Hyperchem程序安装

Hyperchem程序安装
1、将文件解压,在系统盘外的其他盘创建一名为hyperchem的文件夹
2、点击HyperChem6Evaluation.exe
按提示进行。当出现
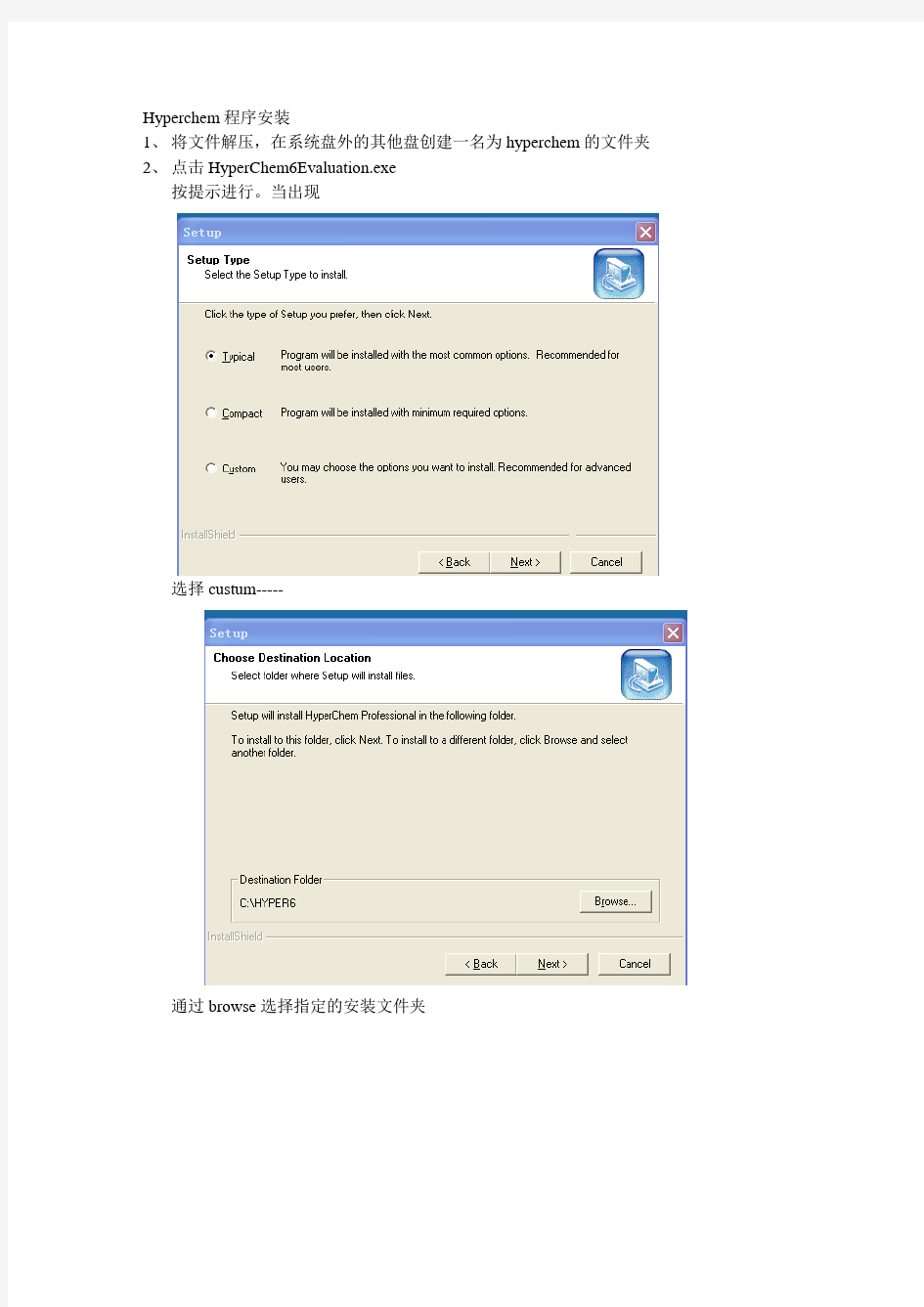
选择custum-----
通过browse选择指定的安装文件夹
当出现
不要按确定而是关闭窗口;当出现
不要按确定而是关闭窗口;当出现
不要按确定而是关闭窗口;
当出现
若你的计算机原已有acrobat reader 软件,则选否,否则选是。
当出现
记住不要作任何勾选,按finish
3、进行程序crack
方法:进入hyperchem安装文件夹,打开programs文件夹,
进入hyperchem软件解压包中的crack文件夹,将其中的所有文件选定并复制,并粘贴到上述programs文件夹。当出现是否复盖时选“是”。将programs文件夹中的绿色chem.exe图标发送到桌面快捷方式。OK!
