用material studio 画晶胞及参数设置
materials studio操作手册

materials studio操作手册Materials Studio是一款功能强大的材料模拟软件,广泛应用于材料科学、化学、物理等领域。
本手册旨在向初学者介绍Materials Studio 的基本操作方法,帮助读者快速上手和熟练使用该软件。
一、软件介绍Materials Studio是由Accelrys公司开发的一款材料模拟软件,提供了多种计算和模拟工具,包括材料结构建模、分子动力学模拟、密度泛函理论计算等。
软件界面简洁直观,操作相对简单,适合初学者学习和使用。
二、软件安装1. 下载Materials Studio安装包,双击运行安装程序。
2. 按照安装向导的提示进行安装,并选择安装路径。
3. 安装完成后,打开软件,输入许可证信息进行激活。
三、材料结构建模1. 打开Materials Studio,点击菜单栏的“建模”选项。
2. 在“建模”界面中,选择所需的建模工具,如“晶体构建”、“分子段构建”等。
3. 根据需要输入所需的参数,如晶体的晶面、晶格常数等。
4. 完成结构建模后,保存并命名该模型。
四、模拟计算1. 在Materials Studio主界面,点击菜单栏的“计算模拟”选项。
2. 在“计算模拟”界面中,选择所需的计算方法,如分子动力学模拟、能带计算等。
3. 根据需要输入所需的参数,如温度、压力、模拟时间等。
4. 点击“开始计算”按钮,等待计算结果的生成。
五、数据分析与可视化1. 根据计算结果,在Materials Studio主界面选择“后处理与分析”选项。
2. 在“后处理与分析”界面中,选择所需的分析工具,如晶体结构分析、能带分析等。
3. 输入相应的参数和选择所需的分析方法。
4. 运行分析工具后,生成分析结果,并通过可视化方式展示。
六、参数优化1. 在Materials Studio主界面,选择“参数优化”选项。
2. 在“参数优化”界面中,选择所需的优化算法,如遗传算法、全局优化算法等。
建立晶体模型的步骤
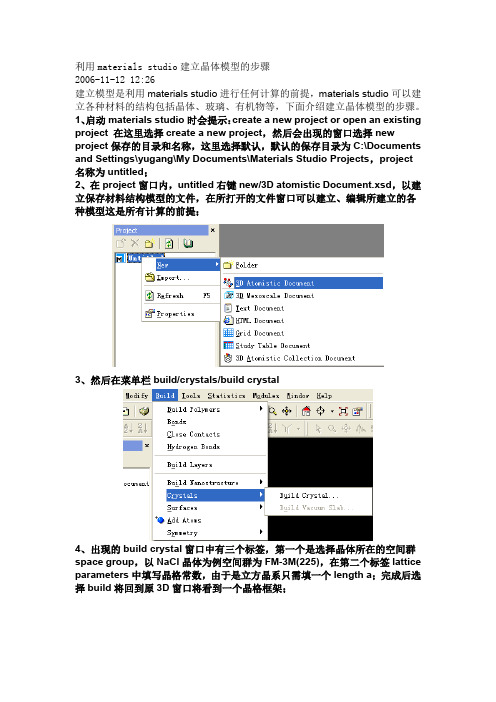
利用materials studio建立晶体模型的步骤2006-11-12 12:26建立模型是利用materials studio进行任何计算的前提,materials studio可以建立各种材料的结构包括晶体、玻璃、有机物等,下面介绍建立晶体模型的步骤。
1、启动materials studio时会提示:create a new project or open an existing project 在这里选择create a new project,然后会出现的窗口选择new project保存的目录和名称,这里选择默认,默认的保存目录为C:\Documents and Settings\yugang\My Documents\Materials Studio Projects,project名称为untitled;2、在project窗口内,untitled右键new/3D atomistic Document.xsd,以建立保存材料结构模型的文件,在所打开的文件窗口可以建立、编辑所建立的各种模型这是所有计算的前提;3、然后在菜单栏build/crystals/build crystal4、出现的build crystal窗口中有三个标签,第一个是选择晶体所在的空间群space group,以NaCl晶体为例空间群为FM-3M(225),在第二个标签lattice parameters中填写晶格常数,由于是立方晶系只需填一个length a;完成后选择build将回到原3D窗口将看到一个晶格框架;5、通过工具栏或者build/add atom 出现添加原子窗口,首先添加Na,坐标a、b、c为0,再添加Cl原子a、b、c坐标为(0,0.5,0)这样,NaCl晶体就建立起来了6、在3D模型文件窗口右键出现的菜单选择display style窗口选择显示模式,选择ball and stick7、完成后NaCl晶体模型为说明:在建立模型输入原子坐标是只输入不同原子的一个坐标,这里的不同包括种类不同和位置不同,之所以只需输入一个是因为先选择了空间群,在空间群的限制下,与该坐标相对应的对称位置都被添加上原子。
