Risk management for IVDs-part 3
美国微生物检查要求及微生物监控体系

其m=中3,大M于=1C00)0o个。p/也yg的r有i样g一品h些t数企2不业0超0运4过用-3数2个0理,1统大1计于A技1s0术p00评o个s估/eg其的P偏样ty离品的L数程t0d个度. (。n=13,
克。官方也规定了对与食品接触的台面强制性实施李斯特氏菌 官方检查。要求加工厂实验室可使用国际认可的方法对食品接 触的表面的环境表面进行单核增生李斯特氏、李斯特氏属或类 似李斯特氏其中一种进行检查。但官方确认和调查检验中采用 的方法应为农业部实验室的方法检测单核增生李斯特氏。在从 事调查过程中食品安全局还会使用脉冲凝胶电泳技术,并使用 CDC(疾病控制中心)的数据库进行鉴定。
美官方对肉类及产 品的微生物监控要求
Evaluation only. eated with Aspose.Slides for .NET 3.5 Client Profile 5.2.0
Copyright 2004-2011 Aspose Pty Ltd. 山东出入境检验检疫局:梁成珠
提纲
美国负责监管肉类微生物的官方机构 美国官方机E构va与lu青ati岛on局on食ly品. 实验室对禽 eated w肉ith及A产sp品ose微.S生lid物es检fo测r .N依E据T 3.5 Client Profile 5.2.0 UCSDoApyFriSgIhSt对20出04口-2国01肉1 A类sp及os产e P品ty的L微td.生
LIA+
FDA指南-ISO-10993-1-生物相容性的风险管理

FDA指南-ISO-10993-1之生物相容性的风险管理
FDA建议将风险评估作为生物相容性部分的第一个文件,应清楚地总结关于风险评估的结论,并解释识别的生物相容性风险与降低风险的信息之间的关系,以及识别任何剩余的知识缺口,然后识别降低剩余风险的生物相容性测试或其他评估。
此外,应解释在生物相容性测试或其他评估中识别到的任何毒性和不良反应。
关于生物相容性信息的文件化方法,FDA提供了如下示例表格作为参考,也可使用其他方法。
生物相容性的风险管理包括三部分:评估器械风险、识别潜在风险、识别可用的信息及降低风险。
1、评估器械风险
从以下几个方面进行风险评估:
(1)材料的成分;
(2)生产过程;
(3)器械的临床使用;
(4)材料暴露的频次和持续时间。
2、识别潜在风险
潜在的风险包括以下三方面:
(1)化学毒性;
(2)器械物理特性引起的不可接受的生物反应;
(3)生产和处理对器械的理化性质的影响。
3、识别可用的信息及降低风险
应识别风险相关的现有的可用信息,并识别剩余的知识缺口。
FDA建议适用时应包括以下信息:
(1)文献和其他公开可获得的信息;
(2)临床经验;
(3)动物研究经验;
(4)医疗器械标准;
(5)之前已被FDA评审的器械。
美国生物药上市后风险管理及对我国的启示

美国生物药上市后风险管理及对我国的启示作者:李梦颖王峻霞蒋蓉来源:《中国药房》2021年第07期摘要目的:借鉴美国生物药上市后风险管理经验,为我国生物药上市后风险管理提供参考。
方法:通过研究美国FDA发布的指南文件、网站信息等资料,对美国生物药上市后风险管理进行分析,并以英夫利昔单抗为例介绍其具体实施情况,总结其管理特点并提出对我国生物药上市后风险管理的启示及相关建议。
结果与结论:美国生物药上市后的风险管理主要包括“风险评估与降低策略(REMS)”“上市后研究和临床试验制度”两方面,其中后者包括上市后要求(PMR)和上市后承诺(PMC)两类。
以英夫利昔单抗为例,该药自1998年8月被美国FDA批准上市后,其生产厂家于2009年向FDA提交了REMS并获批,并先后5次提出上市后研究和临床试验。
可知美国生物药上市后风险管理是由FDA通过出台具体指南,鼓励多角色参与风险管理,实现与患者的有效沟通,对生物药风险进行持续监管,以降低生物药的使用风险。
对于生物药而言,我国尚未制定系统、具体的实施细则和指南,在上市后风险管理方面仍存在欠缺。
建议我国可借鉴美国对生物药上市后风险管理的措施和制度,尽可能吸纳利益相关方参与上市后管理,通过与患者进行有效沟通以提升患者用药风险意识,并且进一步完善上市后研究管理制度,保障患者用药安全。
关键词美国;生物药;上市后风险管理;启示中图分类号 R951;S859.79+7 文献标志码 A 文章编号 1001-0408(2021)07-0776-06ABSTRACT OBJECTIVE: To learn from the experience of post-marketing risk management of biopharmaceuticals in the United States, and to provide reference for post-marketing risk management of biopharmaceuticals in China. METHODS: By studying guidance documents and website information issued by FDA, the risk management of biopharmaceuticals after marketing in the United States was analyzed. Taking infliximab as an example, the specific implementation situation was introduced, the management characteristics were summarized, and the enlightenment and relevant suggestions were put forward for the risk management of biopharmaceuticals after marketing in China. RESULTS & CONCLUSIONS: The post-marketing risk management of biopharmaceuticals in the United States mainly includes two aspects as “risk evaluation and mitigation strategy (REMS)” and “post-marketing study and clinical trials system”. The latter includ ed post-marketing requirement (PMR) and post-marketing commitment (PMC). Taking infliximab as an example, since it was approved