4、点击桌面快捷方式绿色chem.exe图标即可运行了。
分子动力学软件选择
分子动力学软件选择 There are widely used packages like AMBER, CHARMm and X-PLOR https://www.360docs.net/doc/ec13901637.html,/amber/amber.html https://www.360docs.net/doc/ec13901637.html,/ https://www.360docs.net/doc/ec13901637.html,/ CHARMm and X-PLOR both use the same forcefield. Amber's is different. If you're Wintel-bound, you could try Hyperchem, which has a free downloadable demo: https://www.360docs.net/doc/ec13901637.html,/products/hc5_features.html It has a nice structure build capability (the other packages have powerful languages, but can be intimidating to new users). OpenSource adherents can find a wealth of free packages at SAL, an excellent site: https://www.360docs.net/doc/ec13901637.html,/Z/2/index.shtml My personal favourites are MMTK, EGO and VMD/NAMD. I compiled a list of free and commerical programs at https://www.360docs.net/doc/ec13901637.html,/chemistry/soft_mod_en.html modeling in solution is possible e.g. with these programs (to the best of my knowledge): commercial: AMSOL, GROMOS, Titan free: GAMESOL, GROMACS, MOIL, OMNISOL, Tinker You find links to all of these programs at https://www.360docs.net/doc/ec13901637.html,/chemistry/soft_mod_en.html PAPA (计算粒状物料的三维并行分子动力学计算程序) 【URL】http://www.ica1.uni-stuttgart.de/Research/Software_P3T/papa.html 【作者】 ICA 1 Group, Institute of Computer Applications (ICA) of the University of Stuttgart 【语言版本】 English 【收费情况】免费
分子模拟软件简介
3D分子图形显示工具 (RasMol and OpenRasMol)(免费) AMBER (分子力学力场模拟程序) autodock (分子对接软件)(免费) GROMACS (分子动力学软件)(免费) GULP (General Utility Lattice Program)(免费) NIH分子模拟中心的化学软件资源导航(Research Tools on the Web) X-PLOR (大分子X光晶体衍射、核磁共振NMR的3D结构解析)(免费) 高通量筛选软件PowerMV (统计分析、分子显示、相似性搜索 等)(免费) 化合物活性预测程序PASS(部分免费) 计算材料科学Mathub C4:Cabrillo学院化学可视化项目以及相关软件(免费) Databases and Tools for 3-D Protein Structure Comparison and Alignment(三维蛋白质结构对比)(免费) Democritus (分子动力学原理演示软件) DPD应用软件cerius2(免费) EMSL Computational Results DataBase (CRDB) MARVIN'S PROGRAM (表面与界面模拟)(免费) XLOGP(计算有机小分子的脂水分配系数)(免费) 量子化学软件中文网 美国斯克利普斯研究院:金属蛋白质结构和设计项目(免费) https://www.360docs.net/doc/ec13901637.html,/(免费) 3D Molecular Designs (蛋白质及其他3D分子物理模型快速成型技术) 3D-Dock Suite Incorporating FTDock, RPScore and MultiDock (3D 分子对接)(免费)
HyperChem 程序及其应用
HyperChem 程序及其应用 一、HyperChem 程序的运行环境 HyperChem 程序包,用C++写源程序,具有工作站、微机等不同的版本,作为教学示例,我们向大家介绍适用于微机运行的程序版本。 可以通过网络选购HyperChem程序包,也可以免费下载演示版本以供学习只用,现在该公司提供的最新版本为V6.0。 该公司的网址为:https://www.360docs.net/doc/ec13901637.html, 该公司与98年诺贝尔奖金得主Pople的关系可以参看其网页有关介绍。 1、软件环境 Windows95、Windows98或Windows2000系统。 2、硬件环境 486以上的微机,内存应在8M以上,硬盘至少有32M以上自由空间。 为了能够以最佳方式显示分子图像,最好有VGA以上显示器。 3、程序安装 使用该程序应注意程序版权(注册)。 安装程序默认子目录为:C:\hyper6 安装完成后,该目录可以看到如下文件,其中,绿色烧杯为执行程序图标。 有关该程序的使用说明、参考手册等全套文档均可免费获得。 二、程序基本使用方法 我们以演示版本为例,说明该程序的基本使用方法。 1、启动程序 在屏幕上,双击绿色烧杯可以得到如下画面:
