图示表明:ATRP的基本原理其实是通过一个交替的“活化—去活”可逆反应使得体系中游离基浓度处于极低,迫使不可逆终止反应被降低到最低程度,而链增长反应仍可进行,从而实现“活性”聚合[10]。由于在这种聚合反应中,只是将自由基活性种的浓度加以控制,链终止和链转移被极大地抑制了,所以这种聚合反应只能是可控聚合或“活性”聚合,而不是真正的活性聚合。同时,在这种可控聚合反应中包含着卤原子从卤化物到金属络合物(盐)、再从金属卤化物转移到自由基这样一个反复循环的原子转移过程,加之反应活性种为自由基,所以称为原子转移自由基聚合。由于已有实验证明某些基团也可发生类似的转移自由基反应,故王锦山等把这样一种反应称为“原子(基团)转移自由基聚合”[11]。 ATRP研究大致可以分成两个体系:一个是美国Carnegie-Mellon
自由基聚合
2.自由基聚合 2.1引言 连锁聚合 根据聚合反应机理分类,聚合反应可以分为 逐步聚合 连锁聚合反应需要活性中心,单体在活性中心上反应形成大分子。活性中心可以是自由基,也可以是阴、阳离子。活性中心的性质与化合物共价键断裂的方式有关。 共价键有两种断裂方式:均裂和异裂 均裂: 共价键上一对电子分属于两个基团,这种带独电子的基团呈电中性,称作自由基或游离基。 异裂: 共价键上一对电子全部归属于某一基团,形成阴离子或负离子,则另一缺电子基团称作阳离子或正离子。 自由基、阴离子、阳离子都有可能成为活性中心,可打开烯类单体或羰基单体中的π键,或使环状单体的σ键断裂开环,使之链引发和链增长,分别成为自由基聚合,阴离子聚合,阳离子聚合,和配位聚合,实际上配位聚合也属于离子聚合的范畴。 Eg: 自由基聚合: 2.2连锁聚合的单体 单体能否聚合,须从热力学和动力学两方面考虑,热力学上能聚合的单体还要求有适当的引发剂、温度等动力学条件,才能保证一定的聚合速度。从热力学考虑可以进行连锁聚合的单体有: 2.2.1适合连锁聚合的单体 大致可以分为三类: 1.含有碳碳双键的烯类单体:包括单烯类、共轭二烯类,甚至炔烃。其中:
单烯类:乙烯基单体中的碳碳双键中π键可以均裂也可以异裂,因此可以进行自由基聚合或离子聚合。具体选择哪种聚合方式,由取代基的性质决定。 共轭二烯类:如苯乙烯,丁二烯,异戊二烯等单体处于共轭体系,在外界的影响下,双键的电子云易流动,诱导极化。因此单体既可以进行自由基聚合,也可以进行离子聚合。 2.羰基化合物如HCHO,CH3CHO,甚至酮类。 Eg: HCHO 羰基的双键有极性,使氧原子带有部分负电荷,而碳原子则带有部分正电荷。 3.杂环化合物 羰基化合物和杂环化合物的极性较强,一般不能自由基聚合,只适合于离子聚合。因此实际上只有碳碳双键的烯类单体可以进行自由基聚合,但也不是所有的都行,其取代基的性质有很大影响。 2.2.2取代基对于乙烯类单体聚合能力的影响。 除了取代基的种类和性质外,取代基的数量和体积也颇有影响,概括起来,分电子效应和位阻效应两个方面。电子效应又有诱导(极性)效应和共轭效应之分。乙烯基单体取代基的诱导效应和共轭效应能改变双键的电子云密度,并且对所形成的活性种的稳定性也有影响,因此决定着对自由基,阴、阳离子聚合的选择性。 1.无取代基时 乙烯结构对称,偶极矩为零,对进攻试剂选择性差。(目前只有两种聚合途径,在高温高压下可进行自由基聚合;在低压下可进行配位聚合。) 2.一取代乙烯 1)取代基为供电基团 供电基团有:烷氧基,烷基、苯基、乙烯基等 它可以(1)使碳碳双键电子云密度增加,有利于阳离子进攻,生成碳阳离子。 (2)使生成的阳离子增长种共振稳定。(碳阳离子生成后,由于供电子基团的存在,使电子云密度缺少的情况有所改善,体系的能量有所降低,碳阳离子的稳定性有所增加。)例如: 从诱导效应来看:烷氧基使双键电子云密度下降,理应进行阴离子或自由基聚合。 从共轭效应看:氧上未共用电子对能和双键形成P-π共轭,使双键电子云密度增加。 一般情况下,共轭效应占主动,所以是碳碳双键上电子云密度增加。同时又因为烷氧基的共轭,使正电荷不单单集中在碳阳离子上,而分散在碳氧两个原子上,使形成的
活性可控自由基聚合
活性/可控自由基聚合 在20世纪50、60年代,自由基聚合达到了它的鼎盛时期。但由于存在链转移和链终止反应,传统自由基聚合不能较好地控制分子量及大分子结构[1]。1956年美国科学家Szwarc等提出了活性聚合的概念[2],活性聚合具有无终止、无转移、引发速率远远大于链增长速率等特点,与传统自由基聚合相比能更好地实现对分子结构的控制,是实现分子设计、合成具有特定结构和性能聚合物的重要手段。但离子型活性聚合反应条件比较苛刻、适用单体较少,且只能在非水介质中进行,导致工业化成本居高不下,较难广泛实现工业化。鉴于活性聚合和自由基聚合各自的优缺点,高分子合成化学家们联想到将二者结合,即可控活性自由基聚合(CRP)或活性可控自由基聚合。