by FDA in August 1998, its manufacturer submitted REMS to FDA in 2009 and obtained approval, and proposed post-marketing studies and clinical trials for five times. It can be seen that FDA has issued specific guidelines for post-marketing risk management of biopharmaceuticals to encourage multi-role participation in risk management, realize effective communication with patients, and continuously supervise the risk of biopharmaceuticals, so as to reduce the risk of the use of biopharmaceuticals. For biopharmaceuticals, China has not yet formulated systematic and specific implementation rules and guidelines, and there is still lack in post-marketing risk management. It is suggested that China can learn from the measures and system of post-marketing risk management of biopharmaceuticals in the United States, involve stakeholders in post-marketing management,enhance patients’ awareness of drug use risks through effective communication, and further improve the post-marketing research management system to guarantee patients’ safety of drug use.KEYWORDS United States; Biopharmaceuticals; Post-marketing risk management; Enlightenment自1982年全球首个生物技术药物重组胰岛素上市以来,经过近40年的发展,目前已有细胞因子、重组酶和激素、单克隆抗体、融合蛋白、基因治疗药、细胞治疗产品和基因工程疫苗等200多种生物药上市[1],这些生物药在癌症和遗传性疾病等诸多严重疾病的治疗中发挥着重要且关键的作用。
《质量风险管理程序》

目录1 目的 (2)2 范围 (2)3 依据标准、法规与指南 (2)4 术语 (2)5 职责 (2)6 程序 (3)7 文件发放范围及数量 (7)8 附录 (7)9 记录(附后) (7)10 文件变更历史 (7)受控级别:机密1 目的规范公司产品质量风险评价的启动、评估、控制、通报和回顾,减少产品质量风险,确保产品质量。
2 范围本程序适用于公司内与质量有关的风险管理。
3 依据标准、法规与指南《药品生产质量管理规范》《兽药生产质量管理规范》ICH Q7ICH Q94 术语4.1 产品生命周期(product lifecycle):一个产品从初期研发、批准上市前后、直到产品终止的所有阶段。
4.2 质量风险管理(quality risk management):贯穿产品生命周期的药品质量风险的评估、控制、通报和回顾的系统化过程。
4.3 风险(risk):危害发生的可能性及其严重性的组合。
4.4 风险认可(risk acceptance):接受风险的决策。
4.5 风险分析(risk analysis):和被确定的危害源有关的风险的分析。
4.6 风险评估(risk assessment):对信息进行综合处理的系统化过程,支持风险管理过程中的风险决策。
4.7 风险通报(risk communication):在决策者和其他利益相关者之间关于风险和风险管理方面信息的交换或共享。
4.8 风险控制(risk control):实施风险管理决策的行为。
4.9 风险评价(risk evaluation):用定性或定量的方法,将被评估的风险与即定的风险标准进行比较,以确定风险的显著性。
4.10 风险鉴定(risk identification):根据风险提问或问题描述,系统地使用信息来鉴定潜在危害源。
4.11 风险管理(risk management):系统地运用质量管理的政策、程序和方法进行风险评价、控制和通报。
4.12 风险降低(risk reduction):采取措施减少危害发生的可能性和严重程度。
口服固体制剂 GMP 实施指南

目录
目录
1 简介 .............................................................................................................................................. 1 2 质量管理....................................................................................................................................... 3 2.1 概论 ................................................................................................................................... 3 2.2 风险管理............................................................................................................................ 4 2.3 产品质量回顾.................................................................................................................... 9 2.4 自检 ............................................................