点击 Try进入工作区窗口 窗口各部分功能简介 标题名称:最大、最小化、退出按钮 菜单条: FILE、EDIT、BUILD、SELECT、DISPLAY、DATABASE、 SETUP 、COMPUTE、CANCEL、SCRIPT、HELP 工具条:
工作 区: 状态 行: 2、打开已存在的数据文件 File-Open
Display- Labels可以选择原子、化学键等标记方式: Dispay-Rendering
HyperChem软件中Script的常用语句注解
HyperChem软件中Script的常用语句注解 HyperChem软件中Script的常用语句注解 1. 计算方法及参数设定 calculation-method item 计算方法设定 item: MolecularMechanics, SemiEmpirical, AbInitio, DFT molecular-mechanics-method item 分子力学方法设定 item: mm+, amber, bio+, opls, amber94, charmm22, tndo optim-max-cycles x 优化最大迭代次数x设定 optim-convergence goal 优化收敛目标goal设定 optim-converged返回是否收敛(true/false) optim-algorithm item 优化算法设定 item: PolakRibiere, NewtonRaphson, etc. assign-basisset item abinitio计算基组设定 item: 3-21G, STO-3G, etc. semi-empirical-method item 半经验计算方法设定 item: am1, pm3, extendedhuckel, cndo, indo, zindos, mndod, tndo, etc. accelerate-scf-convergence true/false 是否加速SCF收敛 scf-convergence goal SCF收敛目标goal设定 max-iterations n SCF计算的最大迭代次数n设定 2. 计算结果输出 omsgs-to-file filename 将信息输出到filename文件 append-omsgs-to-file filename 将信息追加到filename文件 omsgs-not-to-file 关闭输出文件 query-response-has-tag yes/no 提取的信息回显开关 query-value HSV 提取系统数据结构中的信息并单行输出 HSV: current-file-name, coordinates, dipole-moment, dipole-moment-components total-energy, heat-of-formation, scf-binding-energy, scf-core-energy, scf-electronic-energy start-logging HyperChem运行记录开启 stop-logging HyperChem运行记录关闭 export-property-file filename 将分子轨道信息写入filename文件 3. 分子结构及其文件操作 file-format item 定义分子结构文件格式 item: hin, mol, zmt, ent, skc, xyz, ml2 open-file filename 读入filename分子结构文件 write-file filename 将分子结构写入filename文件 menu-build-add-hydrogens 给分子加氢 menu-build-model-build 给分子加氢并规格化 do-optimization 进行优化操作 do-single-point 进行单点计算 do-molecular-dynamics进行分子动力学操作
化工软件介绍
换热器:HTRI和HTFS都是国际著名的换热器工艺计算软件,你需要了解其意思方可用于实际 1. ASPEN 2. PROII 3. CHEMCAD 4. HYSYS 5. ECSS 所列软件中,1,2,4是工程公司常用软件,3,5一般在国内高校用用,不适合工程设计,至于1,2,4哪个更好,其实还要看具体的工艺过程, 比如HYSYS,在油气工程领域就有着极高的精度和准确性,HYSYS仅仅是石化中使用较多,HYSYS买给Honeywell已经改名叫UniSim了 ASPEN是设计院使用更为合适一些,但价格昂贵,不过可以计算固体、燃烧等模块是HYSYS,PRO/II不能比拟的,比较全面,但界面比较凌乱。你要模拟个氮肥装置,恐怕还是aspen的好, 单体设备的校核型核算,PRO2又是个最简单最合适的选择,PR方程精度较高(其实也就是HYSYS主推的),PRO/II价格便宜,简单多了. 搞天然气开采的可能只能选HYSYS,但ASPEN并不是不好啊. 至于CHEMCAD、和青岛ECSS等软件,实际工程中均使用不大,ASPEN把HYSYS的公司也收购了。实际上最终就是ASPEN和PRO/II谁能当老大。难受的是,这些均为国外软件,青岛ECSS软件是中国国产的唯一一套,使用起来又无法同国外相比。惨! 常用的工艺计算软件化工工艺设计涉及大量的计算,主要的有工艺流程的模拟,管道水力学计算,公用工程管网计算,换热器设计计算,容器尺寸计算,转动设备的计算和选型,安全阀泄放量和所需口径的计算,**泄放系统,控制阀Cv计算和选型,等等。这些计算过程通常都有专用的商业软件或者是工程公司自行开发的软件或者计算表格。大的设计公司通常也会指定公司用于以上设计过程的软件或经过确认的表格。下面就我的经验来看看常用的一些软件。 1. 工艺流程模拟: ASPEN Plus Pro II HYSYS 2. 管道水力学计算: 通常是工程公司自备的EXCEL表格,没必要使用专用软件。当然,也可以自己编制,一般来说使用CRANE手册提供的公式就足够了。 两相流的水力学计算相当复杂,自己编制费力不讨好,用公司内部经过验证的表格就可以了。 3. 公用工程管网计算 我用过Pipe 2000,肯塔基大学教授的出品,包括Gas 2000, Water2000, Steam 2000等一系列。Pipenet也是不错的选择。 有人用SimSCI的InPlant。没用过,有用过的朋友可以介绍一下。 4. 换热器设计计算 HTRI HTFS 这两个软件都可以。常见的介质用HTRI更好,因为它的物性数据是经过实验得到的。HTFS 使用了ASPEN或HYSYS的物性数据,很多都是计算得到的,所以精度可能稍差。