CRP可以合成具有新型拓扑结构的聚合物、不同成分的聚合物以及在高分子或各种化合物的不同部分链接官能团,适用单体较多,产物的应用较广,工业化成本较低。目前实现“活性”/可控自由基聚合可分以下几种途径:(1)稳定“活性”自由基聚合(SFRP);(2)原子转移自由基聚合(ATRP);(3)可逆加成-断裂链转移聚合(RAFT)。 一、稳定“活性”自由基聚合(SFRP) SFRP属于非催化性体系,是利用稳定自由基来控制自由基聚合。其机理是按照下面的可逆反应进行:外加的稳定自由基X·可与活性自由基P·迅速进行失活反应,生成“休眠种”P-X,P-X能可逆分解,又形成X·及活性种自由基P·而链增长。有研究表明,使用烷氧胺作引发剂效果好[3]。 反应体系中的自由基活性种P·可抑制在较低的浓度,这样就可以减少自由基活性种之间的不可逆终止作用,从而聚合反应得到控制。稳定自由基X·,主要有TEMPO(2,2,6,6-四甲基-1-哌啶氮氧自由基)和CoⅡ·,TEMPO属于稳定的有机自由基;CoⅡ·属于稳定的有机金属自由基。氮氧稳定自由基这类体系聚合的一大特点是聚合工艺较简单,可合成一些具有特殊结构的大分子,如树枝-线状杂化结构、聚苯乙烯嵌段共聚物等[4,5],其缺点是氮氧自由基的价格较贵,合成困难,只适用于苯乙烯及其衍生物,并且聚合慢,温度需在110℃~140℃之
可逆加成-断裂链转移(RAFT)聚合概述与最新研究进展
可逆加成-断裂链转移(RAFT)聚合概述与最新研究进展 摘要可逆加成-断裂链转移(RAFT)聚合是一种十分重要的“活性”自由 基聚合方法。这种聚合方式被人们发现以来,RAFT聚合被化学和材料界广泛应用于聚合物的设计和合成上。本文对RAFT聚合的产生、反应机理等做了简要描述,并综述了其最新研究进展。 关键词RAFT聚合“活性”自由基聚合链转移剂 前言 活性聚合最早由美国科学家Szwarc于1956年提出。所谓活性聚合是指那些不存在任何使聚合链增长反应停止或不可逆转副反应的聚合反应。经历了60年的发展,活性聚合已从最早的阴离子聚合扩展到其它典型的链式聚合:如阳离子(1986年),自由基(1993年)等,并在人们的生产和生活中产生了巨大影响。活性聚合是高分子发展史上最伟大的发现之一。 活性聚合中依引发机理的不同,分为阴离子活性聚合、阳离子活性聚合、活性自由基聚合、配位活性聚合等。活性自由基聚合较其它几种聚合方式可聚合的单体多,反应温度范围较宽,能采用的溶剂种类和聚合方法多[1],因而引起了化学和材料界的极大重视。 活性自由基聚合依据其方法可分为引发转移终止(Iniferter)法,稳定自由基聚合(SFRP,NMP)法,原子转移自由基聚合(ATRP)法[2]和可逆加成-断裂链转移聚合(RAFT)法[3]。其中Iniferter法的缺点是聚合过程难以控制,所得聚合物的相对分子质量与理论值偏差较大,相对分子质量分布较宽;NMP的主要缺点表现在需要使用价格昂贵氮氧自由基,而且氮氧自由基的合成较为困难;ATRP 的劣势则表现在当聚合一些能与过渡金属催化剂形成配位键的单体(如丙烯酸)时的控制力不强,而且较难除去金属离子和催化剂,此外还需要较为苛刻的反应条件(对除氧的要求较高)[4]。相比而言,可逆加成-断裂链转移聚合(RAFT)法有着其它几种无法比拟的优点(如反应条件温和、适用单体范围广等),使得“活性”自由基聚合技术的发展又向前迈进了一步[5]。 1RAFT聚合概述 1.1RAFT聚合的提出 1998年,Rizzardo E.等人在第37届国际高分子学术讨论会上提出了一种新的CRP方法即可逆加成-断裂链转移自由基聚合(RAFT)[6]。他们以二硫代酯类化合物为链转移剂,通过增长自由基与二硫代酯类化合物的可逆链转移反应,实现控制聚合体系中增长自由基浓度,达到“活性”/可控的目的。 RAFT技术几乎是在同时被澳大利亚联邦科学与工业研究组织(CSIRO)的Rizzardo课题组和法国的Charmot等人发现和申请专利的。Charmot等人将他们的发现命名为通过磺酸盐交换的大分子设计(MADLX),他们的专利仅仅限制在磺
自由基聚合机理以及四种常见共聚物
自由基聚合机理 烯类单体的加聚反应多属连锁聚合,连锁聚合反应由链引发、链增长、链终止等基元反应组成,各步的反应速率和活化能相差很大。连锁聚合链引发形成活性中心(或称活性种),活性中心不断与单体加成而使链增长(单体之间并不反应),活性中心的破坏就是链终止。自由基、阳离子、阴离子都可能成为活性中心引发聚合,故连锁聚合又可分为自由基聚合、阳离子聚合、阴离子聚合和配位聚合等,其中自由基聚合产物约占聚合物总产量的60%。 热力学上能够聚合的单体对聚合机理的选择是有差异的,如氯乙烯只能自由基聚合、异丁烯只能阳离子聚合、MMA可以进行自由基聚合和阴离子聚合、苯乙烯则可按各种连锁机理聚合。 自由基聚合产物约占聚合物总产量60%以上,其重要性可想而知。