临床试验中风险管理计划

临床试验中风险管理计划英文回答:Risk management is a crucial aspect of clinical trials as it helps to identify, assess, and mitigate potential risks that could impact the safety and efficacy of the trial. A well-developed risk management plan is essential to ensure that the trial is conducted smoothly and that the rights and well-being of the participants are protected.One of the key components of a risk management plan is the identification of potential risks. This involves a comprehensive analysis of all aspects of the trial, including the study design, investigational product, and study population. By identifying potential risks, such as adverse events or protocol deviations, appropriate measures can be put in place to minimize their occurrence.Once the risks have been identified, they need to be assessed in terms of their likelihood and potential impact.This can be done through a risk assessment matrix, which assigns a score to each risk based on its severity and probability. Risks that are deemed to be high in severity and probability require immediate attention and mitigation strategies.Mitigation strategies are actions taken to reduce the likelihood or impact of a risk. These strategies can include additional training for study staff, implementing stricter monitoring procedures, or modifying the study protocol. For example, if there is a risk of protocol deviations due to lack of understanding among study staff, additional training sessions can be organized to ensurethat all staff members are well-informed about the protocol requirements.Monitoring and communication are also important aspects of risk management. Regular monitoring of the trial allows for early detection of any potential risks or deviations from the protocol. This can be done through site visits, data review, or participant interviews. Effective communication between all stakeholders, including the studysponsor, investigators, and ethics committee, is crucial to ensure that any identified risks are addressed promptly and appropriately.In addition to proactive risk management, it is also important to have a plan in place for managing risks that do occur. This includes procedures for reporting and documenting adverse events, as well as strategies for managing any potential harm to participants. For example, if a participant experiences a serious adverse event, there should be clear guidelines on how to provide appropriate medical care and support.Overall, a well-developed risk management plan is essential for the successful conduct of clinical trials. It helps to ensure the safety and well-being of the participants, as well as the integrity and validity of the trial results.中文回答:风险管理是临床试验中至关重要的一环,它有助于识别、评估和减轻可能影响试验安全性和疗效的潜在风险。
日本药品质量再评价_佐藤淳子中文版

会 分事项(溶出试验方法公示3个月以内)。
理协 6. 结果公布【第五步】 ① 提交资料确认为合理时,包括部分变更申请获得批 量管 准时,进行再评价判定。同时公布溶出试验方法。
质
药
医
国
中
16
会
日本橙皮书主要记载的事项分(1)
药
1、药效组栏
制
仿 记载再评价对象的药品所属药效群分类的编号。
2、有效成分栏
医 • 对溶出性不同等的品种的批准另有整理。
国
中
25
会
Orange book?
分
药
制 • Orange book是“医疗用医药品品质情报集”的 仿 别称
会 • 刊登质量再评价的结果 协 • 每次通知质量再评价结果时发行
管理 • 到2011年发行了32本(2011年3月版)
• 在医疗现场,为方便日常业务使用,提供仿制药
会
具体程序和步骤(3)
分
药
制 4. 溶出试验方法公示【第四步】
仿 ① 由厚生劳动省通过书面形式对“溶出试验方法(草 会 案)”进行评估,如果草案方法可行,则进行“溶
协 出试验方法”公示。原研企业向仿制企业提供参照
批次。
理
管 ② 地方药检所根据公示的试验方法实际进行试验操作,
检验公示的试验方法的文字表述是否可妥当实施试
量管 点。
质
药
医 有必要鼓励仿制药的使用
国
中
6
会
为鼓励使用仿制药
分
药
1. 确保能稳定供应
制
仿 2. 收集信息,完善供应体制 协会 3. 强化生产管理和质量管理
理 4. 在审批阶段考虑专利情况
管 5. 考虑和药品价格关联
IVD行业国外原料主要供应商
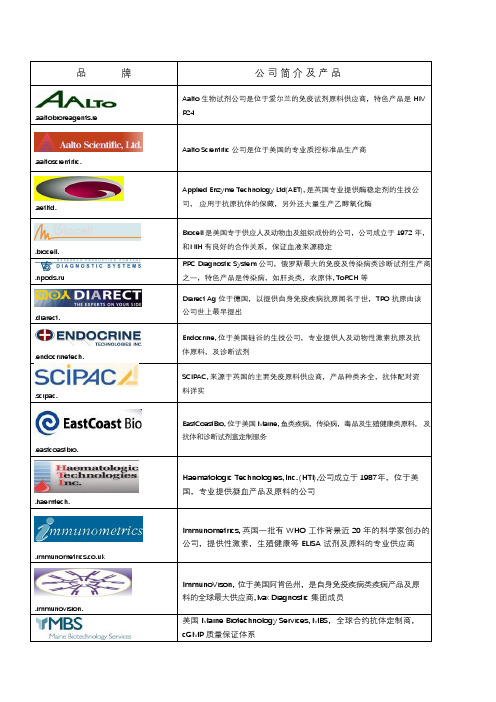
.aaltobioreagents.ie .aaltoscientific..aetltd..biocell..npods.ru.diarect..endocrinetech..scipac..eastcoastbio..haemtech. .immunovision..mainebiotechnology. .operon.es.equitech-bio..quadfive..promeddx..seracare..chemogen..modiquest..seramon..midlandbio..capricornproducts. .instruchemie.nl.sheffield-products. .biogenes.de.biocheckinc..biospacific..bioprocessinginc..fitzgerald-fii..microbix..inventdiagnostica.de .biomarket.fi.calbioreagents..xema-medica..scrippslabs..silverlakeresearch..ssi.dk.virostat-inc..virusys..oycus..accessbiologicals. .anshlabs..arlingtonscientific..auditmicro..brt-us..cardinalbiologicals. .diasource.be.diazyme..dsitaly..icllab..immunoreagents. .magsphere.丹麦提供诊断试剂盒和抗体、抗原和血清,有特色的产品是 CE认证的NGAL诊断试剂盒,MBL试剂盒重症监护和止血,临床化学仪器,试剂盒日本提供诊断试剂盒产品的公司,特色产品是低密度LDL 和胱抑素C 试剂盒。