催化燃烧VOCs的三种过渡金属催化剂的活性比较_郭建光
华南理工大学学报(自然科学版)第32卷第5期Journal of South China University of Technology Vol .32 No .5 2004年5月 (Natural Science Edition ) May 2004 文章编号:1000-565X (2004)05-0056-04 催化燃烧VOCs 的三种过渡金属催化剂的活性比较* 郭建光1 李 忠1 奚红霞1 何余生1 王伯光2 (1.华南理工大学化学工程研究所,广东广州510640;2.广州市环境检测中心站,广东广州510030) 摘 要:利用浸渍法将过渡金属硝酸盐溶液沉积在γ-Al 2O 3上,通过焙烧制得三种过渡金属氧化物催化剂CuO /γ-Al 2O 3、CdO /γ-Al 2O 3和NiO /γ-Al 2O 3.通过催化燃烧销毁乙醇、丙酮和甲苯的实验,对催化剂活性进行了评价.实验结果表明,在催化剂作用下,三种挥发性有机化合物VOCs 催化燃烧的起燃温度和完全燃烧温度都明显低于它们的燃点,其中 CuO /γ-Al 2O 3催化剂的催化活性优于CdO /γ-Al 2O 3和NiO /γ-Al 2O 3催化剂,它对丙酮、乙醇和甲苯的催化起燃温度分别是180,190和230℃,另外对于催化燃烧VOCs ,挥发性气体分子的极性越大就越容易被氧化. 关键词:过渡金属;催化剂;催化燃烧;挥发性有机物中图分类号:TQ 032.41 文献标识码:A 收稿日期:2003-10-20 *基金项目:国家自然科学基金资助项目(20176012);教育部博士点基金项目(20020561010);广东省科技攻关计划项目 作者简介:郭建光(1978-),男,博士生,主要从事环境与化学工程的研究.E -mail :jgguo @scut .edu .cn 挥发性有机化合物(VOCs )是一类主要的空气 污染物,给人类的生命和健康以及环境带来严重的危害[1,2].目前VOCs 的处理技术主要有吸附[3,4]、吸 收、冷凝[5]、膜分离[6]、光催化降解[7]、生物降解[8] 、等离子体技术[9,10]和催化燃烧技术等.催化燃烧技术最显著的优点就是能够在很低的浓度(小于1%)下进行操作,以及相对于热力燃烧而言具有更低的操作温度.这些优点和节能的特性使之成为目前最有应用前景的VOCs 销毁方法之一.在催化燃烧技术中,催化剂是关键,其性能的优劣对销毁效率和能耗有着决定性的影响.在催化燃烧中用得较多的催化剂是贵金属催化剂,如Pt ,Pd ,Au 和Rh 等是典型的贵金属催化剂[11~13].这类催化剂通常负载在载体上,其活性高,选择性好,但是价格昂贵,并且容易中毒.因此寻找价格低廉、催化活性较好的催化剂是催化燃烧技术得以广泛应用的关键.本研究利用浸渍法将Cu ,Cd ,Ni 三种过渡金属的硝酸盐溶液浸渍在γ-Al 2O 3颗粒上,经过焙烧制备得到三种金属氧化物催化剂,通过对乙醇、丙酮和甲苯这3种VOCs 气体 进行催化燃烧,来考察反应的起燃温度和完全燃烧温 度,从而比较三种催化剂的催化活性. 1 实验 1.1 实验材料和仪器 乙醇、丙酮和甲苯为分析纯;硝酸铜、硝酸镉和硝酸镍为化学纯,γ-Al 2O 3(美国Merck 公司).FA /JA 系列上皿电子天平(上海精密科学仪器有限公司天平仪器厂);DF -101B 集热式恒温磁力搅拌器(浙江乐清市乐成电器厂);GC112型气相色谱仪(上海仪器分析总厂);ASAP2010M 快速比表面测定仪(美国Micr omet -rics 公司);流量控制器(北京七星华创电子股份有限公司);温度显示仪(福建百特工控公司). 1.2 实验方法 1.2.1 催化剂的制备 将40~60目的γ-Al 2O 3颗粒分别加入到浓度均为0.5mol /L 的Cu (NO 3)2、Cd (NO 3)2和Ni (NO 3)2溶液中,然后浸渍12h ,过滤、干燥,最后在500℃下焙烧5h ,就可以制得CuO /γ-Al 2O 3、CdO /γ-Al 2O 3和NiO /γ-Al 2O 3三种负载金属氧化物的催化剂. 1.2.2 催化剂的比表面积和孔结构分析 采用ASAP2010M 快速比表面积和孔径测定仪对所用催化剂进行比表面积和比孔容进行表征.实验条件:催化剂在300℃下脱气3h ,在-195.82℃条件下液氮吸附.吸附等温线采用B ET 二参数方程
利用Hyperchem软件进行分子结构构建及性质计算
利用Hyperchem软件进行分子结构构建及性质计算 实验目的 1.初步了解分子模型方法的原理和应用。 2.学习使用Hyperchem软件构建简单的分子并使用适当方法优化结构。 3.学习使用Hyperchem软件计算简单分子的几何和电子性质。 实验原理 化学的学习使我们认识了许多分子的分子式及二维结构,如何得到分子的三维结构,以及分子在空间的几何特征和电子特征,则可以借助于理论计算的工具和方法去模拟计算。 HyperChem软件是HyperCube公司开发的Windows界面程序。是常用的分子设计和模拟软件。它可以应用于构建简单及复杂的分子模型并进行综合计算与分析。分子构建过程可以通过熟练各个菜单及工具栏的操作来实现,计算和分析需要我们了解常用的计算方法。在本实验室中我们需要了解一下计算方法,这些方法位于HyperChem的Setup菜单下。 (1)分子力学(Molecular Mechanics)方法:分子力学又叫力场方法,目前广泛地用于计算分子的构象和能量。适用于超大规模体系,超低精度计算。 分子力学的基本假设:玻恩-奥本海默近似,原子核的运动与电子的运动可以看成是独立的;分子是一组靠各种作用力维系在一起的原子集合。这些原子在空间上若过于靠近,便相互排斥;但又不能远离,否则连接它们的化学键以及由这些键构成的键角等会发生变化,即出现键的拉伸或压缩、键角的扭变等,会引起分子内部应力的增加。每个真实的分子结构,都是在上述几种作用达到平衡状态的表现。分子力学从几个主要的典型结构参数和作用力出发来讨论分子结构,即用位能函数来表示当键长、键角、二面角等结构参数以及非键作用等偏离“理想”值时分子能量的变化。不同的分子力场方法采用不同的势能函数。MM+:适用于有机分子的计算。Amber:适用于有机分子、蛋白质和核酸等大分子的计算。 (2)半经验计算(Semi-empirical)方法:是求解HF(Hartree-Fock)方程时采用各种近似,或者直接使用拟合的经验参数来近似求解自洽场。采用单电子近似,完全不考虑双电子作用而挑选的等效单电子Hamilton算符。适用于大规模体系,低精度计算。其中AM1和PM3方法都适用于基态分子的计算。