materialstudio 计算晶体

近年来,随着计算机技术的飞速发展,计算材料科学成为了材料科学领域中备受瞩目的一个分支。
其中,MaterialStudio计算晶体作为其中的一种重要工具,被广泛应用于材料设计和研究中。
本文将从不同角度探讨MaterialStudio计算晶体的相关内容,以帮助读者更全面地理解这一主题。
一、MaterialStudio计算晶体的基本原理MaterialStudio计算晶体是基于密度泛函理论(DFT)和分子动力学(MD)的计算方法,结合了巨大的晶体数据库和先进的计算技术,可以高效地进行晶体结构的预测和计算。
其原理是通过在计算机上模拟原子之间的相互作用以及结构的动态演化,从而得到材料的结构、力学性能、热学性质等重要信息。
二、MaterialStudio计算晶体的应用领域1.材料设计和优化MaterialStudio计算晶体可以帮助研究人员在计算机上进行大规模的材料结构搜索和优化,以找到具有特定性能的理想材料。
这对于材料设计和新材料的发现具有重要意义。
2.材料性能预测通过MaterialStudio计算晶体,可以准确地预测材料的物理性质,如电子结构、能带结构、光学性质等,对于材料的性能分析和预测具有重要意义。
3.晶体缺陷和界面研究MaterialStudio计算晶体能够模拟材料中的缺陷和界面等微观结构,从而研究材料的稳定性、断裂行为以及晶界对材料性能的影响,为材料改进和优化提供重要参考。
三、MaterialStudio计算晶体在材料科学中的意义MaterialStudio计算晶体的出现极大地促进了材料科学领域的发展和进步。
它不仅提供了一种高效、准确的方法来研究材料的结构和性能,也为材料的设计和优化提供了全新的思路和途径。
通过MaterialStudio计算晶体,研究人员能够更深入地理解材料的微观机制,为新材料的开发和应用提供了有力的支持。
四、个人观点和展望作为一种先进的计算工具,MaterialStudio计算晶体在材料科学领域中具有巨大的潜力和发展空间。
利用软件Materials Studio标定晶体的晶面
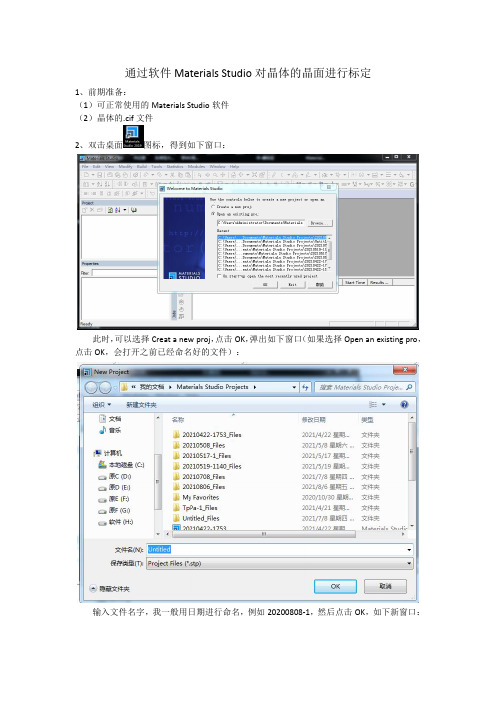
通过软件Materials Studio对晶体的晶面进行标定
1、前期准备:
(1)可正常使用的Materials Studio软件
(2)晶体的.cif文件
2、双击桌面图标,得到如下窗口:
此时,可以选择Creat a new proj,点击OK,弹出如下窗口(如果选择Open an existing pro,点击OK,会打开之前已经命名好的文件):
输入文件名字,我一般用日期进行命名,例如20200808-1,然后点击OK,如下新窗口:
将.cif文件(以TpPa-1的.cif文件为例)拖入软件界面中打开,得到如下窗口:
放大窗口,找到Reflex Tools图标,单击左键,选择“Powder Diffraction”
弹出一个新窗口:
修改角度范围,对于TpPa-1来说,可以将2-Theta:的Min改为3.000,Max 改为30.000,然后点击Calculate.得到新的窗口:
将鼠标光标放到相应的峰下边的绿色短竖线上,单击左键,在窗口的左下角即可看到晶面数据(Reflection:100,2-Theta:4.7)
备注:如果在点击绿色竖短线后左下角不出现晶面数据,则点击View,再单击Status Bar,使其前边带有对号√,即可。
如下图所示:。
用material studio 画晶胞及参数设置

第一种情况: 从程序自带的各种晶体及有机模型中导入体系的晶胞1.打开MS,由file>import>structures>metals\>pure-metals>Fe导入Fe的晶胞。
2.由build>Surfaces>cleave Surfaces打开对话框.在对话框中输入要建立的晶面(hkl),选择position,其中depth控制晶面层数。
3.进入build>Supercell,输入A 、B 、C的值,得到想要的超晶胞。
4.到该步骤,我们已经建立了一个周期性的超晶胞。