产品涉及质控品,转染病,糖尿病,肿瘤,生殖,甲状腺等试剂盒Acris 是一家德国的著名抗体公司,提供近 3 万种各种优质抗体、蛋白及抗体纯化试剂盒,产品X 围涉及免疫学、细胞生物学、细胞神经信号传导、蛋 白组学、肿瘤生物学等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Risk management for IVDsPart 3: Reducing and controlling risks to patientsDonald M. PowersThe first two articles in this series discussed the significance of risks fromIVD devices, emphasized the importance of risk management planning,and explored ways to apply the risk analysis principles of ISO 14971.1,2This third article covers the risk reduction and control phase of ISO14971.3Controlling Risks to PatientsA visit to a modern clinical laboratory provides ample evidence of the risk control practices that IVD manufacturers have employed: electronic monitoring systems, integrated procedural controls, fail-safe error messages, automated calibration, refrigerated onboard reagent storage, bar coded labels, and explicit warning stickers. Whether such risk control features originated from planned risk management activities or corrections of unexpected failures, many of these features were commonplace well before risk management came into vogue.4 Regardless of the motivation, their main purpose is to reduce the risks to patients from incorrect IVD test results.As stated in the preamble to the U.S. quality system regulation, FDA expects manufacturers to reduce risks to an acceptable level.5,6The European Union’s IVD Directive goes further by requiring manufacturers to reduce risks as far as possible, and to select risk control measures in the following order: inherent safety by design, protective measures in the device or the manufacturing process, and information for safety.7The previous articles in this series discussed ways that IVD manufacturers can identify hazards and estimate their risks.2 If risk analysis determines that the risk from a possible failure mode is negligible, further risk reduction is not necessary. For the remaining risks, ISO 14971 outlines a logical sequence of risk control stages that manufacturers must follow to conform to the standard (see Figure 1). These stages are intended to be checkpoints to promote systematic decision-making and ensure that IVD manufacturers document their risk management decisions for future reference. A justification for each decision must be included if the rationale is not obvious.Figure 1. Risk control stages. Source: ISO 14971:2000.Analyzing Risk Control OptionsThe first stage is a comprehensive analysis of risk control options, which enables IVD manufacturers to select the most appropriate control measures for each specific risk. Manufacturers can do little to reduce the severity of harm, since that depends mainly on the medical actions taken in response to the IVD test results. For most IVDs, manufacturers can only reduce the probability that harm will occur.ISO 14971 adopted the three-level hierarchy of risk control options from the IVD Directive, thereby placing the emphasis on mitigating risks through design whenever practicable. To show that the risk control measures were selected according to the prescribed priority hierarchy, IVD manufacturers must document that the options available to reduce each risk were identified, classified according to the hierarchy, and evaluated for technical and economic feasibility.Several standards from the International Organization for Standardization (ISO; Geneva), the International Electrotechnical Commission (IEC; Geneva), the European Committee for Standardization (CEN), and the Clinical and Laboratory Standards Institute (CLSI; Wayne, PA) address safety aspects of IVDs. A few standards (e.g., IEC’s new usability standard for medical devices) integrate elements of the risk management process.8 When appropriate,IVD manufacturers should consider such standards as options to control identified risks, since conforming to a recognized standard generally conveys a presumption of risk acceptability.Inherent safety by design for an IVD assay means its performance characteristics and reliability meet the medical requirements of its intended use. The components that affect specificity, precision, stability, and traceability are the keys to improving analytical performance, while sound hardware and software design features can preventinstrument-generated spurious results and enhance the uptime of electromechanicalsystems. In addition, automation can eliminate the potential for human error, bar coding can ensure the proper identification of patients, samples, or reagent lots, and attention to human factors can simplify procedures and eliminate causes of error.Protective measures reduce the probability that incorrect results will be reported to physicians or patients. Examples of protective measures include fail-safe software, which results in error messages instead of incorrect results, and fault-tolerant systems, which continue to operate correctly after a fault occurs. Instrument-based controls are now commonplace in IVD systems, and quality control testing has been integrated into many assays.9 Other examples of protective measures are in-process tests that detect defects introduced in the manufacturing process and final-release tests that verify conformance to performance specifications.Information for safety is the labeling, which includes operating instructions and other information to avoid risks. Historically, IVD manufacturers have relied heavily on labeling to help clinical laboratories avoid generating hazardous results. However, adding new instructions or precautions without first attempting to eliminate the failure causes or to incorporate protective mechanisms is contrary to the philosophy of ISO 14971. The standard reminds manufacturers that ―information for safety is the least preferred me thod of risk control measure, to be used only when other risk control measures have been exhausted.