量子化学理论与软件介绍
量子化学是应用量子力学的规律和方法来研究化学问题的一门学科。将量子理论应用于原子体系还是分子体系是区分量子物理与量子化学的标准之一。 主要分为:①分子轨道法(简称MO法,见分子轨道理论);②价键法(简称VB法,见价键理论);③密度泛函理论。以下只介绍分子轨道法。 ①分子轨道法:分子体系中的电子用单电子波函数满足Pauli不相容原理的直积(如Slater 行列式)来描述,其中每个单电子波函数通常由原子轨道线性组合得到(类似于原子体系中的原子轨道),被称作分子轨道,分子轨道理论是目前应用最为广泛的量子化学理论方法。 o HF方法:它是原子轨道对分子的推广,即在物理模型中,假定分子中的每个电子在所有原子核和电子所产生的平均势场中运动,即每个电子可由一个单电子函数(电子的坐标的函数)来表示它的运动状态,并称这个单电子函数为分子轨道,而整个分子的运动状态则由分子所有的电子的分子轨道组成(乘积的线性组合),这就是分子轨道法名称的由来。分子轨道法的核心是哈特里-福克-罗特汉方程,简称HFR方程,它是以三个在分子轨道法发展过程中做出卓著贡献的人的姓命名的方程。1928年D.R. 哈特里提出了n个将电子体系中的每一个电子都看成是在由其余的n-1个电子所提 供的平均势场中运动的假设。这样对于体系中的每一个电子都得到了一个单电子方程(表示这个电子运动状态的量子力学方程),称为哈特里方程。使用自洽场迭代方式求解这个方程(见自洽场分子轨道法),就可得到体系的电子结构和性质。哈特里方程未考虑由于电子自旋而需要遵守的泡利原理。1930年,B.A.福克和J.C.斯莱特分别提出了考虑泡利原理的自洽场迭代方程,称为哈特里-福克方程。它将单电子轨函数(即分子轨道)取为自旋轨函数(即电子的空间函数与自旋函数的乘积)。泡利原理要求,体系的总电子波函数要满足反对称化要求,即对于体系的任何两个粒子的坐标的交换都使总电子波函数改变正负号,而斯莱特行列式波函数正是满足反对称化要求的波函数。将哈特里-福克方程用于计算多原子分子,会遇到计算上的困难。C.C.J.罗 特汉提出将分子轨道向组成分子的原子轨道(简称AO)展开,这样的分子轨道称为原子轨道的线性组合(简称LCAO)。使用LCAO-MO,原来积分微分形式的哈特里-福克方程就变为易于求解的代数方程,称为哈特里-福克-罗特汉方程,简称HFR 方程。 o CI方法:组态相互作用(Configuration Interaction)方法。用HF自洽场方法计算获得的波函数和各级激发的波函数为基展开体系波函数。完全的组态相互作用(Full-CI)是指定基组下最精确的方法,但其计算量约以基函数的阶乘规模增加,目前仅限于对小分子作为Benchmark以检测其他方法的可靠性,在实际应用中常采用截断CI方法,如
HyperChem
分子图形与分子模型设计——HyperChem使用简介 厦门大学化学系 2005年3月
HyperChem使用简介 HyperChem是HyperCube Inc.的产品,它具有非常强大的综合计算与分析功能,是优秀的分子图形和分子设计的工具软件之一。HyperChem是运行在Windows系统的分子计算与建模软件,具有量子化学(半经验和从头算)、分子力学、分子动力学、随机动力学、Monte Carlo模拟等计算功能,计算结果可以用三维图形显示。它还提供用户VB、C/C++和FORTRAN等语言的应用程序接口。HyperChem 7.5版本已经推出。图1是HyperChem的工作窗口,最下部是工作状态档。在菜单下面是常用工具档。 图1 HyperChem的工作窗口 HyperChem的操作可以使用鼠标和键盘两种。在工具档从左开始有8个工具图标,当鼠标点图标之后,鼠标在工作区的形状也改变为该图标的形状: 1.绘图工具。鼠标双击该图标可直接进入缺省元素周期表,选择所要绘制元素。 2.选择工具。 3.xy轴方向旋转工具,也可使用键盘的上下左右光标键进行相同的操作。 4.z轴方向旋转工具,也可使用键盘的Home和End键进行相同的操作。 5.xy轴平移工具,也可使用键盘的Shift+上下左右光标键进行相同的操作。 6.z轴平移工具。 7.缩放工具,也可使用键盘的PgUp和PgDn键进行相同的操作。 8.z轴截片工具。 鼠标操作有较为多样:左点击、右点击、左拖拉、右拖拉、左右拖拉、Shift+左点击、Shift+右点击、双击等。一般的旋转和平移操作是使用鼠标的左键进行,当完成了某个基团、分子的选择之后,可以使用右键对所选部分进行旋转和平移操作。 HyperChem的详细操作将结合具体的实例进行讲解。以下通过对HyperChem 5.1的菜单命令的介绍,说明它的主要功能和使用方法。 一、File 1.New (Ctrl+N):新建一个沿尚未命名文件。此时的工作窗口是空白。 2.Open (Ctrl+O):打开一个已存在的分子结构文件。可打开的文件格式如下:HyperChem (HIN)、Tripos MOL2 (ML2)、MDL MOL (MOL)、Brookhaven PDB (ENT)、Isis Sketch
分子图形软件——结构可视
