microrna研究进展

生命科学
Chinese Bulletin of Life Sciences
第17卷 第3期2005年6月
Vol. 17, No.3
Jun, 2005
microRNA 研究进展
华友佳,肖华胜*
(生物芯片上海国家工程研究中心,上海 201203)
摘 要:小分子RNA 家族中的一员——microRNA ,是一段非常短的非编码RNA 序列,对多种生物学过程起调控作用。本文试从microRNA 的结构特点、合成及作用机制和功能等方面对microRNA 的研
究进展作一个简单回顾。
关键词:microRNA(miRNA); Drosha ;Dicer ;siRNA 中图分类号:Q522 文献标识码:A
Progresses on the microRNA study
HUA You-Jia, XIAO HUa- Shang*
(National Engineering Center for Biochip at Shanghai, Shanghai 201003 , China)
Abstract: microRNA, one of the small molecular RNA family, is a very small section of non-coding RNA sequence,which can regulate several biological processes. This review tries to have a brief introduction on the progresses of microRNA study, such as the structure, the biogenesis, processing and the function Key words: microRNA (miRNA );Drosha ;Dicer ;siRNA
文章编号 :1004-0374(2005)03-0000-00
收稿日期:2004-12-16;修回日期:2005-02-23
基金项目:“863”项目功能基因组和生物芯片重大专项“2002AA2Z 2002”资助
作者简介:华友佳(1981—),男,在读博士生;肖华胜(1970—),男,副研究员,*通讯作者。
作为Science 2002年十大科技突破的第一名——microRNA 成为生物学研究的一大焦点。它在生物的发育时序调控和疾病的发生中起到非常重要的作用。研究发现,它与siRNA 有很多相似之处,但也有很大不同。对microRNA 的研究将会对转录后基因调节领域的发展产生深远影响。1 microRNA 的概念与特征
microRNA(miRNA)是一类长度很短的非编码调控单链小分子RNA ,约20~24 nt(少数小于20 nt 的),由一段具有发夹环结构的长度为70~80 nt 的单链RNA 前体(pre-miRNA)剪切后生成。它通过与其目标mRNA 分子的3'端非编码区域(3'-untranslated region, 3' UTR)互补匹配导致该mRNA 分子的翻译受到抑制。miRNA 基因以单拷贝、多拷贝或基因簇等多种形式存在于基因组中,而且绝大部分定位于基因间隔区。其转录独立于其他基因,并不翻译
成蛋白质,而是在体内代谢过程中起到多种调控作用。miRNA 在各个物种间具有高度的进化保守性,并且在茎部的保守性更强;但在环部可以容许更多的突变位点存在。
最早发现的miR NA 是在线虫中的let -7[1]和lin-4 [2]。运用多种实验方法以及生物信息学方法,已经找到了数百种miRNA(表1)。
许多miRNA 在表达上具有阶段性和组织特异性[3],例如在拟南芥中,miR-157在幼苗中高表达,miR-171则在花中高表达[4]。这种特异性对miRNA 调控功能的作用有重要意义。
表1 模式生物的miRNA 种类数
模式生物人 小鼠 大鼠 果蝇线虫 拟南芥miRNA
207
214
186
78
116
111
2生命科学第17卷
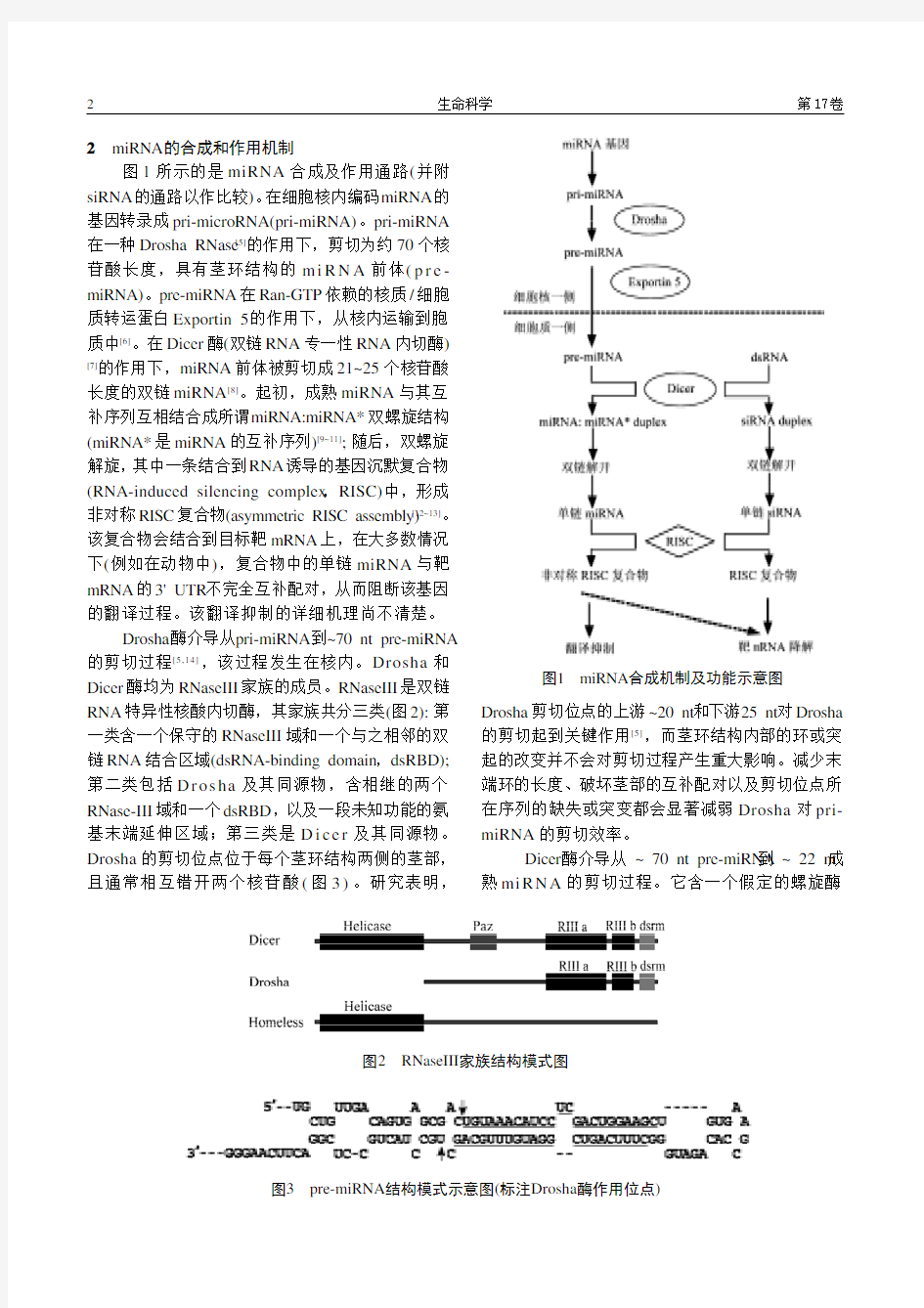
2 miRNA 的合成和作用机制
图1所示的是miRNA 合成及作用通路(并附siRNA 的通路以作比较)。在细胞核内编码miRNA 的基因转录成pri-microRNA(pri-miRNA)。pri-miRNA 在一种Drosha RNase [5]的作用下,剪切为约70个核苷酸长度,具有茎环结构的m i R N A 前体(p r e -miRNA)。pre-miRNA 在Ran-GTP 依赖的核质/细胞质转运蛋白Exportin 5的作用下,从核内运输到胞质中[6]。在Dicer 酶(双链RNA 专一性RNA 内切酶)[7]
的作用下,miRNA 前体被剪切成21~25个核苷酸长度的双链miRNA [8]。起初,成熟miRNA 与其互
补序列互相结合成所谓miRNA:miRNA* 双螺旋结构(miRNA*是miRNA 的互补序列)[9~11]; 随后,双螺旋解旋,其中一条结合到RNA 诱导的基因沉默复合物(RNA-induced silencing complex ,RISC)中,形成非对称RISC 复合物(asymmetric RISC assembly)[12~13]。该复合物会结合到目标靶mRNA 上,在大多数情况下(例如在动物中),复合物中的单链miRNA 与靶mRNA 的3' UTR 不完全互补配对,从而阻断该基因的翻译过程。该翻译抑制的详细机理尚不清楚。
Drosha 酶介导从pri-miRNA 到~70 nt pre-miRNA 的剪切过程[5,14],该过程发生在核内。Drosha 和Dicer 酶均为RNaseIII 家族的成员。RNaseIII 是双链RNA 特异性核酸内切酶,其家族共分三类(图2): 第一类含一个保守的RNaseIII 域和一个与之相邻的双链RNA 结合区域(dsRNA-binding domain ,dsRBD);第二类包括D r os ha 及其同源物,含相继的两个RNase-III 域和一个dsRBD ,以及一段未知功能的氨基末端延伸区域;第三类是D i c e r 及其同源物。Drosha 的剪切位点位于每个茎环结构两侧的茎部,且通常相互错开两个核苷酸(图3)。研究表明,
Drosha 剪切位点的上游~20 nt 和下游25 nt 对Drosha 的剪切起到关键作用[5],而茎环结构内部的环或突起的改变并不会对剪切过程产生重大影响。减少末端环的长度、破坏茎部的互补配对以及剪切位点所在序列的缺失或突变都会显著减弱Drosha 对pri-miRNA 的剪切效率。