‖ Risk management experts allow a risk reduction of no more than one level on the probability scale from labeling or training. Although mandatory training programs designed to reduce risks can sometimes achieve greater risk reduction, training alone is seldom effective in eliminating errors.10IVD manufacturers often overestimate the ability of laboratory quality control (QC) procedures to detect incorrect test results. When delegating risk controls to clinical labs, design engineers must make assumptions with caution. While a manufacturer’s QC recommendations may provide helpful advice, there is no assurance that labs will follow them. In order to qualify as a protective measure, a QC procedure would have to be an integral part of the assay procedure. The IVD guidelines in annex H of the second edition of ISO 14971, which has recently been advanced to a final draft international standard, make it clear that an IVD manufacturer’s recommended QC procedures, as well as recommended confirmatory tests, are only considered information for safety.11Warnings also have limited effectiveness as a risk reduction measure and should be considered as a last resort. The pres sure to avoid calling attention to an assay’s shortcomings in a package insert has all too often led to vague, innocuous statements that would not pass an objective test for effectiveness. The revised ISO 14971 will include a new annex titled―Information for Safety and Information about Residual Risk,‖ which provides guidance on using information for safety as a risk control measure and promoting true awareness when disclosing residual risks.11Implementing Risk Control MeasuresThe risk control implementation stage begins once an IVD manufacturer decides which options yield the greatest risk reduction. Often, more than one risk control approach is needed. Manufacturers have to verify each risk control measure twice, first to document that it was implemented correctly, and then to verify or validate its effectiveness in reducing the risks to the desired level.IVD manufacturers should set up risk management documentation to ensure clear traceability from each identified hazard and hazardous situation to the associated risk control measures. While manufacturers are able to verify some hardware- and software-based risk controls individually through fault injection techniques, a means of testing and evaluating fault-tolerant systems,12 most will demonstrate the cumulative risk reduction from multiple control measures during design validation.Evaluating and Disclosing Residual RisksRisks are rarely eliminated completely. Risks that remain after a manufacturer has implemented all risk control measures are called residual risks, which must meet the acceptability criteria in the risk management plan. Confirming that the residual risks from each hazardous situation have been reduced to an acceptable level is a pivotal checkpoint in the risk control process.If the acceptability criteria are met, then manufacturers must determine what information to include in the labeling to disclose the residual risks. They must also consider the comprehension level of end-users and the detail that users need in the labeling to make informed decisions. If the acceptability criteria are not met, then risk reduction efforts continue until further reduction is not technically or economically feasible. If the risk acceptability criteria are still not met, an IVD manufacturer must either abandon the project or justify it based on a risk-benefit analysis.ISO 14971 recognizes that end-users need to be aware of the residual risks in order to manage them. Such disclosure involves providing the information necessary to understand the residual risks and minimize exposure to the hazards.In IVD labeling, the information in the ―Limitations of the Method‖ section is an example of residual risk disclosure. This part of the labeling contains information such as drug interferences that the IVD manufacturer could not eliminate. Disclosure of drug interferences is not a risk control, since laboratories do not have any practical means to monitor the presence of drugs in specimens. However, in the spirit of ISO 14971, IVD manufacturers should opt to disclose these residual risks only after their efforts to eliminate the interferences prove unsuccessful. The information in the ―Performance Characteristics‖ section is another example of residual-risk