如果要做周期性计算,则应选择build>Crystals>buil d vaccum slab,其中真空层通常选择10埃以上。
如果建立团簇模型则选择build>Symmetry>Non-periodic Structure,去掉模型的周期性,并跟据自己的实际需要删除部分原子,得到想要的团簇模型。
5.在表面插入分子时通过菜单栏上的几个小图标添加即可。
第二种情况: 手动建模,优点是可控制晶格常数。
6.首先从文献中查到晶体的晶格常数的实验值。
7.打开build>Crystals>build crystals,可见到对话框。
在对话框中选择空间群与点群,然后在Lattice Parameter中设置晶胞基矢的长度及夹角。
8.然后打开build>Add atom,从对话框中输入坐标。
这里只需输入几个有代表性的原子的坐标,不必全部输入。
在坐标输入前首先在option页面中选择coordinate system,或者分数坐标或者卡迪尔坐标。
9.以下步骤重复2-5步。
10.需要注意的是,采取什么样的团簇并不是任意的。
原因是很多模型构造出来后在优化过程中往往不收敛。
要避免这个问题的办法是查阅文献,参考文献上模型进行选取,因为它们的模型通常是经过试验证实收敛的。
material-studio-入门教程:构建晶胞以及forcite动力学模拟

动力学计算步骤:一、构造分子1:File-new-3D atomistic document-重命名2:构建分子-右键改Display style-adjust hydrogen加氢-clean3:优化分子-moduces-forcite-calculation-几何优化more里面可以改参数-改任务名称-Run二、构建晶胞点开优化好的分子-modules-amorphous cell-construction-添加指定分子-改number,密度,number of configurations调成1-setup-forcefield-compass-Construct三、使分子有序排列,构建超级晶胞先构建一个分子的晶胞-bulid- symmetry –supercell-扩大a,b-保存四、将不同晶胞合并将两个晶胞的参数改成一样后,Bulid-bulid layer-打开所选晶胞-layer details 调节方向-bulid五、动力学模拟计算第一步:Forcite-calculation-dynamic-more-NVT,number of steps和flame output every一样;温度Anderen;Energy中forcefield选择compass-Run。
第二步:Forcite-calculation-dynamic-more-NPT,压力选择0.0001Gpa(常压),number of steps和flame output every一样;thermostat选择Anderen;barostat选择Berendsen;Energy 中forcefield选择compass-Run六、分析参数Modules-forcite-analysis1:径向分布。
2:密度分布。
Materials studio的应用实验内容

Materials Studio 实验资料说明:1、所有实验结果请保存在F盘下,文件夹名不能出现中文。
自己的实验结果复制后请删除。
2、实验报告不要封面。
3、实验报告首页顶端写明:班级、学号、姓名实验报告内容包括:实验项目名称;每一任务的实验要求内容;每一任务的验步骤;按实验要求所得的实验结果。
任务一建立结构模型是Materials studio的基础,进一步的计算和分析大多都需要在模型的基础上进行内容:建立四方结构P4mm的钛酸钡模型步骤:1、把电脑时间调整为2006年1月(为了方便findit软件使用)2、打开findit软件,在元素周期表里面点选Ba、Ti、O,下面大方框内出出现(Ba and Ti andO)字样,点选大方框左边的exclusive AND然后search3、点击Space Group,按该列排列,拖动上一栏滚动条至Space Group为P4mm,选择一个Ba1 O3 Ti1的条目,则下面一栏出现该条目内容其中Unit Cell 条目为晶胞的参数及夹角最下面Atom下的内容为各原子相对坐标4、打开Materials studio,Create a new proj,命名为学号5、菜单View-》Explorers-》properties explorer,打开参数对话框(在屏幕左下),此对话框可以看到点选的原子或键的参数。