Dicer 酶介导从 ~ 70 nt pre-miRNA 到 ~ 22 nt 成熟mi R N
A
的剪切过程。它含一个假定的螺旋酶
图1 miRNA 合成机制及功能示意图
图3 pre-miRNA 结构模式示意图(标注Drosha
酶作用位点
)
图2 RNaseIII 家族结构模式图
3
第3期
华友佳,等:micro RNA 研究进展
(Helicase)区域、一个DUF283域、一个PAZ(Piwi-Argonaute–Zwille)域、相继的两个RNaseIII 域(RIII a 和RIII b)和一个dsRBD [7](图4)。与其他RNase III 家族的酶相比,Dicer 多了PAZ 域,这个区域对它的功能至关重要[15~17]。Dicer 酶的PAZ 域可识别Dr os ha 剪切产物的末端,并将第二个RNa seIII
(RNaseIII b)定位在miRNA 前体的茎部[18]。Dicer 家族中那些PAZ 域缺失的成员不会介导miRNA 的成熟过程,但是依然具有剪切酶的活性,这点类似于Drosha 酶。在Dicer 的作用过程中,对于双链有效的剪切需要二聚化的RNaseIII 域。这是由于依据已知的RNase III 结构,功能催化位点仅在RNaseIII 二聚体的交界处形成。这就可以理解为何Drosha 家族和Dicer 家族的两个RNaseIII 域均在空间上前后紧密相邻。由于Dicer 包含与Drosha 相似的结构,因此也可以部分替代后者的功能[5],但效率较低。
有趣的是,在细胞核内也观测到Dic er 的存在,但它不参与miRNA 通路。Drosha 剪切产生的pre-miRNA 必须由转运蛋白Exportin-5转移至核外,再由那里的Dicer 继续剪切过程。这说明Dicer 对miRNA 通路的作用依赖于细胞质环境,但其机制尚不清楚。
在miRNA:miRNA* 双螺旋中,只有其中一条单链可以选择性结合到R I S C 上去而成为成熟miRNA ,随后另一条立即被降解。尽管从理论上来说,成熟miRNA 的产生是随机选择的结果,但由于两条链的稳定性有所不同,导致机会的不等。
图5是miRNA:miRNA* 双螺旋的结构,两条链的3'端均有2个游离核苷酸(2-nuc leot ide 3'overhangs)。此外,它的两条链是不完全配对的:miRNA 链上靠近5'端有一个不与miRNA*链相应位置配对的小突起。这个小突起显著地减弱了miRNA 链5'端的稳定性。由于成熟miRNA 的产生总是趋向于选择更不稳定的5'端[12~13],因此,miRNA 链
被选中的机会要大大多于miRNA*链(大约100倍)。结果往往是双链解开后miRNA 链结合到RISC 中以
作靶识别,而miRNA*链则迅速被降解。这样极度不对称的选择性的好处是,不会因为miRNA*链结合到RISC 中的比例过多而显著降低microRNA 对翻译的抑制效率。另外,成熟miRNA 5'端的8~10 nt 在与靶mRNA 的结合中比后12~14 nt 更为重要,这可能与5'端的小突起有关。反之,在极少的情况下,miRNA ∶miRNA* 双螺旋的两条链具有相似的稳定性,其分别结合到RISC 上的概率也相似,这已经为实验所证实[12]。同样地,如果改变siRNA 双链的结构,使两条链的稳定性有差异,则5′端更不稳定的那条链将更有可能结合到R I S C 上去[12~13]。
3 miRNA 功能意义
在植物中,大多数的miRNA 介导其靶mRNA 的降解。例如,最近发现的mir-196即介导其靶基因Hoxb8的mRNA 剪切[19]。植物中的miRNA 与相应的靶mRNA 近似完全配对,并且互补区域散布在靶mRNA 的转录区域内而非仅仅局限在3' UTR ,使得miRNA 会结合到包括编码区域在内的多个位点上去,从而直接降解mRNA 而非抑制其翻译。图1虚线箭头所示的即为这一功能。唯一例外的是mir-172,它在拟南芥的花朵发育中介导翻译抑制。与动物miRNA 不同的是,mir-172的互补位点与其靶基因APETALA 2(AP2)的互补位点落在编码区域而非3' UTR [20]。可以发现,植物中的miRNA 功能与siRNA 的功能非常相似。
在动物中,针对数种模式生物(人、小鼠、果蝇、线虫)的miRNA 所做的研究已经非常深入。最早发现的miRNA 基因是线虫中的lin-4,它编码一段22 nt 长度的RNA 片段,调控其靶基因lin-14[21~22]和lin-28[23]的表达,对线虫幼虫发育起重要的调节作用。第二个发现的是let-7,调控其靶基因lin-41[1,24]和hbl-1[25~26]的表达。在小鼠中的组织特异性筛选揭示了大约20种脑部特异性miRNA ,其中mir -124是含量最丰富的脑部特异性miRNA [27~28]。在果蝇中,mir-14抑制细胞死亡并参与脂肪代谢[29]。一些脑部特异性miRNA 在类维生素A 酸诱导的胚胎肿瘤细胞神经系统分化中处于上游调控的位置。在线虫中,lsy-6介导化学感受器左/右不对称表达[30]。此外,还猜测miRNA 在突触连接过程中起作用,例如在记忆形成过程中保证输入特异性的假想机制[31]。
图4 Dicer 域结构模式图(
线虫
DCR-1)
图5 miRNA ∶miRNA* 双螺旋
结构示意图
4生命科学第17卷
一些m i R N A所处的位点很特殊,例如mir-175的基因编码序列就位于与Waisman综合症相关的基因组区域[32],Waisman综合症是早期肇端帕金森病以及X染色体偶联智力缺陷的一种形式。
[参 考 文 献]
[1]Reinhart B J, Slack F J, Basson,M, et al. The 21-nucleotide
let-7 RNA regulates developmental timing in Caenorhabditis elegans.Nature, 2000, 403: 901~906
[2]Lee R C, Feinbaum R L, Ambros, V. The C.elegans
heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14.Cell, 1993, 75: 843~854
[3]Lee R C, Ambros V. An extensive class of small RNAs in
Caenorhabditis elegans. Science, 2001, 294: 862~864 [4]Reinhart B J ,Weinstein E G ,Rhoades M W, et al.