高压聚乙烯、聚氯乙烯、聚苯乙烯、聚四氟乙烯、聚醋酸乙烯酯、聚丙烯酸酯类、聚丙烯腈、丁苯橡胶、丁腈橡胶、氯丁橡胶、ABS树脂等聚合物都通过自由基聚合来生产。本节将对自由基链式聚合反应作较详细的讨论。 自由基聚合的基元反应 烯类单体的自由基聚合反应一般由链引发、链增长、链终止等基元反应组成。此外,还可能伴有链转移反应。现将各基元反应及其主要特征分述如下。 1 链引发 链引发反应是形成单体自由基活性种的反应。用引发剂引发时,将由下列两步组成:(1)引发剂I分解,形成初级自由基R?; (2)初级自由基与单体加成,形成单体自由基。 单体自由基形成以后,继续与其他单体加聚,而使链增长。 比较上述两步反应,引发剂分解是吸热反应,活化能高,约105~150kJ/mo1,反应速
率小,分解速率常数约10-4~10-6s-1。初级自由基与单体结合成单体自由基这一步是放热反应,活化能低,约20~34kJ/mo1,反应速率大,与后继的链增长反应相似。但链引发必须包括这一步,因为一些副反应可以使初级自由基不参与单体自由基的形成,也就无法继续链增长。 有些单体可以用热、光、辐射等能源来直接引发聚合。这方面的研究工作不少,苯乙烯热聚合已工业化;紫外光固化涂料也已大规模使用。 2 链增长 在链引发阶段形成的单体自由基,仍具有活性,能打开第二个烯类分子的π键,形成新的自由基。新自由基活性并不衰减,继续和其他单体分子结合成单元更多的链自由基。这个过程称做链增长反应,实际上是加成反应。 为了书写方便,上述链自由基可以简写成,其中锯齿形代表由许多单元组成的碳链骨架,基团所带的独电子系处在碳原子上。 链增长反应有两个特征:一是放热反应,烯类单体聚合热约55~95kJ/mol;二是增长活化能低,约20~34KJ/mol,增长速率极高,在0.01~几秒钟内,就可以便聚合度达到数千,甚至上万。这样高的速率是难以控制的,单体自由基一经形成以后,立刻与其他单体分子加成,增长成活性链,而后终止成大分子。因此,聚合体系内往往由单体和聚合物两部分组成,不存在聚合度递增的一系列中间产物。 对于链增长反应,除了应注意速率问题以外,还须研究对大分子微观结构的影响。在链增长反应中,结构单元间的结合可能存在“头-尾”和“头-头”或“尾-尾”两种形式。经实验证明,主要以头-尾形式连接。这一结果可由电子效应和空间位阻效应得到解释。对一些取代基共轭效应和空间位阻都较小的单体聚合时头-头结构会稍高,如醋酸乙烯酯、偏二氟乙烯等。聚合温度升高时,头-头形式结构将增多。
活性自由基聚合
活性自由基聚合 摘要:阐述了活性自由基聚合的产生背景和基本概念,介绍了活性自由基聚合的分类,描述了原子转移自由基聚合的研究进展。 关键词:活性自由基聚合 1.活性自由基聚合的基本思想 活性自由基聚合的核心思想是抑制增长自由基浓度,减少双基终止的发生。由高分子化学知识可知,链终止速率与链增长速率之比可用下式表示:[1] 通常kt/kp为104~105,假定体系中单体浓度为1mol/L,则: 当然,自由基活性种浓度不可能无限制地降低,一般来说,[P*]在10- 8mol/L左右,聚合反应的速率仍很可观。在这样的自由基浓度下,R t/R p≈10-4~10-3,Rt相对于R p就可忽略不计,所谓的活性自由基聚合的“活性”就在这里。自由基浓度的下降必然降低聚合反应速度,但由于链增长反应活化能高于链终止反应活化能,因此提高聚合反应温度不仅能提高聚合速率(因为能提高k p),而且能有效降低k t/k p比值,从而抑制链终止反应的进行。
这里需要解决两个问题:一是如何从聚合反应开始直到反应结束始终控制如此低的反应活性种浓度;二是在如此低的反应活性种浓度下,如何避免聚合物的聚合度过大(DP n=[M0]/[P*]=1/10-8=108)。 解决这两个问题的方法是在聚合体系中加入数量可人为控制的反应物X,此反应物X不能引发单体聚合,但可与自由基P*迅速作用而发生钝化反应,生成一种不会引发单体聚合的“休眠种”P-X。而此休眠种在聚合反应条件下又可均裂成增长自由基P*及X,如下式表示:[2] 这样体系中存在的自由基活性种的浓度将取决于3个参数:反应物X的浓度、钝化速率常数k d和活化速率常数k a,其中反应物X的浓度是人为可控的,所谓的可控活性自由基聚合的“可控”就在这里。另外研究表明,如果钝化反应和活化反应的转化速率足够快(不小于链增长速率),则在活性种浓度很低的情况下,聚合物的分子量将不由P*而由P-X的浓度决定。
自由基聚合习题参考答案