6、菜单file-》new-》3D atomistic7、菜单Build-》crystals-》Build crystal8、Enter Group 输入四方向钛酸钡对应的点群p4mm,Lattic Parameters选项卡下面输入上面查得的晶胞参数,对于立方晶系,a=b≠c,夹角都是90°9、菜单build-》add atoms(或者点工具栏上按钮),点击Option选项卡,去掉Test for bonds…前面的勾10、点击Atom选项卡,点击elemelt 后面的省略号按钮,在元素周期表中选择Ba;并按照上面(步骤3)查到的相对坐标输入,Add11、对比右下角的三维坐标轴方向,观察所加入原子的位置及坐标12、依次加入其它原子,(加入O原子的坐标有两行,如第一行坐标的z≠0,可能造成生成的图形少一个O原子)13、菜单Build-》bonds-》calculate 生成键,14、右键Display Style 选择ball andstike,生成球棒图15、通过工具栏上的3D viewer Rotation mode按钮(箭头按钮后面)旋转所生成的图形,并观察,(方向键也可以实现旋转)16、如果旋转时图形闪烁,有的角度看不到,可通过菜单Tools-》Option-》Graphis选项卡,关闭硬件加速(前面打勾)。
MaterialsStudio建模操作详细步骤(本人原创)

MaterialsStudio建模操作详细步骤(本⼈原创)第2章Materials Studio建模2.1界⾯常⽤操作2.1.1 Materials Studio的启动从Windows“启动”菜单中选择“程序”Accelrys Materials Studio 4.0| Materials Studio。
如果在桌⾯上有Materials Studio图标,也可以通过双击图标来启动Materials Studio。
在启动Materials Studio时,⾸先会出现⼀个所谓的欢迎界⾯(Welcome to Materials Studio),必须创建⼀个新的项⽬或从对话框中载⼊⼀个已经存在的项⽬。
注意:如果是第⼀次打开Materials Studio,会看到⼀个叫做Materials Studio ⽂件关联的对话框,如果出现这种情况,按照提⽰点击OK按钮即可。
2.1.2 创建项⽬在欢迎界⾯对话框上选择创建⼀个新的项⽬,然后点击OK。
然后会出现新建项⽬对话框,选择要存储⽂件的位置并且键⼊“tiejifeijinghejin”作为⽂件名,然后点击OK。
此时的项⽬管理器如图2-1所⽰:图2-1 Project 界⾯Materials Studio对中⽂⽀持不好,命名时最好⽤英⽂字母,可以右击点Rename,进⾏重命名。
2.1.3 输出图像可以将3D Atomistic⽂件显⽰的图像作为位图输出,输出的图像可以包含到其它⽂件中。
位图图像被存储为.bmp格式,可以使⽤简单的位图编辑器⽐如Windows的画图进⾏编辑。
从菜单栏中选择File | Export...显⽰Export对话框。
点击Export as type⽂本框右侧的选项箭头,从下拉列表中选择Structure Bitmap (*.bmp)。
⼀旦选择了位图格式,Options...按钮就被激活了。
点击Options...按钮以显⽰Bitmap Export Options对话框。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第一种情况: 从程序自带的各种晶体及有机模型中导入体系的晶胞
1.打开MS,由file>import>structures>metals\>pure-metals>Fe导入Fe的晶胞。
2.由build>Surfaces>cleave Surfaces打开对话框.