MicroRNAs in plants. Genes Dev, 2002, 16(13):1616~1626 [5]Lee Y, Ahn C, Han, J, et al. The nuclear RNase III Drosha
initiates microRNA processing. Nature, 2003, 425: 415~419
[6]Lund E, Guttinger S, Calado A, et al. Nuclear export of
microRNA precursors. Science, 303: 95~98
[7]Bernstein E, Caudy A A, Hammond S, et al. Role for a
bidentate ribonuclease in the initiation step of RNA interference.Nature, 2001, 409: 363~366
[8]Lee Y, Jeon K, Lee J T, et al. MicroRNA maturation: stepwise
processing and subcellular localization. EMBO J, 2002, 21: 4663~4670
[9]Hutv ag ner G , McLachlan J, Pasquinelli A E, et al. A cellular
function for the RNAinterference enzyme Dicer in the matu-ration of the let-7 small temporal RNA. Science, 2001, 293: 834~838
[10]Grishok A, Pasquinelli A, Conte D, et al. Genes and mecha-
nisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell, 2001106: 23~24
[11]Ketting R F, Sylvia E J, Bernstein F E, et al. Dicer functions
in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans.Genes Dev, 2001, 15: 2654~2659
[12]Schwarz D S, Hutvagner G, Du T, et al. Asymmetry in the
assembly of the RNAi enzyme complex. Cell, 2003, 115: 199~208
[13]Khvorova A, Reynolds A, Jayasena S D Functional siRNAs
and miRNAs exhibit strand bias. Cell, 2003, 115: 209~216 [14]Zeng Y, Cullen B R. Sequence requirements for microRNA
processing and function in human cells. RNA,2003,9: 112~123 [15]Lingel A, Simon B, Izaurralde E, et al. Structure and nucleic-
acid binding of the Drosophila Argonaute 2 PAZ domain.
Nature,2003, 426:465~469
[16]Song J J, Liu J, Tolia N H, et al. The crystal structure of the
Argonaute 2 PAZ domain reveals an RNA binding motif in
RNAi effector complexes. Nature Struct Biol, 2003, 10: 1026~1032
[17]Yan K S, Yan S, Farooq A, et al. Structure and conserved
RNA binding of the PAZ domain. Nature, 2003, 426: 468~674 [18]Carmell M A, Hannon G J. RNase III enzymes and the
initiation of gene silencing. Nature Struct Mol Biol, 2004, 11: 214~118
[19]Yekta S, Shih I H, Bartel D P. MicroRNA-directed cleavage
of HOXB 8 mRNA.Science, 2004, 304: 594~596
[20]Chen X A MicroRNA as a translational repressor of
APETALA 2 in Arabidopsis flower development. Science, 2004, 303: 2022~2025
[21]Lee R C, Freinbaum R L, Ambros V. The C. elegans
heterochronic gene lin-4 encodes small RNAs with antisense complimentarity to lin-14. Cell, 1993, 75: 843~854 [22]Wightman B, Ha I, Ruvkun G. Posttranscriptional regula-
tion of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans.Cell , 1993, 75: 855~862
[23]Moss E G, Lee R C, Ambros V. The cold shock domain
protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell, 1997, 88: 637~646 [24]Slack FJ, Basson M, Lin Z, et al. The lin-41 RBCC gene acts
in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor.Mol Cell, 2000, 5: 659~669
[25]Abrahante JE, Daul AL, Li M, et al. The Caenorhabditis
elegans hunchback-like gene lin-57/hbl-1 controls develop-mental time and is regulated by microRNAs. Dev Cell, 2003, 4: 625~637
[26]Lin SY, Johnson SM, Abraham M, et al. The C aenorhabditis
elegans hunchback homolog, hbl-1, controls temporal pat-terning and is a probable microRNA target. Dev Cell, 2003, 4: 639~650
[27]Lagos-Quintana M, Rauhut R, Lendeckel W, et al. Identifi-
cation of tissue-specific microRNAs from mouse. Curr Biol, 2003, 4: R42
[28]Sempere LF, Freemantle S, Pitha-Rowe I, et al. Expression
profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol, 2004, 5: R13
[29]Xu P, Vernooy S Y, Guo M, et al. The Drosophila microRNA
Mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol, 2003, 13: 790~795
[30]Johnston R J, Hobert O. A microRNA controlling left/right
neuronal asymmetry in Caenorhabditis elegans. Nature, 2003, 426: 845~49
[31]Martin K C, Kosik K S. Synaptic tagging – who’s it? Nat Rev
Neurosci, 2002, 3: 813~820
[32]Dostie J, Mourelatos, Yang M, et al. Numerous microRNPs
in neuronal cells containing novel microRNAs. RNA, 2003, 9: 180~186