第3章自由基聚合-习题参考答案 1、判断下列单体能否进行自由基聚合并说明理由 H2C CHCl H2C CH H2C CCl2H2C CH2H2C C H2C CHCN H2C C(CN)2H2C CHCH3F2C CF2ClHC CHCl H2C C CH3 COOCH3H2C C CN COOCH3 HC CH OC CO O 答: (1)可以。Cl原子的诱导效应为吸电性,共轭效应为供电性两者相抵,电子效应微弱,只能自由基聚合。 (2)可以。为具有共轭体系的取代基。 (3)可以。结构不对称,极化程度高,能自由基聚合。 (4)可以。结构对称,无诱导效应共轭效应,较难自由基聚合。 (5)不能。1,1—二苯基乙烯,二个苯基具有很强的共轭稳定作用,形成的稳定自由基不能进一步反应。 (6)可以。吸电子单取代基。 (7)不可以。1,1双强吸电子能力取代基。 (8)不可以。甲基为弱供电子取代基。 (9)可以。氟原子半径较小,位阻效应可以忽略不计。 (10)不可以。由于位阻效应,及结构对称,极化程度低,难自由基聚合 (11)可以。1,1-双取代。 (12)可以。1,1-双取代吸电子基团。 (13) 不可以。1,2-双取代,空间位阻。但可进行自由基共聚。 2、试比较自由基聚合与缩聚反应的特点。
答: 自由基聚合:(1)由链引发,链增长,链终止等基元反应组成,其速率常数和活化能均不等,链引发最慢是控制步骤。 (2)单体加到少量活性种上,使链迅速增长。单体-单体,单体-聚合物,聚合物-聚合物之间均不能反应。 (3)只有链增长才是聚合度增加,从一聚体增加到高聚物,时间极短,中间不能暂停。聚合一开始就有高聚物产生。 (4)在聚合过程中,单体逐渐减少,转化率相应增加 (5)延长聚合时间,转化率提高,分子量变化较小。 (6)反应产物由单体,聚合物,微量活性种组成。 (7)微量苯酚等阻聚剂可消灭活性种,使聚合终止。 缩聚反应:(1)不能区分出链引发,链增长,链终止,各部分反应速率和活化能基本相同。 (2)单体,低聚物,缩聚物中任何物种之间均能缩聚,使链增长,无所谓活性中心。 (3)任何物种之间都能反应,使分子量逐步增加,反应可以停留在中等聚合度阶段,只在聚合后期才能获得高分子产物。 (4)聚合初期,单体缩聚成低聚物,以后再由低聚物逐步缩聚成高聚物,转化率变化微小,反应程度逐步增加。 (5)延长缩聚时间分子量提高,而转化率变化较小。 (6)任何阶段都由聚合度不等的同系缩聚物组成。 (7)平衡和基团非等当量可使缩聚暂停,这些因素一旦消除,缩聚又可继续进行。 3、解释下列概念: 歧化终止,偶合终止,引发剂效率,笼蔽效应,诱导效应,自动加速现象,诱导期,聚合上限温度,悬浮聚合,乳液聚合,增溶作用,临界胶束浓度,胶束,种子乳液聚合, 答: 歧化终止:链自由基夺取另一自由基的氢原子或其他原子终止反应。 偶合终止:两链自由基的独电子相互结合成共价键的终止反应。 引发剂效率:引发剂在均裂过程中产生的自由基引发聚合的部份占引发剂分解总量的分率,
原子转移自由基聚合理论
(1)ATRP介绍 王锦山等[1]采用1-苯-1-氯乙烷作为引发剂,氯化亚铜和联吡啶(bpy)的络合物作为催化剂,在130℃下引发苯乙烯(St)的本体聚合,反应3h产率可达95%。理论分子量和实验值符合较好。为了验证反应的自由基机理,比较了所得聚合物与一般自由基聚合所得聚合物的立构规整度,发现两者比较一致。并且当加入第二单体丙烯酸甲酯时,成功实现了嵌段共聚,具有明显的活性聚合特征。由此他们提出了原子转移自由基聚合(ATRP)。 ATRP是以简单的有机卤化物为引发剂、过渡金属配合物为卤原子载体,通过氧化还原反应,在活性种与休眠种之间建立可逆的动态平衡,从而实现了对聚合反应的控制。 聚合原理 引发阶段,处于低氧化态的转移金属卤化物Mt n,从有机卤化物R-X中吸取卤原子X,生成引发自由基R·及处于高氧化态的金属卤化物Mt n+1-X,自由基R·可引发单体聚合,形成链自由基R-M n·。R-M n·可从高氧化态的金属配位化合物Mt n+1-X中重新夺取卤原子而发生钝化反应,形成R-M n-X,并将高氧化态的金属卤化物还原为低氧化态的Mt n。增长阶段,R-M n-X与R-X一样(不总一样)可与Mt n发生促活反应生成相应的R-M n·和Mt n+1-X,R-M n·与R-M·性质相似均为活性种,同时R-M n·和Mt n+1-X又可反过来发生钝化反应生成R-M n-X和Mt n,则在自由基聚合反应进行的同时始终伴随着一个自由基活性种与大分子卤化物休眠种的可逆转换平衡反应。 由此可见,ATRP的基本原理其实是通过一个交替的“促活—失活”可逆反应使得体系中的游离基浓度处于极低,迫使不可逆终止反应被降到最低程度,从而实现可控/“活性”自由基聚合。 引发剂 ATRP聚合体系的引发剂主要是卤代烷RX(X=Br,C1),另外也有采用芳基磺酰氯、偶氮二异丁腈等。RX的主要作用是定量产生增长链。α-碳上具有诱导或共轭结构的RX,末端含有类似结构的大分子(大分子引发剂)也可以用来引发,形成相应的嵌段共聚物。另一方面,R的结构应尽量与增长链结构相似。卤素基团必须能快速且选择性地在增长链和转移金属之间交换。Br和Cl均可以采用,采用Br的聚合速率大于Cl[2]。 金属催化剂及配体 第一代ATRP催化剂为CuX(其中X为Br,Cl),此后有人采用了RuⅡ,RhⅡ,NiⅡ,FeⅡ,ReⅤ等过度金属卤化物[3]。而最早采用的配位剂是联二吡啶(bpy),后来有了dNbipy,PMDETA,BDE,BPMODA和Me6TREN等高活性的催化剂配