disclosure. With such information, a lab director can make informed decisions about whether a particular assay will be useful for its intended medical purpose.The details about how IVD manufacturers control specific risks are becoming more important with the advent of equivalent quality control.13,14 This concept allows a laboratory to build on a manufacturer’s risk controls and design a holistic quality control program. This requires knowing not only the circumstances that can lead to incorrect results but also the internal control mechanisms that can detect or suppress them.15,16Although in principle an IVD manufacturer is responsible for deciding what information is necessary to include in the product labeling, in practice much of the content has already been decided by labeling regulations and standards.17–20 Several examples of prescribed information for safety are intended to help laboratories avoid common use errors (see Table I).Table I. Information for safety to avoid potentially hazardous use errors. Source: ISO/DIS 14971:2005,annex H.Performing Risk-Benefit AnalysisA risk-benefit analysis is required when efforts to reduce residual risks to acceptable levels have failed, yet the IVD manufacturer believes the product is still worthwhile. Unfortunately, while methods for estimating risks with reasonable confidence exist, a standardized approach to estimate the benefits has not been developed. ISO 14971 acknowledges thatrisk-versus-benefit decisions are largely matters of judgment by experienced and knowledgeable individuals. Some guidance for conducting risk-benefit analyses is included in annex D.6 of the draft second edition of ISO 14971.Risk-benefit analyses of individual residual risks are difficult and provide at best only enough information to decide whether to proceed with a design project. Having reduced each individual risk in an iterative manner until it is either acceptable or irreducible, any residualrisks that remain outside the acceptability criteria require a decision whether to proceed with the project. If it is obvious that an individual risk is irreducible and will jeopardize an IVD manufacturer’s ability to meet the overall acceptability criteria or to provide a product with medical benefits that outweigh its overall residual risks, the development project may be stopped. Outside clinical consultation often helps in making this judgment. If the answers are not obvious, the project will usually continue.Addressing Risks Resulting from Risk Control MeasuresOccasionally, implementing risk control measures creates new hazardous situations. For example, a proliferation of warning labels can confuse laboratory operators and desensitize them to hazard warnings. Similarly, an overly complicated set of instructions may be difficult to follow without errors. There are also abundant examples in the recall literature in which actions intended to reduce risks inadvertently created new hazards. IVD manufacturers must review the results of the risk control phase to avoid increasing risks.In some cases, IVD manufacturers may introduce a new hazardous situation intentionally as the lesser of two risks. For example, an instrument programmed to give an error message when a fault occurs is trading off the risk from a delayed result against the risk from a possible incorrect result. The actual degree of risk depends on how the test results are used. For self-testing glucose monitors, if a diabetic were not able to perform blood glucose tests in a timely manner because the meter malfunctioned, there is a chance of accidental overmedication with insulin, leading to hypoglycemic shock. While an error message may seem clearly preferable to an incorrect glucose result, manufacturers must estimate the risks created by a delay in availability of the results and evaluate them against the risk acceptability criteria.On the other hand, a delay in the availability of routine hospital laboratory test results while an automated instrument undergoes repairs is less likely to cause serious harm. Labs anticipate the need for service calls and are expected to have backup plans for lab tests that are urgent. Acceptability of the risks from delayed results is determined in each case by a risk assessment.Verifying Completeness of Risk ControlA verification checkpoint in the risk control process was added as a safeguard against possible overlooking of any necessary risk control measures in the rush to launch a new product. IVD manufacturers are required to verify that they have adequately addressed all hazardous situations identified in the risk analysis phase and any new ones created by the risk control measures.ISO 14971 also requires manufacturers to record the results of all risk control activities in the risk management file, including each decision to take or not take