植物基因功能研究方法的新进展
植物基因功能诠释研究方法的新进展 (东北农业大学,150030) 摘要:本文通过阅读大量的文献,总结了植物基因功能注释研究方法的最新进展。对每种方法的原理及优缺点做了综述,拟供初学者和作相关研究者参考。 关键词:基因功能;研究方法;新进展 基因组研究应该包括两方面的内容:以全基因组测序为目标的结构基因组学(struc tural genomics)和以基因功能鉴定为目标的功能基因组(functional genomics)。结构基因组学代表基因组分析的早期阶段,以建立生物体高分辨率遗传、物理和转录图谱为主。功能基因组学代表基因分析的新阶段,是利用结构基因组学提供的信息系统地研究基因功能,它以高通量、大规模实验方法以及统计与计算机分析为特征。功能基因组学(functional genomics)又往往被称为后基因组学(postgenomics),它利用结构基因组所提供的信息和产物,发展和应用新的实验手段,通过在基因组或系统水平上全面分析基因的功能,使得生物学研究从对单一基因或蛋白质的研究转向多个基因或蛋白质同时进行系统的研究。[1,2]这是在基因组静态的碱基序列弄清楚之后转入基因组动态的生物学功能学研究。研究内容包括基因功能发现、基因表达分析及突变检测。基因的功能包括:生物学功能,如作为蛋白质激酶对特异蛋白质进行磷酸化修饰;细胞学功能,如参与细胞间和细胞内信号传递途径;发育上功能,如参与形态建成等采用的手段包括经典的减法杂交,差示筛选,cDNA代表差异分析以及mRNA差异显示等,但这些技术不能对基因进行全面系统的分析。新的技术应运而生,包括基因表达的系统分析,cDNA微阵列,DNA芯片等。鉴定基因功能最有效的方法是观察基因表达被阻断或增加后在细胞和整体水平所产生的表型变异,因此需要建立模式生物体。 自华大基因启动“千种动植物基因组参考序列谱构建计划”和“千种植物转录组研究”以来,已完成水稻、黄瓜、马铃薯、白菜等植物的基因组序列图谱绘制,并通过对大豆的重测序研究建立了高密度分子标记图谱。这将是21世纪生命科学研究的重要领域。[3]本文将对研究基因功能的新技术及其新进展作一综述。 1 利用生物信息学方法分析基因的功能 生物信息学是利用生物信息学和电子技术(互联网技术)寻找并克隆新的未知功能的基因,着重于技术和操作层面,利用生物信息学对新基因进行电子克隆,及克隆该新基因的序列后对其进行简单的功能分析,如基因的编码区、启动子区、内含子/外显子、翻译启始位点和翻译终止信号预测,基因的同源比对,编码的氨基酸辨识蛋白质,蛋白质的物理性质,蛋白质的二级/三级结构、特殊局部结构以及功能预测等[4]。 1.1 通过序列比对预测基因功能
植物叶绿体发育研究进展
Botanical Research 植物学研究, 2018, 7(6), 627-633 Published Online November 2018 in Hans. https://www.360docs.net/doc/1b6443299.html,/journal/br https://https://www.360docs.net/doc/1b6443299.html,/10.12677/br.2018.76077 Advances in Research on Plant Chloroplast Development Baohua Jin, Qianwen Ge Zhejiang Normal University, Jinhua Zhejiang Received: Nov. 14th, 2018; accepted: Nov. 23rd, 2018; published: Nov. 30th, 2018 Abstract Chloroplasts are important organelles for photosynthesis of green plants, and they are highly concerned by researchers. At present, there is a certain research basis for the structure and func-tion of chloroplasts, but the molecular mechanism of chlorophyll metabolism and regulation in specific chloroplast development remains to be further studied. This article summarizes the de-velopment of chloroplasts, the synthesis of chlorophyll and catabolism, in order to better promote the understanding of chloroplasts. Keywords Chloroplast, Photosynthesis, Chlorophyll 植物叶绿体发育研究进展 金宝花,葛倩雯 浙江师范大学,浙江金华 收稿日期:2018年11月14日;录用日期:2018年11月23日;发布日期:2018年11月30日 摘要 叶绿体是绿色植物进行光合作用的重要细胞器,其备受研究学者的关注。目前对叶绿体的结构、功能等已有一定的研究基础,但具体叶绿体发育中叶绿素的代谢过程及调控的分子机制仍有待深入研究。 本文从叶绿体的发育、叶绿素的合成以及分解代谢等方面进行概述,以期能更好地促进人们对叶绿体的了解。
野木瓜及其同属植物的研究进展
综述: 野木瓜属植物的研究进展 摘要:本文综述了木通科野木瓜属植物的研究进展。概述了野木瓜植物的化学成分、药理性质及质量控制的研究。为野木瓜属植物的进一步药用开发和药源扩展提供参考。 关键词:野木瓜属植物;化学成分;药理性质;质量控制 一.野木瓜本草及基源 “野木瓜”名称始见于明·朱棣的《救荒本草》[1],云:“野木瓜,一名八月,又名杵瓜。出新郑县山野中。蔓延而生,妥附草木上。叶似黑豆叶,微小光泽,四五叶搌生一处,结瓜如肥皂大,味甜。古代本草中记载野木瓜属植物的只有清·吴淇俊的《植物名实图考》[1],曰:“五爪金龙产南安,横根抽茎,茎叶俱绿。就近生小枝,一枝五叶,分布如爪;叶长二寸许,本宽四五分,至末渐肥。复出长尖,细纹无齿,根褐色,硬如萆。”。。。。 1. 野木瓜种属的归类及分布 1.1野木瓜种属的归类 野木瓜[1]Stauntonia系木通科( Lardizabalacea) 9个属的其中一属,该属植物通常为常绿木质藤本,大部分全株及果实皆可入药。其性微苦,平。具镇痛、治咳嗽和痢疾之功,主治风湿痹痛,腰腿疼痛,头痛,牙痛,痛经,跌打伤痛,肠胃炎等症。多数种类为药食两用植物,浆果多汁味甜, 可生食、制果酱和酿酒, 种子榨油, 供食用和工业用。 该属约20余种植物, 我国有23种(2个亚种),本属植物分两个亚属,即有蜜腺状花瓣存在的野木瓜亚属和无蜜腺状花瓣的无瓣亚属。野木瓜亚属有:Stauntonia chinensis DC.、翅野木瓜Staun tonia decora(Dunn)C. Y. Wu.、三叶野木瓜Stauntonia brunoniana Wall. ex Hemsl.、斑叶野木瓜Stauntonia maculata Merr.牛藤果Stauntonia elliptica Hemsl. 野木瓜无瓣亚属按其花药顶端附属体的形状又
植物功能组研究进展
程论文(作业)封面(2011 至2012 学年度第 2 学期)课程名称:_ ___ 课程编号:___________ 学生姓名:__ ________ 学号:_______ 年级:__ ___________ 任课教师: _ ____________ 提交日期:年月日成绩:__________________ 教师签字:__________________ 开课---结课:第周---第周评阅日期:年月日