可控自由基聚合技术在合成高分子材料中的应用探究
可控自由基聚合技术在合成高分子材料中的应用探究1500 一、摘要:本文主要是说明了可控自由基聚合技术在合成高分子材料中的应用,然后具体的分析了线型聚合物的合成、接枝聚合物的合成、接枝聚合物的合成、无机/聚合物复合材料的制备,并对其未来的价值进行重要的论述。 关键词:可控自由基聚合;合成;材料 二、线型聚合物的合成 线型聚合物的合成主要包括两个方面嵌段共聚物和梯度共聚物。所谓的嵌段共聚物就是序列规整的聚合物中研究最多应用也最广泛的一类聚合物物质。通过可控自由基聚合可以得到AB型、ABC型、ABA型等多嵌段型的嵌段共聚物。然而对实现ATRP的方法可有两种方式,一方面是先把第一种单体的均聚物制备完成,然后直接把第二种单体加入就可;另一方法是先得到含有卤原子的大分子引发剂,然后再把第二种单体聚合引发,从而得到了二嵌段共聚物。梯度共聚物就是作为一类结构精密的新型共聚物,它具有嵌段和无规共聚物的多种优点 ,是作为一种特别有效的高分子共混增容剂。通常是采用不含有链终止反应的聚合技术是制备梯度共聚物的前提条件,且这种技术是作为梯度共聚物最佳技术方法。然而,因为各共聚单体的竞聚率存在着很大的差别,所以在梯度共聚物的制备上一般是依据所用单体的不同和制备要求而选择不同的加料方法,通常的加料方法主要有批量法和半批量法。 三、接枝聚合物的合成 关于接枝聚合物的合成主要是可控自由基聚合技术,通常大部分所采用ATRP技术合成梳状聚合物主要有两种方法途径:大分子单体技术和大分子引发技术。通过一些侧链比较均一的梳状聚合物利用大分子单体技术制得。哈丽丹·买买提等在纤维素氯化锂/N,N-二甲基乙酰胺(DMAc)均相溶液中,利用氯乙酰氯与纤维素发生均相酰化反应生成纤维素氯乙酸酯,再通过溶解DMAc中用氯化亚铁催化剂引发甲基丙烯酸丁酯,制备出纤维素/甲基丙烯酸丁酯接枝共聚物。 郑兴良等合成了两亲性接枝共聚物PtBA-g-PPEGMEMA,在对抗肿瘤药物方面的阿霉素进行了负载,最终通过试验表明该体系是有缓释特征的。林先凯等是通过以N,N-二甲基甲酰胺作溶剂材质、氯化亚铜/三( N,N-二甲基氨基乙基) 胺为催化配位体系,利用ATRP在商用PVDF 粉末上直接接枝,制备出PVDF-g-PNIPAAm 共聚膜。张洪文等是通过表面引发ATRP在 酯薄膜表面接枝了由γ-甲基丙烯酰氧基丙基三甲氧基硅烷和甲基丙烯酸甲酯形成的共聚物,最终得到提高基质材料的疏水性能。 四、支化聚合物的合成 3. 1星形聚合物 通过ATRP 技术制备星形聚合物的方法主要有先臂后核”和“先核后臂”两种方法。“先臂后核”法就是优先使用ATRP 制备出带有活性末端基的均聚物,这种均聚物然后再与多官能团化合物进行相互的反应得出多臂星形聚合物。然而“先核后臂”法就是利用多官能团的引发剂作用进行单体的ATRP。陈建芳等就是通过原子转移自由基偶联法得到了星形杂臂苯乙烯-甲基丙烯酸甲酯共聚物( PS-PMMA)和多臂星形聚苯乙烯 ( S-PS)。 4. 2聚合物刷 聚合物刷其实是一种比较特殊的高分子结构,其突出特征就是在特定基质的表面或界面上具
原子转移自由基聚合概述
原子转移自由基聚合概述 1.引言 “活性”/可控自由基聚合不同于传统意义上的自由基聚合反应。它克服了分子量及其分布不可控,难以合成嵌段聚合物等缺陷,做到了分子量可控,分子量分布较窄,聚合物结构可控等一系列要求。这类聚合反应主要是有效降低了增长活性中心的浓度,抑制了双基终止的发生,延长了自由基的寿命和分子量的统一性;使用快引发的方式,保证不同分子链同时增长。目前大致有以下几种不同的机理得到了较为深入地研究:基于引发-转移-终止剂(Initiator-chain transfer-terminator)的活性自由基聚合(Iniferter法)、基于氮氧稳定自由基的活性自由基聚合(Living nitroxide-mediated stable free radical polymerization-SFRP)、原子转移自由基聚合(Atom transfer radical polymerization-ATRP)、基于可逆加成碎裂链转移剂的活性自由基聚合(Living radical polymerization in the presence of reversible addition-fragmentation chain transfer-RAFT)和退化转移自由基聚合(degenerative transfer process-DT)等等。 