action, every review andverification check discussed above, and the results of the risk reduction activities. This requirement reinforces the need for IVD manufacturers to establish a logical documentation system at the outset, with the necessary traceability to show that they have addressed all hazardous situations and reduced all risks to acceptable levels.Evaluating Residual-Risk AcceptabilityEven after all the individual risks have been mitigated, there is still the question of whether the overall risk of using the IVD device will be acceptable. After documenting the reduction of each individual residual risk, an IVD manufacturer compiles the information in a way that reveals the overall residual risk. The overall residual risk is the risk that remains after a manufacturer has implemented all risk control measures. The risk of using the device may not be acceptable because of the cumulative probability of serious harm from manylow-probability failure modes. In that case, an IVD manufacturer must apply further risk control measures to make the overall residual risk acceptable.Estimating overall residual risk is more challenging than estimating risks from specific device failures, and up to now, little guidance has been available to help IVD manufacturers in evaluating it. A discussion of several possible approaches has been added to the second edition of ISO 14971 as annex D. Two tools described in this annex are particularly applicable to IVDs. In event tree analysis, multiple individual risks from a specific sequence of events can be considered together to determine if the overall residual risk is acceptable. In fault tree analysis, the combined probability of harm from several hazardous situations can be estimated from their individual probabilities. The output of a cause-consequence analysis, a blend of fault tree and event tree analysis, is a diagram for documenting and communicating relationships between risks and their initiating causes.21For well-understood IVD assays, confirming that the overall residual risk is acceptable by performing a detailed risk-by-risk comparison to a comparable assay on the market may suffice. If manufacturers use this approach, they must ensure that information about risks of the comparable assay is up-to-date. Current product labeling and performance data can often be obtained from manufacturers’ Web sites or from clinical laboratories. Peer-reviewed data from product evaluations and clinical experience are found in laboratory journals. In addition, a wealth of adverse-event experience, recalls, and summaries of product safety and effectiveness is available on the FDA Web site.22Another approach especially useful for evaluating the overall residual risk of new IVD systems is to engage application specialists (i.e., laboratory technologists) who are familiar with using similar devices. An important caveat is that they must not have been directly involved in developing the device being evaluated.The application specialists evaluate the system in a representative clinical lab environment to confirm the acceptability of its overall residual risk, specifically considering the characteristicsrelated to safety identified in the risk analysis (e.g., performance, usability, and reliability). In addition, the specialists review the operating instructions and other essential labeling information to identify instructions that are difficult to follow, excessive reliance on warnings, and conflicting requirements. Based on their professional knowledge and experience, they would judge whether the overall residual risk of the system is acceptable.What if the overall residual risk is judged not acceptable according to the criteria in the risk management plan, and further risk reduction is not practicable? An IVD manufacturer has to decide whether the potential medical benefits outweigh the overall residual risk.Risk-benefit analyses of overall residual risk are not as common for IVDs as they are for life-sustaining medical devices. Manufacturers must spend considerable energy in developing a defensible comparison of overall residual risk with the overall benefit that the IVD device provides. Quantifying the medical benefits of an IVD assay with an unacceptable potential for incorrect results is difficult. When the overall residual risk falls outside management’s risk acceptability criteria, IVD manufacturers usually abandon plans to commercialize the device.If an IVD manufacturer pursues a risk-benefit analysis, the decision process is similar to that used for analyzing the individual risks. If the evidence supports a conclusion that the medical benefits outweigh the overall residual risk, the overall residual risk is judged acceptable. Otherwise, the manufacturer has no other choice but to abandon the product.ConclusionA key element