植物的功能基因组学研究进展 摘要:基因组研究计划包括以全基因组测序为目标的结构基因组学和以基因功能鉴定为 目标的功能基因组学两方面的内容。目前基因功能鉴定的方法主要有:基因表达的系统分析(SAGE) 、cDNA 微阵列、DNA(基因) 芯片、蛋白组技术以及基于转座子标签和T-DNA 标签的反求遗传学技术等。本文对上述各种技术的优缺点以及它们在植物基因功能鉴定中的应用进行了综述。 关键词:功能基因组学; 基因表达的系统分析;cDNA 微阵列;DNA 芯片;蛋白组 以拟南芥和水稻为代表的植物基因组研究已取得了迅速的进展,到目前为止,占拟南芥基因组(100Mb) 近三分之一的DNA 序列已被测定并在GenBank 数据库中登记注册,预期到2001 年通过全球合作将完成拟南芥全基因组的序列测定工作。随着植物基因组计划的实施和进展,GenBank 中累积了大量的未知功能的DNA 序列,如何鉴定出这些基因的功能将成为基因组研究的重点课题, 因此, 基因组研究应该包括两方面的内容: 以全基因组测序为目标的结构基因组学(structural genomics) 和以基因功能鉴定为目标的功能基因组研究, 后者往往又被称为后基因组研究。功能基因组研究的内容是利用结构基因组所提供的信息, 发展和应用新的实验手段系统地分析基因的功能〔1 〕。目前人类和酵母的功能基因组研究已经全面展开, 尤其是对已完成全基因组测序的酵母来说, 其功能基因组研究任务更加紧迫。植物的基因组研究虽然起步较晚, 但由于吸取了人类基因组研究中积累的一些经验, 所以进展也相当迅速, 对植物功能基因组学的研究目前也已经受到重视, 在1998 年12月出版的最新一期Plant Cell (10 :1771) 和Plant Physiol . (118 :713) 上均编发了关于植物功能基因组学研究的编者按, 并由Bouchez 和Hofte (1998) 〔2 〕综述了植物尤其是拟南芥功能基因组学研究的现状, 本文在此基础上综述了目前植物功能基因组学研究中使用的主要技术手段以及最新的研究进展。 1 基因功能的含义 基因的功能主要包括: 生物化学功能, 如作为蛋白质激酶对特异的蛋白质进行磷酸化修饰; 细胞学功能, 如参与细胞间和细胞内的信号传递途径; 发育上的功能, 如参与形态建成等。目前,获得一段DNA 序列的功能信息的最简单的方法是将该DNA 序列与GenBank 中公布的基因序列进行同源性比较,如利用BLASTn 和BLASTx 两种软件分别进行核苷酸和氨基酸序列同源性比较等。同源性比较的结果大体可以分为如下类型: 与生化和生理功能均已知的基因具同源性; 与生化功能已知的基因具同源性, 但该基因的生理功能未知;与其它物种中生化和生理功能均未知的基因具同源性; 虽与生化和生理功能均已知的基因具同源性, 但对该基因功能的了解尚不深入, 仍停留在表观现象上。上述同源性检索分析方法仅仅为该DNA 片段的功能提供了间接的证据,对基因功能的直接证据还需要实验上的数据。Bouchez 和Hofte (1998)〔2 〕将所需要的实验证据归纳如下: (1) 通过研究基因的时空表达模式确定其在细胞学或发育上的功能, 如在不同细胞类型、不同发育阶段、不同环境条件下以及病原菌侵染过程中mRNA 和/ 或蛋白质的表达的差异等。(2) 研究基因在亚细胞内的定位和蛋白质的翻译后调控等。(3) 利用基因敲除(knock - out) 技术进行功能丧分析或通过基因的过量表达(转基因) 进行功能获(gain2of2function) 分析,进而研究目的基因与表型性状间的关系。(4) 通过比较研究自发或诱发突变体与其野生型植株在特定环境条件下基因表达的差异来获取基因功能的可能信息。 2 植物的表达序列标记(EST) 与基因组大规模测序 通过从cDNA 文库中随机挑取的克隆进行测序所获得的部分cDNA 的5′或3′端序列称为表达序列标记( EST) ,一般长300~500bp 左右, 利用EST作为标记所构建的分子遗传图
植物研究进展论文
研究生课程论文 题目:PCR-DGGE在真菌研究中的应用 学院生命科学学院 课程名称植物学科研究进展 专业年级植物学2014级 学号 20141069 姓名成斌 2015年 1月 20日
PCR-DGGE在真菌研究中的应用 成斌 (河北大学生命科学学院河北保定071002) 摘要:变性梯度凝胶电泳(DGGE)在真菌研究中技术应用的主要步骤:从样品中直接提取真菌DNA,选取5′端含GC夹的特异性引物对18S rDNA或IST序列等的部分片段进行扩增,得到合适的目的DNA片段,并在变性梯度的聚丙烯酰胺凝胶中进行电泳,使不同来源的真菌DNA 片段有效分离,再进行各种分类分析。 关键词:PCR-DGGE;变性梯度凝胶;真菌;引物;DNA 丛枝菌根真菌(Arbuscular mycorrhizal fungi, AMF)在自然界分布广泛,能够与80%以上的陆生植 物形成共生体[ 1 - 2 ]。它能够提高植物抗旱性、抗病性,促进生长,提高产量,改善作物矿质营养,被誉为“生物肥料”[ 3 ]。由于AM真菌至今仍然不能被纯培养,给菌种鉴定、遗传学以及群落生态学研究等带来不少难题[ 4 ]。随着分子生物学技术的不断发展,各种分子技术已被应用到AM真菌研究中。目前,国外已在这一领域进行了大量的探索和研究,而国内在该领域研究则进展缓慢[ 5 ]变性梯度凝胶电泳(Denaturing Gradient Gel Electrophoresis,DGGE)是在含有浓度线性递增变性剂的聚丙烯酰胺凝胶电泳中,将具有不同碱基序列而长度相似的双链DNA分离[8]。变性梯度凝胶电泳技术是由Fischer和Lerman于1979年最先提出的用于检测DNA突变的一种电泳技术[ 7 ]。后来,该技术逐渐被应用于微生物生态学研究,并证实了这种技术在研究自然界微生物群落结构变化、遗传多样性和种群差异方面具有明显的优越性,并且该方法能够较准确地反映出环境样品中优势种 群的动态变化规律[ 8 - 9 ]。目前,在原核生物生态学研究中DGGE技术已发展的较为成熟。然而,将其应用于真核生物生态学研究的报道,并不多见[ 5 ]。 1 样品DNA的提取 从样品中提取DNA 的产率,直接决定了DGGE 条带的代表性[14]。DNA产率低,其条带的代表性就差。真菌分布广泛,种类繁多,为获得较高的DNA 提取产率,DNA 提取方法也需要根据样品的特性具体分析,寻找针对性较强的处理方法。 1.1 土壤样品中真菌DNA 的提取 对于土壤样品DNA的提取,通常会采用改良后的Bead-Beating 法[5]。具体方法是:称取10 g土样,
芸薹属植物比较基因组学研究进展
植物学通报Chinese Bulletin of Botany 2007, 24 (2): 200?207, https://www.360docs.net/doc/1b6443299.html, 收稿日期: 2006-05-26; 接受日期: 2006-08-26 * 通讯作者。E-mail: yuanbeauty@https://www.360docs.net/doc/1b6443299.html, .专题介绍. 芸薹属植物比较基因组学研究进展 李媛媛, 傅廷栋, 马朝芝* 华中农业大学作物遗传改良国家重点实验室, 武汉 430070 摘要 芸薹属(Brassica )植物是双子叶植物比较基因组学研究的重点对象。经过十几年的研究, 芸薹属植物比较基因组学研究已取得很大进展。宏观共线性和微观共线性两个层次的研究均发现, 芸薹属植物之间以及芸薹属和拟南芥之间都存在广泛的共线性, 表明拟南芥信息在芸薹属中具有重要应用价值。芸薹属作物基因组内存在着多个拷贝的共线性区域, 支持二倍体芸薹属作物起源于多倍体祖先的假设。 关键词 芸薹属, 比较基因组, 拟南芥, 宏观共线性, 微观共线性 李媛媛, 傅廷栋, 马朝芝 (2007). 芸薹属植物比较基因组学研究进展. 植物学通报 24, 200?207. 比较基因组学(comparative genomics)又称比较遗传学, 是指在不同物种之间利用共同的标记构建图谱或对不同物种基因组相应部分(或全部)区域进行测序, 比较它们之间的基因数目、相对位置、结构关系等, 以揭示不同物种之间的基因家族成员数目和排列顺序的异同。一般来讲, 比较基因组学主要包括两个方面: 基于遗传图谱的宏观共线性和基于物理图谱或测序的微观共线性。目前, 禾本科植物的比较基因组研究最为透彻,而芸薹属(Brassica )植物则是双子叶植物比较基因组学研究的重点对象。从20世纪90年代至今, 经过十几年的历程, 芸薹属植物比较基因组学研究已在宏观共线性和微观共线性两方面都取得了较大进展。 1 芸薹属植物基因组概况 芸薹属是十字花科(Cruciferae)植物中最重要的一个属,包含许多有重要经济价值的油料、蔬菜和饲料作物。从细胞遗传学角度讲, 芸薹属栽培种包括白菜(B. rapa ;AA , 2n = 20)、甘蓝(B. oleracea ; CC , 2n = 18)和黑芥(B. nigra ; BB , 2n = 16) 3个二倍体基本种以及甘蓝型油菜(B. napus ; AACC , 2n = 38)、芥菜型油菜(B.juncea ; AABB , 2n = 36)和埃塞俄比亚芥(B. carinata ; BBCC , 2n = 34) 3个四倍体复合种。种间人工合成的研究结果表明, 白菜、甘蓝和黑芥为3个基本染色体种,它们通过相互杂交和自然加倍而形成了现在的四倍体种,这就是著名的禹氏三角(U, 1935)。通过对核DNA 含量的计算, 推测二倍体芸薹属基因组约为拟南芥基因组(125 Mb)的3-5倍, 而四倍体芸薹属基因组则是拟南芥基因组的10倍左右(Bennett and Sm ith, 1976;Arumuganathan and Earle, 1991)。 2 芸薹属植物比较遗传图谱 比较遗传作图是利用一个种的基因或者基因的部分片段或者遗传标记, 通过遗传学的方法在其它的物种中寻找其同源序列及构建相应的遗传标记图。芸薹属植物比较遗传图谱研究可对芸薹属植物之间的结构、亲缘关系及其进化演变提供分子水平的证据; 特别是芸薹属和拟南芥的比较遗传作图, 将大大增加芸薹属中可供利用的遗传标记。近年来, 芸薹属植物之间以及芸薹属植物与拟南芥之间的比较遗传作图研究都取得了一些重要结果。 2.1 芸薹属植物之间的比较作图 芸薹属不同种基因组的比较研究首先是在白菜和甘蓝之
植物基因组学的的研究进展
基因组学课程论文 题目:植物基因组学的的研究进展姓名:秦冉 学号:11316040