在这些不同的实现“活性”/可控自由基聚合的方法当中,原子转移自由基聚合是目前最有希望实现工业化的一种方法。 2.原子转移自由基聚合概述 原子转移自由基聚合是1995年由卡内基梅隆大学Matyjaszewski课题组提出的一种“活性”/可控自由基聚合新机理Wang, J-S; Matyjaszewski, K. Controlled/"living" radical polymerization. Atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117: 5614–5615.。在同一年,日本京都大学的泽本光南(Mitsuo Sawamoto)教授也在同时期独立发表了金属催化的活性自由基聚合Kato, M; Kamigaito, M; Sawamoto, M; Higashimura, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(triphenylphosphine)ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules. 1995, 28: 1721–1723.,其本质就是原子转移自由基聚合。原子转移自由基聚合主要是依靠大分子活性中心与卤素原子在催化剂的参与下形成增长活性中心(活性种)和与卤素可逆终止的大分子链自由基(休眠种)之间的平衡来控制聚合的。它可以抑制链终止反应,控制聚合速度以保证同时增长,最终达到控制分子量及分布,并实现大分子结构设计的。 2.1.原子转移自由基聚合体系组成 原子转移自由基聚合的体系有以下几个组份。 单体。原子转移自由基聚合适用的单体种类比较多,并无太大限制,最好有可以稳定自由基的基团。但是每一种单体的聚合速率相差较多,需要通过其他因素的控制来调控Matyjaszewski, Krzysztof; Xia, Jianhui. Atom Transfer Radical Polymerization. Chemical Reviews. 2001, 101 (9): 2921–90.。 溶剂。常用的溶剂如甲苯、二甲苯、氯仿、N,N-二甲基甲酰胺、二甲基亚砜、水等都可以使用。有些体系直接用单体做本体聚合。 引发剂。聚合要做到活性可控,就要求引发既有较快的引发速率,使所有大分子链几乎在同一时间开始增长来保证分子量窄分布。同时,由于原子转移自由基聚合的机理,一般使用有机卤代物做引发剂,最常用的是卤代烷。溴代烷和氯代烷都可以较好的控制聚合物的分子量,但是溴代烷有更强的活性。同时,一定的引发剂结构可制备不同结构的聚合物。如多卤代烷支链的引发剂可制备星型聚合物。 催化剂。催化剂是ATRP中的重要组份。它既决定了反应速率又一定程度上决定了产品分子量的分布。若催化剂投料较少,则活性种浓度较高,有利于加快反应速率。但会使双基终止等副反应增加,但不利于制备分子量窄分布的聚合物。 最初的催化剂体系是卤化亚铜/联吡啶体系,反应体系是非均相体系。后来在联吡啶上引入油溶性长链,变为均相催化体系,并且有史以来第一次在自由基聚合中获得近似单分散的聚合物。为了开发较为便宜且反应速率较快的催化体系,后来又出现了Fe、Ru、Ni体系,而配体开始用高催化活性的多胺、亚胺等代替。 配体。配体与催化剂形成络合物,以解决催化剂在有机相中的溶解问题。不同配体对此问题
可控自由基聚合
可控自由基聚合制备用于光固化薄膜领域的定向低聚物结构 By Jon Scholte, Soon Ki Kim, and C.Allan Guymon 简介 光聚合是一个急速扩张的领域,它在工业上有许多方面的应用。由于光固化环氧树脂有良好光学清晰度与优异的附着性能,因此工业上经常使用到这类型树脂。虽然光固化环氧树脂以硬度闻名,但是其脆性很大以至在很多领域都不适合使用。最近的研究表明,混合型环氧丙烯酸酯树脂的整体硬度不但能达到纯的环氧树脂的水平,而且在更短的辐射时间里能达到更高的转化率。通常需要阳离子引发剂与自由基引发剂复配使用来使得复合体系中的双组分均能达到足够的转化率,因此这大大限制了它的使用范围。环氧树脂机械性能的改性主要是通过加入预聚物来实现。传统预聚物是由一系列分子量和聚合度的化合物组成,如果能准确控制预聚物的结构,这将会非常有用。能通过可控自由基聚合技术来得到这种可控结构的预聚物,氮氧调控自由基聚合就是其中一种。这项技术使用高度稳定的氮氧自由基去调控活性聚合物链的增长,因此产物保持较低的分散指数。另外,预聚物的分子量和结构能通过反应投料的改变来控制,这得益于NMP体系中产生的自由基有长的活性周期。 对预聚物结构的控制能实现材料的相分离,这对某些领域来说是非常有用的。先前的研究表明,在环氧树脂中引入多官能化的星型聚合物后,光固化聚合物网络的机械性能,包括冲击强度,得到了提高。