of the last IVD design review prior to a new product launch is a comprehensive review of all the risk management activities that have been undertaken. An IVD manufacturer must verify three things: proper execution of the risk management plan, acceptability of the overall residual risk, and t he entire organization’s readiness to carry out its ongoing risk management duties during the life of the device. The documented summary of this review becomes the high-level risk management report required for conformance to ISO 14971.The revised ISO standard no longer requires the risk management report to document traceability of hazards to risk controls. This requirement was eliminated because it caused the final report to become unwieldy, particularly for complex systems that require extensive risk analyses. Nevertheless, IVD manufacturers must still demonstrate traceability in the risk management file. A guidance document from the Global Harmonization Task Force presents an example of an efficient risk management summary table that IVD manufacturers can use to document how the individual risks were controlled for a given device, and the status of implementation and verification of the risk control measures.23 Such a document will be an invaluable tool for continuing to manage risks through a product’s life cycle.The next installment of this series will discuss the production and postproduction monitoring stages of the risk management process, and explore how these aspects of the riskReferences1. DM Powers, ―Risk Management for IVDs, Part 1: Planning and Documenting the Risk Management Process,‖ IVD Technology 12, no. 2 (2006): 28–33.2. DM Powers, ―Risk Management for IVDs, Part 2: Assessing Risks to Patients from Incorrect Test Results,‖ IVD Technology 12, no. 3 (2006): 24–31.3. ISO 14971:2000, ―Medical Devices: Application of Risk Management to Medical Devices‖ (Geneva: International Organization for Standardization).4. DR Wallace and DR Kuhn, ―Lessons from 342 Medical Device Failures,‖ National Institute of Standards and Technology Web site (Gaithersburg, MD: 1999 [cited 23 March 2006]); available from Internet:/wallace/hase99.pdf.5. Federal Register 61 FR:52601–52662, October 7, 1996.6. FDA, ―Guide to Inspections of Quality Systems,‖ FDA Web site (Rockville, MD: August 1999 [ci ted 23 March 2006]); available from Internet: /ora/inspect_ref/igs/qsit/QSITGUIDE.PDF.7. ―Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on In Vitro Diagnostic Medical Devices,‖ Official Journal of the European Communities L331 (1998).8. IEC/CD 62366, ―Medical Devices: Application of Usability Engineering to Medical Devices‖ (Geneva: International Electrotechnical Commission, 2004).9. FD Lasky, ―Technology Variations: Strategies for Assuring Quality Results,‖ Laboratory Medicine 36, no. 10 (2005): 617–620.10. ME Maddox, ―Human Factors: Designing Medical Devices to Minimize Human Error,‖ Medical Device & Diagnostic Industry 19, no. 5 (1997): 166–178.11. ISO/DIS 14971:2005, ―Medical Devices: Application of Risk Management to Medical Devices,‖ 3rd ed. (Geneva: International Organization for Standardization).12. MC Hsueh, TK Tsai, and RK Iyer, ―Fault Injection Techniques and Tools,‖ Computer 30, no. 4 (1997): 75–82.13. MB Bandy, ―CLIA Quality Control Requirements,‖ IVD Technology 11, no. 2 (2005): 24–29.14. DM Powers and T Roscoe, ―QC for the Future,‖ IVD Technology 11, no. 9 (2005): 22–27.15. L Ochs, ―QC for the Future: CLSI Standard Development and Option 4 Proposal,‖ Laboratory Medicine 36, no.10 (2005): 639–640.16. DM Powers, ―Laboratory Quality Control Requirements Should Be Based on Risk Management Principles,‖ Laboratory Medicine 36, no. 10 (2005): 633–638.17. Code of Federal Regulations, 21 CFR 809.10.18. EN 375:2001, ―Information Supplied by the Manufacturer with In Vitro Diagnostic Reagents for Professional Use‖ (Brussels: European Committee for Standardization).19. EN 591:2001, ―Instructions for Use for In Vitro Diagnostic Instruments for Professional Use‖ (Brussels: European Committee for Standardization).20. ISO 18113, ―Clinical Laboratory Testing and In Vitro Diagnostic Test Systems—In Vitro Diagnostic Medical Devices—Information Supplied by the Manu facturer (Labeling)‖ (Geneva: International Organization for Standardization).21. Center for Chemical Process Safety, Guidelines for Hazard Evaluation Procedures with Worked Examples, 2nd ed. (Hoboken, NJ: Wiley, 1992).22. ―Databases on the FDA Website,‖ FDA Web site (Rockville, MD: [cited 23 March 2006]); available from Internet: /search/databases.html.23. ―Implementation of Risk Management Principles and Activities within a Quality Management System,‖ Global Harmonization Task Force Web site (Brussels: 2005 [cited 23 March 2006]); available from Internet: /sg3/inventorysg3/sg3n15r82005.pdf.Copyright ©2006 IVD Technology。