化学结构可采用多种表示方法。分子的二维平面结构、三维空间结构和分子表面结构等表示法都属于化学结构的可视化表示方法,它们使用计算机图形技术直观、形象地表示分子结构。化学结构的可视化表示采用了大量计算机图形学的技术,现已发展成为一门新兴的边缘学科——分子图形学。分子图形学采用计算机图形技术处理分子结构的显示和操作。分子建模技术是分子图形学的一个重要的分支,它包括三维分子结构的构建、操作及其物理化学性质的表示,任何一个分子建模软件都需要以分子图形作为交互界面。分子图形学为化学工作者的研究工作提供了很大的方便,也对深入认识化合物的结构和化学反应的本质带来了可能,尤其对于复杂的生物大分子更是如此。分子图形学是物理化学、有机化学、生物化学和医药化学等基础研究及技术开发的常用研究工具。现今许多公司开发了速度快、图形质量高、功能强、价格低且具有友好用户界面的分子图形软件。 化学结构的表示 任何一门科学都具有自己的表述语言,化学学科也不例外。原子和分子是化学研究的主要对象,而原子和分子又是不可见的微观物体,目前人类发现的化学物质已经超过2300万种以上,科学和有效的化学结构表示是化学家面临的重要的问题。化学结构的表示有:命名法、线型编码法、二维结构、三维结构、表面结构等方法。化合物结构的可视化表示具有表示简单、直观和容易记忆的优点,因此化学物质结构图(结构式)常作为化学家的共同表述语言。随着化学研究的深入,大多数化学物质的结构已经清楚,描述分子最好、最准确的方法就是绘制分子结构式。分子结构的绘制即是化学教育的重要内容,也是化学研究的重要工具。化学结构的表示要具体说明组成分子的原子数目、原子种类、各原子间的相对位置和连接性,这些化学结构信息可以使用图形方式表示,也可使用结构代码的命名法表示。化学结构的表示应该是准确、简洁和单义的,并且可以方便地进行计算机的存储、检索和显示。 由于化合物有很多同分异构体,使用分子式无法准确地表示其结构。无机和有机化合物采用系统的命名法是IUPAC(International Union of Pure and Applied Chemistry)法,它给化合物定义唯一的名称,但其表达冗长、复杂且难以记忆。为了适应计算机检索的要求,有关科学工作者正在大力研究和设计化学结构信息的表示方法。化合物结构线性代码是采用线性顺序数字、字母的字符串表示化学物结构,对线型编码已开展了大量研究工作,制定了许多线型编码方法,例如WLN(Wiswesser Line Nation)法、ROSDAL(Representation of Organic Structure)、SLN(Sybyl Line Notation)法、SMILES(Simplified Molecular Input Line Entry Specification)法等。目前WLN 命名法已近于过时。SMILES是一种应用广泛的相当重要的表示法,它可以将复杂的有机结构以字符串方式表示,已经广泛使用于结构数据库搜索中。许多著名的化学结构检索数据库都可使用SMILES字符串进行检索。 虽然线型编码可用于化学数据库的输入、存储和检索,但线性结构代码缺少直观性,需要专门学习其编码规则。许多用户进行化学结构检索时,更愿意使用二维化学结构的输入方式。使用二维图形表示化学结构是化学家最常用的方式之一,这些结构图形形象地代表了分子模型,使得分子表示更直观。在二维结构图形中,原子以元素符号表示,以线条表示化学键,然而二维化学结构只能简单地表示分子中化学键的拓扑结构。三维化学结构的表示还需要分子中原子在三维空间的位置信息,它可形象、逼真地表示分子的真正结构。如果在分子的表面增加了更复杂的分子性质信息(例如静电势等),则化学结构可以使用分子三维表面结构图形表示。图8-1 是苯基丙氨酸(phenylalanine)的各种化学结构表示。
ChemWindow6.0在3D分子模型绘制中的应用
ChemWindow6.0在3D分子模型绘制中的应用 摘要主要介绍了如何使用ChemWindow 6.0中SymApps 6.0程序绘制三维分子模型,并结合具体实例,对SymApps 6.0的特点和功能进行了探讨。 关键词ChemWindow 6.0 SymApps 6.0 三维分子模型绘制 常用的3D分子结构绘制软件有CS Chem3D、 HyperChem、WebLab ViewerPro和RasMol等,而很少有人用到ChemWindow 6.0中的SymApps 6.0程序,该程序也能将绘制的2D分子结构显示成三维图形,还可以计算给出键长、点群等信息,可制作分子三维旋转动画,也可将图形拷贝至Word、Powerpoint 中,提高文档及多媒体课件的质量。本文结合实例,对如何在ChemWindow 6.0中绘制3D分子模型进行了介绍。 1 应用ChemWindow 6.0绘制3D分子模型的步骤 (1)打开ChemWindow 6.0窗口,绘制目标分子的二维分子结构图[1]。 (2)用“selection”箭头或者执行“EditSelect All”命令,将所绘制的2D分子结构图选中。需要注意的是这一步是绘制3D分子模型所必须的,否则无法打开SymApps程序窗口。 (3)建立目标分子的3D结构图。执行“Other SymApps”命令,即可打开SymApps程序窗口。在打开的窗口中就可以发现2D分子模型被转化成了“Wireframe”形式的结构图(如图1所示为乙烯分子的“Wireframe”形式结构图)。 (4)结构合理化。对由2D结构产生的3D结构,须运行“Compute 3—D Structur”命令,使其结构合理化,否则在绘制稍微复杂分子结构(如桥环化合物)的时候,会产生错误的结构图。 (5)3D结构的形式。分子的三维结构有4种类型,分别为Wireframe,Sticks,Ball&Sticks和Spacefill,可依次按按钮进行转换,其形式依次为图2中的1、2、3、4所示。
计算化学教程01Gaussian和Hyperchem使用
双击在跳出的对话框中选择相应的元素,如下图选择了C,同时把对话框底部Allow Ions 选项选上,这样就可以任意画键。然后右上角关闭对话框。 意结构图 选择按钮,旋转图形成近平面,如左图 们同平面三个原子连接起来。如下7. 删除键的方法: 鼠标在Draw模式 下放至要删除的键 的中部,点击右键。 Hyperchem搭建初步构型 的粗模型,在键长和键角上可能很不合理,我们可通过中的几何优化功能,粗略优化一下构型。如下选择菜单Compute → 在跳出的对话框中选择Ok,程序就会对刚才的模型进行优化。这样就得到了化学上相对合理构型,如右。(比较一 下可以看出,程序对键长自动作了一定的调整) 用Hyperchem 9. 保存构型。选择菜单File →Save,保存为 10. 用Gaussview打开,C5.mol 如右, 在Gaussview中可以进行一定的修改。打 开时注意选择*.mol 扩展名。