此外,已有研究表明,当体系中加入了非反应性的低聚物,自由基光聚合会引发相分离。在这,我们提出加入环氧官能团改性的丙烯酸预聚物来增强环氧树脂,同时建立了玻璃化转变温度模型。通过简单的NMP法合成线性多官能预聚物。此外,我们还证实线性预聚物骨架上的反应性官能团会影响到聚合物网络的强度。 实验 新型预聚物的合成用到了BlocBuilder RC-50,它用来引发和调控自由基。在2L的反应容器中加入BlocBuilder RC-50,丙烯酸正丁酯以及等质量的乙酸乙酯作为溶剂。通过三步法加入不同的投料制得末端功能化的预聚物。第一步用到的原料是丙烯酸正丁酯和脂环族的环氧甲基丙烯酸酯。第二步加入的投料为丙烯酸正丁酯,最后一步加入的原料跟第一步一样。这个设计方案使得环氧基团能保留在单体的末端部位。类似的,制备无规预聚物的反应容器中最初装有BlocBuilder RC-50,丙烯酸正丁酯和乙酸乙酯,单一的投料只用了丙烯酸正丁酯和环氧族甲基丙烯酸酯。合成后,乙酸乙酯和残留的单体通过旋蒸的方式除去,通过GPC 来测定分析产物的分子量与分子量分布。表1的信息包括预聚物的目标重均分子量与官能团数目。薄膜的制备用到了不同浓度的预聚物以及交联剂(3,4-环氧环
“活性”可控自由基聚合
“活性”/可控自由基聚合 熊鹏鹏2010214110 摘要: 自由基聚合是生产高分子量聚合物的重要方法, “活性”/ 可控自由基聚合综合了自由基聚合和离子聚合的优点, 使自由基聚合具有可控性。本文对目前可以实现“活性”/ 可控自由基聚合的途径和各自机理进行介绍, 指出应该重视对“活性”/可控自由基聚合的研究。关键词: “活性”/可控自由基聚合; 稳定自由基; 可逆加成-裂解链转移; 原子转移; 引发转移终止剂;退化转移。 自由基聚合是工业上和实验室中生产高分子量聚合物的重要方法, 该法具有可聚合的单体种类多、反应条件宽松、以水为介质、容易实现工业化生产等优点, 但也存在着缺陷, 如自由基聚合的本质( 慢引发, 快速链增长, 易发生链终止和链转移等) 决定了聚合反应的失控行为,其结果常常导致聚合产物呈现宽分布, 分子量和结构不可控, 有时甚至会发生支化、交联等,从而严重影响聚合物的性能, 此外, 传统的自由基聚合也不能用于合成指定结构的规整聚合物。 鉴于离子聚合和配位聚合可以很好地控制聚合物结构, 而能不能控制自由基聚合体系则成为当前的研究热点, 但近年来从离子聚合和可控有机自由基反应的研究进展来看, 答案是肯定的。 就聚合反应而言, 要合成具有确定结构的聚合物, 则要求所有的链应同时引发, 增长相似, 这就需要快速引发, 在聚合结束前增长链应保持活性, 链转移和链终止的效应可以忽略, 而自由基聚合的本质( 慢引发, 快终止) 与之正好相反。所以实现可控自由基聚合要基于
以下三个原则: 1) 自由基体系中的增长反应应对自由基敏感, 终止反应对自由基浓度的敏感度次之。这样, 在自由基浓度很低时, 链增长反应与终止反应的速率比才足够高, 才能合成出分子量很大的聚合物。 2) 增长链的浓度必须比初始游离自由基的浓度高得多, 在整个反应过程中所有的链均需保持活性, 且游离自由基与高浓度休眠链处于动态平衡之中, 这种持续自由基效应对任何控制自由基反应来说都是最重要的。 3) 引发迅速, 增长链( 休眠链和活性链总和) 浓度保持稳定, 使用与大分子休眠链结构相类似的引发剂。目前已经发现有五种途经可以实现“活性”/ 可控自由基聚合, 下面分别论述其机理和研究现状。 1稳定“活性”自由基聚合( SFRP) SFRP属于非催化性体系, 是利用稳定自由基来控制自由基聚合。其机理是按照下面的 可逆反应进行: 外加的稳定自由基X·可与活性自由基P·迅速进行失活反应, 生成“休眠 种”P-X, P- X能可逆分解, 又形成X·及活X 性种自由基P·而链增长。有研究表明, 使用烷 氧胺作引发剂效果好。 反应体系中的自由基活性种P·可抑制在较低的浓度, 这样就可以减
自由基聚合题库
? 1. 目前,悬浮聚合发主要用于生产( )。
A. PVC、PVDC C. PE
正确答案:A.
B. PS D. PP
? 2. 下列单体中可进行自由基、阴离子、阳离子聚合反应的是( )。
A. 氯乙烯 B. 苯乙烯 C. 乙烯 D. 醋酸乙烯 正确答案:B.
? 3. 聚乙烯醇的单体是( )。
A. 乙烯醇 B. 乙醇
C. 乙醛
D. 醋酸乙烯酯
正确答案:D.
? 4. 典型乳液聚合中,主要引发地点是在 ( )。
A. 单体液滴 B. 胶束 C. 水相 D. 单体液滴和胶束 正确答案:B.
? 5. 过硫酸钾引发剂属于( )。
A. 氧化还原引发剂 B. 水溶性引发剂 C. 油溶性引发剂 D. 阴离子引发剂 正确答案:B.
? 6. 在自由基聚合中,若初级自由基与单体的引发速度较慢,则最终聚合速率与单体浓 度呈( )级关系。
A. 1 C. 2
正确答案:B.