植物基因组学的的研究进展 摘要:随着模式植物——拟南芥和水稻基因组测序的完成,近年来关于植物基因组学的研究越来越多。本文主要对拟南芥、水稻2种重要的模式植物在结构基因组学、比较基因组学、功能基因组学等领域的研究进展以及研究所使用的技术方法进行简单介绍。 关键词:植物;基因组学;研究进展 The recent progress in plant genomics research Abstract: With the completion of genome sequencing ofthe model plant-- Arabid opsis and rice,more and more researches on plant genomics emerge in recent yea rs. The research progress of the 2 important model plant--Arabidopsis and rice in structural genomics,comparative genomics,functional genomics and technology methods used in this research are introduced briefly in this paper. Keywords:plant; genomics; research advances 前言 基因组是1924年提出用于描述生物的全部基因和染色体组成的概念。1986年由美国科学家Thomas Roderick提出的基因组学是指对所有基因进行基因组作图(包括遗传图谱、物理图谱、转录本图谱)、核苷酸序列分析、基因定位和基因功能分析的一门科学。自从1990年人类基因组计划实施以来,基因组学发生了翻天覆地的变化,已发展成了一门生命科学的前沿和热点领域。而植物基因组研究与其他真核生物和人类基因组研究有很大的不同。首先,不同植物的基因组大小即使在亲缘关系非常近的种类之间差别也很大; 其次,很多植物是异源多倍体,即便是二倍体植物中有些种类也存在较为广泛的体细胞内多倍化( endopolyp loidy)现象[1]。基因组研究主要包括三个层次:①结构基因组学,以全序列测序为目标,构建高分辨率的以染色体重组交换为基础的遗传图谱和以DNA 的核苷酸序列为基础的物理图谱。②功能基因组学,即“后基因组计划”,是结构基因组研究的延伸,利用结构基因组提供的遗传信息,利用表达序列标签,建立以转录图谱为基础的功能图谱( 基因组表达图谱),系统研究基因的功能,植物功能基因组学是当前植物学最前沿的领域之一。③蛋白质组学,是功能基因组学的深入,因为基因的功能最终将以蛋白质的形式体现。 近来,以水稻( Oryza sativa)和拟南芥(Arabadopsis thaliana)为代表的植物基因组研究取得了很大进展,如植物分子连锁遗传图谱的构建,在此基础上,已经在植物基因组的组织结构和基因组进化等方面得到了有重要价值的结论; 植物基因组物理作图和序列测定的研究集中于拟南芥和水稻上; 植物比较基因组作图证实在许多近缘植物甚至整个植物界的部分染色体区段或整个基因组中都存在着广泛的基因共线性,使得我们可以利用同源性对各种植物的基因组结构进行研究、分析和利用。本文主要对拟南芥、水稻2种重要的模
植物细胞融合的研究进展_综述_郭学民
河北科技师范学院学报 第19卷第1期,2005年3月 Jo ur nal o f Hebei N or mal U niver sity of Science&T echnolog y Co llege V o l.19 No1.1M arch2005 植物细胞融合的研究进展(综述) 郭学民1,2,徐兴友1,2,王同坤1,王华芳2,尹伟伦2 (1河北科技师范学院生命科学系,河北秦皇岛,066600;2北京林业大学生物科学与技术学院)摘要:概述了原生质体分离和培养的影响因素,介绍了近年来国内外原生质体培养与融合及杂种细胞、筛选和鉴定的动态。 关键词:细胞融合;原生质体;筛选与鉴定 中图分类号:Q321+.2 文献标识码:A 文章编号:1672-7983(2005)01-0065-05 细胞融合(cy to mixis),亦称细胞杂交(cell fusio n),是指亲本的两个细胞在特定的物理和化学因子处理下合并为一个杂种细胞的过程[1]。植物细胞融合可分为体细胞杂交(somatic hybridizatio n)和配子-体细胞杂交(gameto-somatic hy br idizatio n),前者是指不经过有性过程,而直接由体细胞原生质体融合产生杂种细胞,形成愈伤组织,并再生出植株的过程[2],后者是指性细胞(如小孢子四分体、精子、精细胞、幼嫩花粉、成熟花粉、卵细胞、助细胞和中央细胞等)原生质体和二倍体原生质体融合产生三倍体杂种细胞,形成愈伤组织,并再生出植株的过程[3]。植物细胞融合是植物细胞工程的一个重要分支,是一种突破物种生殖隔离、创造远缘杂种的新途径,原生质体技术还可用于细胞突变体的筛选、细胞器移植和外源DNA的导入。 自1960年Cocking[4]用酶法分离出番茄根原生质体后,Natag a和T akebe[5]1970年首次利用烟草叶分离原生质体,经培养获得再生植株;1975年以色列的Vardi等[6]首次从木本植物Sham onti甜橙珠心组织诱导胚性愈伤组织,并从愈伤组织分离原生质体,经培养通过胚状体再生出植株;在禾本科植物中,除在珍珠谷、紫狼尾草用悬浮细胞为材料,较早获得原生质体再生植株外,直到1985年Fujim ur a[7]等率先在水稻原生质体培养中获得了再生植株,才出现了重大突破。现已从许多种内、种间、属间甚至亚科间的体细胞杂交获得杂种细胞系或杂种植株。随着多种植物原生质体的成功培养和融合技术的不断改进,植物细胞融合获得了巨大成功。植物细胞融合包括原生质体的制备、细胞融合的诱导、杂种细胞的筛选和培养,以及植株的再生和鉴定等环节。 1 原生质体的分离和培养 1.1 起始材料 起始材料及其生理状态对原生质体的制备及其活力有很大的影响。在以往的双子叶植物培养中,大多以叶片为分离原生质体的材料,近年来,起始材料的适用范围有了较大扩展。目前,以愈伤组织、悬浮细胞和体细胞胚为材料制备原生质体是最主要的方式;禾本科植物原生质体培养获得成功的试验,几乎都是用从幼胚或成熟胚诱导形成的胚性愈伤组织或胚性细胞系来游离原生质体。采用这些材料制备原生质体方法简便、产量高、不污染、不易破碎。 1.2 基因型 同一植物不同基因型的原生质体脱分化与再分化所要求的条件不同,所以在相同条件下,不同品种的再生能力不同。王光远和夏镇澳[8]在水稻原生质体培养中曾用26个品种进行组织培养,其中仅有3个品种(粳稻农虎6号、国香1号和上农香糯)能成功地用于原生质体培养,获得再生植株。据统计,小麦获得原生质体再生植株的基因型只有大约10个[9]。基因型的选择在植物原生质体培养中起着重要作用,它不仅影响原生质体的产量和活力,而且还影响植株的再生。Cheng和Veillenux证明芙薯(Solanum phureja)从原生质体培养到愈伤组织形成受2个独立位点的显性基因的调控[10]。因此,现有 收稿日期:2004-03-09;修改稿收到日期:2004-12-12
植物基因组学
1.基因组的结构和变异 2.分子标记连锁图谱构建基因 3.QTL定位的原理和方法 4.QTL精细定位 5.基因和QTL的可隆 5.1插入突变方法 5.2图位克隆的方法(含比较图位克隆) 5.3候选基因法 6.资源评估和利用 7.分子标记辅助选择(含分子设计育种) 8.转基因 8.1转基因体系和实证研究 8.2转基因的生态学安全研究 9.比较基因组 9.1标记水平比较基因组 9.2序列水平的比较研究 9.3性状水平的比较研究 9.4功能比较研究 10.***优势研究 10.1遗传学解释 10.2分子生物学解释 11.分子进化(主要是玉米进化) 12.基于连锁不平衡的关联分析 12.1实证研究 12.2方法学研究 13.基因组研究中的一些新技术运用 13.1DNA芯片技术 13.2 DNA shuffling 13.3Gene Trap 13.4 Gene therapy in plants 13.5 TILLING 技术 1.植物基因组的结构和变异 在越来越多的植物基因组被测完后,该研究的重要性逐渐显现,该方面的文章可以说是汗牛充栋.在玉米方面该领域的大牛是Buckler, ES; Messing, J, Dooner HK, Doebley J ; Gaut, BS. 1. Buckler, E. S., Gaut, B. S. and McMullen, M. D. (2006) Molecular and functional diversity of maize. Curr. Opin. Plant Biol. 9, 172-176 这是关于玉米基因组结构的REVIEW文章,先了解大概,在细读研究文章.其任何2个玉米自交系之间的遗传变异大于人和大猩猩之间的差异的经典论断充分说明玉米变异的广泛性.最近因为人类基因组研究的进展而似乎可以改写. 2.Messing J, Dooner HK. Organization and variability of the maize genome. Curr Opin Plant Biol.