中选择File →Save 保存为C5.gjf,文件用Scite编辑器打开,内容如下 第1行%开头定义chk文件,类似还可以定义内存%mem,%rwf等。 第2行#开头定义计算关键字 第3行空 第4行说明性文字 第5行空 第6行0 指体系电荷,如负1价离子则写-1. 1 为体系多重度= 2s+1 s为体系总自旋第7-11行为原子坐标。 第12空行 用Gaussian优化构型 下面以优化C5为例进行说明: 1. 编辑C5.gjf文件,内容如右 %mem定义内存为4GB %nprocshared=8定义使用CPU 数目 #P B3LYP/6-31+G*定义了优化 Freq。 文件传到计算服务器上。 转换文件格式(注意Linux上区分大小写)5. 运行ps x 查看,如果正常,则显示如下: Gaussian优化构型 传回文件。查看工作是否计算完可以通过类似5步中的ps 录下运行tail C5.out 查看文件末尾信息,如果最后一行显示如下: Normal termination …说明任务正常结束,结果文件可用。否则可能显示Error …等等错误提示。 7. 若任务正常结束,则再用Scite打开传回的文件C5.out,查找Frequencies,找到如个,或后面的好几个均为负值,则说明此结构不稳定,需进行调整。
化工仿真模拟软件使用
化工仿真模拟软件使用 换热器:HTRI和HTFS都是国际著名的换热器工艺计算软件,你需要了解其意思方可用于实际 1. ASPEN 2. PROII 3. CHEMCAD 4. HYSYS 5. ECSS 所列软件中,1,2,4是工程公司常用软件,3,5一般在国内高校用用,不适合工程设计,至于1,2,4哪个更好,其实还要看具体的工艺过程, 比如HYSYS,在油气工程领域就有着极高的精度和准确性,HYSYS仅仅是石化中使用较多,HYSYS买给Honeywell已经改名叫UniSim了 ASPEN是设计院使用更为合适一些,但价格昂贵,不过可以计算固体、燃烧等模块是HYSYS,PRO/II不能比拟的,比较全面,但界面比较凌乱。你要模拟个氮肥装置,恐怕还是aspen 的好, 单体设备的校核型核算,PRO2又是个最简单最合适的选择,PR方程精度较高(其实也就是HYSYS主推的),PRO/II价格便宜,简单多了. 搞天然气开采的可能只能选HYSYS,但ASPEN并不是不好啊. 至于CHEMCAD、和青岛ECSS等软件,实际工程中均使用不大,ASPEN把HYSYS的公司也收购了。实际上最终就是ASPEN和PRO/II谁能当老大。难受的是,这些均为国外软件,青岛ECSS软件是中国国产的唯一一套,使用起来又无法同国外相比。惨! 常用的工艺计算软件化工工艺设计涉及大量的计算,主要的有工艺流程的模拟,管道水力学
计算,公用工程管网计算,换热器设计计算,容器尺寸计算,转动设备的计算和选型,安全阀泄放量和所需口径的计算,**泄放系统,控制阀Cv计算和选型,等等。这些计算过程通常 都有专用的商业软件或者是工程公司自行开发的软件或者计算表格。大的设计公司通常也会指定公司用于以上设计过程的软件或经过确认的表格。下面就我的经验来看看常用的一些软件。 1. 工艺流程模拟: ASPEN Plus ?Pro II ?HYSYS ? 2. 管道水力学计算: 通常是工程公司自备的EXCEL表格,没必要使用专用软件。当然,也可以自己编制,一般来说使用CRANE手册提供的公式就足够了。 ?两相流的水力学计算相当复杂,自己编制费力不讨好,用公司内部经过验证的表格就可以了。 ? 3. 公用工程管网计算 我用过Pipe 2000,肯塔基大学教授的出品,包括Gas 2000, Water2000, Steam 2000等一系列。 ?Pipenet也是不错的选择。 ?有人用SimSCI的InPlant。没用过,有用过的朋友可以介绍一下。 ? 4. 换热器设计计算 HTRI ?HTFS ?这两个软件都可以。常见的介质用HTRI更好,因为它的物性数据是经过实验得到的。 HTFS使用了ASPEN或HYSYS的物性数据,很多都是计算得到的,所以精度可能稍差。 ? 5. 压力容器尺寸计算(长度与内径)
化学工具总结
化学软件 分子模型化 3D分子图形显示工具 (RasMol and OpenRasMol)(免费) AMBER (分子力学力场模拟程序) autodock (分子对接软件)(免费) GROMACS (分子动力学软件)(免费) GULP (General Utility Lattice Program)(免费) NIH分子模拟中心的化学软件资源导航(Research Tools on the Web) X-PLOR (大分子X光晶体衍射、核磁共振NMR的3D结构解析)(免费) 高通量筛选软件PowerMV (统计分析、分子显示、相似性搜索等)(免费) 化合物活性预测程序PASS(部分免费) 计算材料科学Mathub C4:Cabrillo学院化学可视化项目以及相关软件(免费) Databases and Tools for 3-D Protein Structure Comparison and Alignment (三维蛋白质结构对比)(免费) Democritus (分子动力学原理演示软件) DPD应用软件cerius2(免费) EMSL Computational Results DataBase (CRDB) MARVIN'S PROGRAM (表面与界面模拟)(免费) XLOGP(计算有机小分子的脂水分配系数)(免费) 量子化学软件中文网