B. 1.5 D. 不能确定
? 7. 苯醌是常用的分子型阻聚剂,一般用单体的( )就能达到阻聚效果。
A. 1.0%一 0.5% C. 2.0%一 5.0% 正确答案:D.
B. 1.0%一 2.0% D. 0.1%一 0.001%
? 8. ( )的自由基是引发聚合反应常见的自由基。
A. 高活性 B. 低活性 C. 中等活性 D. 无活性 正确答案:C.
? 9. 某工厂用 PVC 为原料制搪塑制品时,从经济效果和环境考虑,他们决定用( )聚合 方法。
A. 本体聚合法生产的 PVC C. 乳液聚合法生产的 PVC
正确答案:C.
B. 悬浮聚合法生产的 PVC D. 溶液聚合法生产的 PVC
? 10. 自由基链转移反应中,不可能包括活性链向( )的转移。
A. 高分子 B. 单体 C. 引发剂 D. 溶剂
? 1. 对于自由基聚合,在其他条件保持不变的前提下升高聚合温度,得到的聚合物的分 子量将( )。
A. 减小 B. 增大 C. 不变 D. 不一定 正确答案:B.
? 2. 在乙酸乙烯酯的自由基聚合反应中加入少量苯乙烯,会发生( )
A. 聚合反应加速 C. 相对分子量降低 正确答案:B.
B. 聚合反应停止 D. 相对分子量增加
? 3. 传统自由基聚合的机理特征是( )。
A. 慢引发,快增长,速终止 C. 快引发,快增长,难终止
正确答案:A.
B. 快引发,慢增长,不中止 D. 慢引发,慢增长,速终止
? 4. 合成丁基橡胶的主要单体是( )。
A. 异丁烯+丁二烯 C. 异丁烯
正确答案:B.
B. 异丁烯+异戊二烯 D. 丁二烯
? 5. 合成橡胶通常采用乳液聚合反应,主要是因为乳液聚合( )。
A. 产品较纯净
B. 易获得高分子量聚合物
C. 不易发生凝胶效应 D. 聚合反应容易控制
可控活性自由基聚合的研究进展
第22卷第2期高分子材料科学与工程Vo l.22,N o.2 2006年3月POLYM ER M AT ERIALS SCIENCE AND EN GINEERING M ar.2006可控活性自由基聚合的研究进展X 郑 璇,张立武 (重庆大学化学化工学院,重庆400044) 摘要:可控活性自由基聚合(CRP)是一种合成具有设计微观结构和窄分子量分布聚合物的方法,原子转移自由基聚合(AT RP)较其它CRP方法具有分子设计能力较强等优点,是应用最广泛的CR P。文中简要介绍了CRP的分类,同时以A T RP为例从单体、引发剂、催化体系等方面讨论了CR P聚合体系的发展。 关键词:可控活性自由基聚合;分类;聚合体系;进展 中图分类号:T Q316.32+2 文献标识码:A 文章编号:1000-7555(2006)02-0016-04 在20世纪50、60年代,自由基聚合达到了它的鼎盛时期。但由于存在链转移和链终止反应,传统自由基聚合不能较好地控制分子量及大分子结构[1]。1956年美国科学家Szwarc等提出了活性聚合的概念[2],活性聚合具有无终止、无转移、引发速率远远大于链增长速率等特点,与传统自由基聚合相比能更好地实现对分子结构的控制,是实现分子设计、合成具有特定结构和性能聚合物的重要手段。但离子型活性聚合反应条件比较苛刻、适用单体较少,且只能在非水介质中进行,导致工业化成本居高不下,较难广泛实现工业化。鉴于活性聚合和自由基聚合各自的优缺点,高分子合成化学家们联想到将二者结合,即可控活性自由基聚合(CRP)或活性可控自由基聚合[1]。CRP可以合成具有新型拓扑结构的聚合物、不同成分的聚合物以及在高分子或各种化合物的不同部分链接官能团,适用单体较多,产物的应用较广,工业化成本较低。 1 C RP的分类 CRP的基本思想是[2]:向体系中加入一个与增长自由基之间存在着偶合-解离可逆反应的稳定自由基,以抑制增长自由基浓度,减少双基终止的发生。目前,各种CRP体系已经发展起来,可分为基于可逆终止和可逆转移机理两类。其中可逆终止机理包括稳定自由基聚合(SFRP)和原子转移自由基聚合(ATRP);可逆转移机理包括可逆加成-断裂链转移(RAFT)活性自由基聚合和退化转移自由基聚合[3]。1.1 稳定自由基聚合[4,5] SFRP属于非催化性体系,是利用稳定自由基来控制自由基聚合。稳定自由基X?,主要有T EM PO(2,2,6,6-四甲基-1-哌啶氮氧自由基)和Co(Ⅱ)?。前者属于稳定的有机自由基,主要可进行苯乙烯及其衍生物的聚合,聚合工艺较简单,可合成一些具有特殊结构的大分子,但氮氧自由基价格较贵,合成困难、聚合速率慢,温度需在110℃~140℃之间。后者属于稳定的有机金属自由基,主要进行丙烯酸酯活性聚合,但得到的聚合物分子量不高,且分子量分布较宽。研究者认为,通过使用新型氮氧自由基,此体系可以扩展到(甲基)丙烯酸和其它单体。其它有机金属化合物或过渡金属盐与自由基可逆络合的活性自由基聚合反应也有报道,如Al、Cr、Rh。 1.2 原子转移自由基聚合[2,5] X收稿日期:2005-01-18;修订日期:2005-05-16 联系人简介:郑 璇(1978-),女,硕士,E-mail:zhengxu an16@https://www.360docs.net/doc/d37142232.html,