在对话框中输入要建立的晶面(hkl),选择position,其中depth控制晶面层数。
3.进入build>Supercell,输入A 、B 、C的值,得到想要的超晶胞。
4.到该步骤,我们已经建立了一个周期性的超晶胞。
如果要做周期性计算,则应选择build>Crystals>buil d vaccum slab,其中真空层通常选择10埃以上。
如果建立团簇模型则选择build>Symmetry>Non-periodic Structure,去掉模型的周期性,并跟据自己的实际需要删除部分原子,得到想要的团簇模型。
5.在表面插入分子时通过菜单栏上的几个小图标添加即可。
第二种情况: 手动建模,优点是可控制晶格常数。
6.首先从文献中查到晶体的晶格常数的实验值。
7.打开build>Crystals>build crystals,可见到对话框。
在对话框中选择空间群与点群,然后在Lattice Parameter中设置晶胞基矢的长度及夹角。
8.然后打开build>Add atom,从对话框中输入坐标。
这里只需输入几个有代表性的原子的坐标,不必全部输入。
在坐标输入前首先在option页面中选择coordinate system,或者分数坐标或者卡迪尔坐标。
9.以下步骤重复2-5步。
10.需要注意的是,采取什么样的团簇并不是任意的。
原因是很多模型构造出来后在优化过程中往往不收敛。
要避免这个问题的办法是查阅文献,参考文献上模型进行选取,因为它们的模型通常是经过试验证实收敛的。
几点说明
1.与高斯相比,dmol3能够计算的体系更大。
如果要研究表面的吸附,而模拟表面的团簇模型又比较大,建议采用dmol3。
如果计算的是局部化学反应,而体系也不是很大,则可以使用高斯。
2.关于是否考虑周期性条件的问题
研究金属表面时,团簇计算方法在前些年由于计算量小曾经被广泛的应用过,直到现在也被很多人在使用着,主要被用来计算吸附和多个分子的共吸附等,即不考虑化学键的断裂。
近年来由于国际上计算能力的提升,人们开始考虑周期性条件,这点从JPCA,JPCB,PRL,PRB,JACS等杂志上刊出的文章里也可以看出,但是计算量要大很多。
需要注意的是,由于团簇计算方法没有考虑周期性,即在k空间里只计算了Γ点,采用该方法计算表面的化学键的断裂(即表面扩散问题等)时有可能受到质疑。
3.在研究表面时,通常把团簇固定,只优化吸附在表面的分子,这一点可以通过菜单栏上的Modify>Cons traint实现。
首先选定团簇中需要固定的原子,然后在下面的对话框中打勾。
同时也可以在Measurement里固定部分键长和键角。
4. 关于计算参数设置
主要有几个参数需要注意
1 对于Electronic页面,需要注意的是Core treatment,对于过渡金属原子通常需要考虑相对论效应,因此一般不使用All Electron方法。
其他几种方法任选。
Basis set应为DNP,Setup下的Quality一般选fine。
为了提高计算速度,一个较好的办法是先用粗糙的Basis set和Quality进行优化,然后再提高精度。
2 还有一个非常重要的选项是Electronic>More>SCF里的Use smearing。
这个关键字有助于加快收敛,但是设的多大往往会产生错误的结果,它也相当于允许的误差范围。
具体设置办法可参考help。
其他的关键字可酌情设置。