美国斯克利普斯研究院:金属蛋白质结构和设计项目(免费) https://www.360docs.net/doc/ec13901637.html,/(免费) 3D Molecular Designs (蛋白质及其他3D分子物理模型快速成型技术) 3D-Dock Suite Incorporating FTDock, RPScore and MultiDock (3D 分子对接)(免费) AMSOL (半经验量子化学计算)(免费) Amsterdam Density Functional (ADF, 第一原理电子结构计算) Bilbao晶体学服务器(免费) BOSS (蒙特卡罗模拟软件) CADPAC (剑桥量子化学计算软件)(免费) Car-Parrinello分子动力学(CPMD, ab-initio分子动力学计算软件)(免费) CHARMM (大分子分子力学模拟计算软件)(部分免费) Chem2Pac package (A computational Chemistry Integrator)(免费) ChemTK Lite (QSAR软件)(免费) Chemweb计算化学在线工具:GAMESS(免费) Clebsch-O-Matic (在线计算器)(免费) Collaborative Computational Projects (协同计算计划) COLUMBUS (量化从头计算分子电子结构程序集)(免费) CrystalMaker Software (晶体结构可视化软件) DL_POLY (分子动力学模拟软件)(免费)
HyperChem基本操作 一、画原子
HyperChem基本操作一、画原子 HyperChem基本操作一、画原子 作者:水云间文章来源:本站原创点击数:571 更新时间:2005-11-11 HyperChem基本操作画原子 1. 打开Element Table对话框。这里有两种方法: 在Build菜单中选择Default Element,或者双击Drawing工具。Default Element对话框允许从周期表中选择缺省元素。 2. 如果单击Properties...按钮,将显示当前选择元素的物理属性。也可以 按下Shift键同时单击元素按钮,结果是一样的。单击OK键,物理属性框 消失。 3. 如果Allow Ions或者Explicit Hydrogens打开(用对勾选择),左键单击 这些选项使其关闭。 4. 在缺省元素列表中选择Carbon,接着关闭元素对话框。缺省元素将设置 为碳。当然也可以把打开的Default Element对话框移走,这样可以看到 HyperChem工作区。当画原子非常多的分子时,这是非常有效的。 5. 左键单击Drawing工具,把指针移到工作区。 6. 左键单击工作区左下角,将出现一个小圈,代表未成键的碳原子。 7. 在工作区不同位置画更多的原子。 画价键 1. 把指针移到刚才画的第一个碳上。 2. 按下鼠标左键。这是价键在第一个原子的位置。 3. 保持鼠标按钮按下的同时拖向工作区的顶端。 4. 放开鼠标按钮。这是价键在第二个原子的位置。一条线代表两个碳 原子之间的价键。 5. 用仍旧停留在价键末端的指针, 用左键拖向工作区右下角。 6. 放开鼠标按钮。这是第三个原子的位置。 7. 在空白工作区画六个价键,形成一个环。 现在你清楚了如何画原子和分子,并且学会了一些基本技巧。 选择原子 在这个练习中,通过选择原子,你可以学到基本的选择技巧。首先必须设置 选择的级别[原子(atoms),基(residues),或分子(molecules)]。 这里设置为原子(atoms)。 1. 左键单击Select菜单。 2. 左键单击选择Atoms。 接下来,关闭Multiple Selections: 1. 左键单击Select菜单。 2. 如果Multiple Selections打开,左键单击来关闭它。 这样当你重新选择一个原子或者一组原子的时候,前面的选择将会取消。 选择原子: 1. 左键单击Selection工具,把指针移到工作区。 2. 左键单击前面创建的原子。这将激活这个原子。可以在Preferences对话
hyperchem使用方法(1)
HyperChem应用 HyperChem应用 水中质子的从头计算 用explicit hydrogens画H3O+: 1. 在Build菜单选择Explicit Hydrogens。 2.选择Allow Ions。 3. 在Display菜单选择Labels打开Labels对话框。 4. 选择Symbols单击OK。 5. 在Build菜单选择Default Element打开周期表,设置氧元素为缺省元素。 6. 左键单击Drawing工具。 画H3O+ 1. 左键单击工作区创建氧原子。 2. 从氧原子画三个价键得到H3O。 在量子力学计算中添加正电荷: 1. 在Setup菜单选择Ab Initio。 2. 单击Options按钮。 3. 设置总电荷(Total charge)值为1,单击OK关闭Options对话框。 4. 单击OK按钮关闭Ab Initio对话框。 这样就得到了H3O+。 选择基组 1. 在Setup菜单选择Ab Initio。 2. 为基组选择Other。 3. 按下Assign Other Basis Set按钮。 4. 从列表中选择4-31G 单击OK。 5. 为基组选择Minimal (STO-3G)。 6. 选择Apply Basis Set接着单击OK,或者直接选择OK关闭Ab Initio对话框。 观察每个原子应用的基组: 1. 在Display菜单选择Labels。 2. 选择Basis Set,单击OK。 最小化能量结构 计算H3O+的几何优化: 1. 从Setup菜单选择Ab Initio。 2. 单击Options按钮,保证Total charge = 1,Spin multiplicity = 1, Spin pairing = RHF,Conver-gence limit = 0.01,Iteration limit = 50,Accelerate conver-gence= Yes,为几何优化选择合适的选项。Single Point only选项在几何优化中不起作用, 因而可以是任何值。 3. 左键单击OK关闭Options对话框和Ab Initio对话框。 4. 从Compute菜单中选择Geometry Optimization。 5. 选择Polak-Ribiere作为几何优化方法,RMS gradient值为0.1,左键单击OK开始几何优化。H3O+优化 结果为键角113.74 度键长0.99 埃。 计算优化后H3O+包括相关能的总能量: 1. 从setup菜单选择Ab Initio,选择Options按钮并选择计算MP2 correlation energy,