紫薇属植物研究进展
园 艺 学 报 2007,34(1):251-256 Acta Horticulturae Sinica 紫薇属植物研究进展 张 洁1,2,王亮生13,张晶晶1,舒庆艳1,高锦明2 (1中国科学院植物研究所北京植物园,北京100093;2西北农林科技大学国家生命科学与技术人才培养基地,陕西杨凌712100) 摘 要:系统回顾了有关紫薇属植物种质资源、栽培繁殖及资源可持续利用等方面的研究进展,突出阐述了花色、花香、花型等重要观赏性状的研究现状,提出了保存种质资源,丰富花色花香,选育优良品 种,研究药用成分是今后的主要研究方向。 关键词:紫薇属;种质资源;花色;育种;药用成分;综述 中图分类号:S68 文献标识码:A 文章编号:05132353X(2007)0120251206 Advances i n Stud i es on Genus L agerstroem ia ZHANG J ie1,2,WANG L iang2sheng13,Z HANG J ing2jing1,SHU Q ing2yan1,and G AO J in2m ing2 (1B eijing B otanical Garden,Institute of B otany,the Chinese A cade m y of Sciences,B eijing100093,China;2The N ational B ase of L ife Science and B iotechnology Education,N orthw est A&F U niversity,Yangling,Shaanxi712100,China) Abstract:The advances in researches on ger mp las m res ources conservati on,cultivati on,p r opagati on and sustainable utilizati on of crape myrtle(genus L agerstroe m ia)were revie wed.I n the meanti m e,the p r ogress of orna mental traits such as p lant shape,fl ower col or,scent and for m were studied.I n the future,it should be e mphasized t o carry out research on conserving core collecti on,enriching fl ower col or and scent,selecting and breeding novel cultivars,and deepening studies on their phar maceutical components. Key words:L agerstroe m ia;Ger mp las m;Fl o wer col or;B reeding;Phar maceutical components;Re2 vie w 紫薇(L agerstroe m ia indica),别名“百日红”、“满堂红”、“痒痒树”,炎夏少花季节开花,花期长达3个多月,在园林绿化中得到广泛的应用,也适用于盆栽和切花观赏,少数种类还是我国及东南亚国家传统的治疗糖尿病和咳嗽等常见多发病的药用植物。 1 资源学研究 紫薇属于桃金娘目千屈菜科(Lythraceae),主要分布在亚洲东部至南部和澳大利亚的北部(陈俊愉,2001)。1795年,查尔斯?林奈为了纪念朋友Magnus von Lagerstr oe m,首次对紫薇属植物命名,因错认为其起源于印度,将其命名为L agerstroe m ia ind ica,但事实上,紫薇属起源于中国的南部和西部(中国科学院中国植物志编辑委员会,1983)。在美国,紫薇被称为crape myrtle(常拼写为‘crape myrtle’或‘crepe2myrtle’),其原因可能是紫薇的褶皱状花瓣像绉纸(crepe paper),叶子像桃金娘科(Myrtaceae)植物M yrtus co mm un is(htt p://dallas1ta mu1edu/woody/c myrtle/comna mes1ht m l)。111 野生种质资源研究 大量文献记载,全世界紫薇属植物约有55种(王献,2004b)。但国际植物名称检索表(I nterna2 ti onal Plant Na mes I ndex)报道有近80种,并详细列出了种、变种、亚变种、起源国及最初文献,同收稿日期:2006-06-27;修回日期:2006-12-12 基金项目:教育部留学回国人员科研启动基金资助项目(2005383) 3通讯作者Author f or corres pondence(E2mail:wanglsh@ibcas1as1cn)
植物抗逆基因研究进展_杨柳
植物抗逆基因研究进展 杨 柳1,2,张振乾1,2,宋继金3,谭太龙1,2,官春云1,2,刘忠松1,2 (1湖南农业大学农学院,长沙410128;2国家油料改良中心湖南分中心,长沙410128; 3芷江县农业局粮油站,湖南芷江419100) 摘 要:干旱、高温、低温、高盐等极端条件对植物生长造成严重的危害,对农业生产造成相当大的影响。为了减轻其不利影响,目前采用分子生物学的方法,在基因组成、表达调控及信号转导等分子水平上认识植物抗逆机理,通过基因工程手段导入抗逆相关基因,改良作物的胁迫抗性。综述了植物抗逆相关基因的克隆、功能验证以及应用等方面的进展。 关键词:植物;抗逆性;基因工程 中图分类号:Q789 文献标识码:A 文章编号:1001-5280(2010)02-0126-04 干旱、盐碱、金属离子和低温等逆境条件严重抑制植物生长发育,会引起植物植株生理生化、形态等方面的变化,甚至死亡。因此,开展抗逆研究、提高作物抗逆能力,能够使作物增产稳产。随着分子生物学的发展,借助分子生物学手段,从基因组成、表达调控及信号转导等方面进行深入研究,揭示抗逆的分子机理,并导入相关基因改良作物的胁迫抗性,近年来取得较大进展,在农作物抗逆育种上展示出了广阔的应用前景。 1 抗旱 当植物耗水大于吸水时,会使组织内水分亏缺,过度水分亏缺的现象称为干旱。旱害则是指土壤水分缺乏或大气相对湿度过低对植物的危害。干旱对植物生产的不利影响主要有:(1)降低细胞含水量,破坏细胞膜系统;(2)增加透性,降低光合作用;(3)使植物的物质代谢紊乱,生长发育迟缓、死亡。干旱胁迫可激活相关基因,如LEA蛋白、抗氧化酶和水孔蛋白等的转录,并导致编码蛋白的积累。植物抵抗旱害的能力称为抗旱性。 1.1 晚期胚胎发生富集蛋白(LEA蛋白) LEA蛋白在植物胚胎发育后期的种子中大量积累,低温、重金属、高盐,特别是干旱等逆境刺激均能诱导其转录和累积。LEA蛋白在逆境中有脱水保护、渗透调节和清除自由基活性等作用[1,2]。LEA蛋白有LEA1~LE A66类,其中LE A1~LEA3与植物抗逆 收稿日期:2010-04-24 作者简介:杨 柳(1982-),女,湖南岳阳人,硕士,从事油菜育种研究。通讯作者:刘忠松。 基金项目:国家科技支撑计划项目(2009BAD A8B01)。性相关。 Cheng等[1]利用基因枪法转化水稻成熟胚愈伤组织,获得转基因水稻株系,分析表明LEA2有较强的抗脱水作用。Stra ub等[3]研究发现,大麦HV A1(LEA3同源蛋白)与种子的干旱脱水有关,且干旱、极端温度及盐胁迫均可诱导其迅速在幼苗中表达。Xu等[4]研究转HV A1基因水稻发现,T2代对快速水分胁迫和高盐的耐受性较强,且与HV A1蛋白积累水平呈正相关,说明H V A1蛋白可防止干旱胁迫对细胞膜的损伤[5],有潜在的抗旱作用[6,7]。 1.2 水分通道蛋白(Aquaporin) 水孔蛋白存在于所有器官组织中,可分为4类[8],同一器官可表达多种不同类型的水孔蛋白[9]。水孔蛋白表达具有组织特异性,并受发育阶段和环境因素的调节。 野生马铃薯水孔蛋白ScP IP2a基因m RN A与果实成熟中细胞的扩张作用相一致[10],冰草M IP2A可能控制水分从木质部薄壁细胞流向木质部导管腔[11]。向日葵液泡水孔蛋白SunTIP7转录物在干旱胁迫下积累,表明其与干旱有关[12]。Kaldenhoff等[13]利用反义基因技术抑制拟南芥质膜PIP1b的表达,PIP1a的表达也被抑制,原生质体透性降低为对照的1/3,而根量增加5倍,说明根量的增加弥补了水孔蛋白数量的不足和导水率的降低。PIP2在拟南芥根皮层中大量表达导致根皮层水导度、渗透作用及伤流降低[14]。 1.3 Rubisco活化酶 Rubisco活化酶表达量的增加也可增强植物的抗旱性能[15]。干旱情况下,植物体内的ATP含量降低, Rubisco活化酶活性也随之降低,为减小干旱造成的不利影响,Rubisco活化酶的表达增强[16]。
植物抗逆性的研究进展1
植物抗逆性的研究进展 摘要:在自然条件下生长的植物,经常会面临干旱、涝害、盐碱、高温、低温、冷害、营养匮乏、重金属污染等非生物胁迫,严重影响了植物的正常生长和发育。而干旱影响了植物的正常生长发育,导致植被减少和草地退化等现象,也是影响农作物产量的主要胁迫因素之一。本文就植物在遭受干旱胁迫时,其生长指标、生理生化指标以及其他酶类变化,对研究中存在的问题及应用前景进行了讨论。 关键词:植物;抗旱机制;生长指标;生理生化指标;基因工程 干旱问题已经成了全国乃至全世界高度重视的问题,它导致了植被的减少、河流断流、土地沙化、沙尘暴频发、湖泊湿地萎缩、草地退化、森林锐减、生物量和生物多样性急剧下降[1],这些都影响了人类赖以生存的生态环境安全和社会的可持续发展,因此,全面深入的研究植物抗旱机制有利于提高植物对水分胁迫的耐受性,提高植物的抗旱能力,解决干旱半干旱地区植被的恢复与重建以及农作物产量低下等问题,同时也有利于选择和培育出耐旱性更强的品种。本文就植物在遭受干旱胁迫时,其生长指标、植物生理生化指标以及其他酶类变化,对研究中存在的问题及应用前景进行了讨论。 1生长指标与抗旱机制 水是植物生长必不可少的环境因素,当植物收到干旱胁迫时,植物的生长指标的变化就能反映出植被的耐旱程度。植物的株高、胸径或基径、冠幅、叶片数量、单株叶面积、叶长、生物量均与干旱胁迫强度呈负相关关系,叶片萎蔫程度、根系发达程度与干旱胁迫强度呈正相关关系。Yin等[2]对海拔不同的青杨与青海杨的干旱胁迫,通过对比他们的早期生长状况"干物质积累含量与水分利用效率等指标来研究植物对干旱胁迫的响应,研究结果表 明,高海拔的青海杨在干旱胁迫过程中采取的是保守的用水方式,具有较强的耐旱性。刘建兵等[3]研究表明,干旱胁迫环境下,不同种源马尾松苗高增长量和高增长百分率逐渐减小,苗木生物量和苗高一样呈现正相关,各个种源马尾松苗木
基因组学研究进展论文
TILLING技术 姓名:罗洁学号:M201071486 院系:生命科学与技术学院 摘要:TILLING技术是一种全新的反向遗传学研究方法,它提供了一种高通量,低成本,规模化和高效筛选化学诱变剂EMS诱发产生点突变的技术。本文简要介绍了TILLING技术的原理和特点,并对其在植物功能基因组学,作物品种改良和在生物进化及检测多态性中的应用作了初步探讨。 Abstract: TILLING technology is a kind of brand-new reverse genetics study method, it provides a high throughput, low cost, scalization and efficient screening chemical mutagen EMS induced produce point mutations of technology. This paper briefly introduced the TILLING technology principle and characteristics of plants, and in its functional genomics, crop cultivar improvement and in biological evolution and testing polymorphism application was discussed 关键词:TILLING技术反向遗传学点突变 前言 随着越来越多的植物的基因组测序的完成,以表型筛选为主的正向遗传学方法正逐步被反向遗传学方法替代而成为功能基因组学研究的主要方法。反义RNA抑制、RNAi和插入突变是植物中常用的用于反向遗传学研究的方法,但这3种技术均需复杂繁琐的植物转基因表达载体构建过程以及组织培养、基因转化和周期极长的转基因植物筛选过程,作为常规方法仅限应用于极少数物种[1]。 基因组靶向定位诱导损伤技术(targeting induced local lesions in genomes,简称TILLING)是美国华盛顿Hutchinson癌症研究中心以Henikoff为首的科学家发展建立的[2]。它将诱发产生高频率点突变的化学诱变方法与PCR筛选技术和Li—Cor公司生产的4300DNA遗传分析系统的双色红外荧光高通量检测技术有效结合,快速有效地从化学诱变剂甲基磺酸乙酯(ethyl methane sulfonate,EMS)诱变产生的突变群体中鉴定出点突变,这一全新的、高通量、低成本的反向遗传学研究方法大大地方便了植物基因组学的研究。在TILLING技术基础上发展起来的Ecotilling(ecotypic TILI ING)技术不用化学诱变,可以直接探知存在于特定群体中某个基因或基因群的多态性(等位基因),识别单碱基突变(SNP)、小片段插入、微卫星重复等,为基因功能研究和生物进化提供重要信息[3]。 最初的TILLING是利用DHPLC对DNA池中的突变进行检测(McCallum et al., 2000a; 2000b)。为了进一步提高这个方法的效率,适应大规模筛选的要求,Colbert 等(2001)对TILLING 作了改进。他们将所获PCR 片段先用特异性识别错配碱基的内切酶酶切异源双链核酸分子,再用变性的序列胶电泳分离酶切产物,用标准的图像处理程序分析点突变的存在。有几种酶已经被用于错配碱基的特异性识别、切割,包括S1 核酸酶(Howard et al., 1999)和T4核酸内切酶Ⅶ(Youil et al., 1996)。目前,使用较多的是S1核酸内切酶家族中的CELI酶,它是一种从芹菜(celery)中分离出的植物所特有的核酸内切酶(Oleykowski et al.,1998)。CELI 酶能够识别错配碱基并在错配碱基的3' 端进行切割,再用变性的序列胶(PAGE)分离酶切产物,能够将超过1kb的PCR扩增产物内的点突变定位在几个碱基对内。因此,改进的TILLING 充分运用了CELI 酶和凝胶电泳技术,从而使其成为一个低成本、高通量的筛选突变基因的技术平台。 TILLING技术的基本原理是:首先是运用化学诱变剂诱发产生一系列的点突变而获得突变群体,提取DNA并把多个待测样品DNA混合建立突变群体及其DNA池,然后利用特异性引物对特定DNA区段进行PCR扩增,通过变性和复性过程得到异质双链。如果有突变发生,那