Random priming
群落生态学 野外植物群落取样和调查和方法

如:假定一位调查者在一块面积大约 为30km×50km的森林中用10m×10m的 样方计数蕨类植物,因为森林面积大, 需要较长的时间走到随机设的样方地 点
但是在每一样方中计数蕨类植物所需 的时间较短。
如果在每一个样方点取一些样方—— 比如可能计数一个40m×40m的网格中 的每一个10m×10m样方,这样可能工 作效率更高
图2 巢式样方法
不同群落类型最小面积经验值
群落类型
地衣群落 苔藓群落 沙丘草原 干草原 草甸 高草地 灌丛 温带森林 热带雨林
群落最小面积
0.1~0.4 m2 1~4 m2 1~10 m2 1~25 m2 1~50 m2 5~50 m2 10~50 m2 200~500 m2 500~4000 m2
野外植物群落取样和调查 方法
邵小明
目录
?取样( sampling ) ?植物群落特征描述 ?植被的环境特征
一、取样
1、基本原理
在群落调查时,由于人力、物力和时间的限制,一般只能抽取 其中的一部分作为样本来获取数据并进行分析,进而推断群落总体 的特征,这个过程称为取样。
为了既保证取样研究的结果能够反映群落总体的特征,又使取样 所花费的人力,物力和时间尽可能少,选择合适的取样方法是至关 重要的。
6)(依据)环境因子取样
群落的分布与某些环境因子有关,比如海拔高度、坡度、坡向和小 地形变化等。因此,该方法要先依某一环境因子设置环境梯度,沿环境 梯度分层(类),后在每层中抽取若干个个体,得到所需要样本的取样 方法。
适用范围:总体中个体的分布样式与环境因子有关。
3、取样单位和大小
<1> 取样单位( sampling unit )
生工一班酶工程试题

一、选择1.酶工程技术是(C)的技术过程A.利用酶的催化作用将底物转化为产物B.通过发酵生产和分离纯化获得所需酶C.酶的生产与应用D.酶在工业上的大规模应用2.核酸类酶是(D)A.催化RNA进行水解反应的一类酶B.催化RNA进行剪接反应的一类酶C.由RNA组成的一类酶D.分子中起催化作用的主要成分是RNA的一类酶3.RNA剪切酶是(B)A.催化其他RNA分子进行反应的酶B.催化其他RNA分子进行剪切反应的R酶C.催化本身RNA分子进行剪切反应的R酶D.催化本身RNA分子进行剪接反应的R酶4.酶的改性是指通过各种方法(A)的技术A.改进酶的催化特性B.改变酶的催化特性C.提高酶的催化效率D.提高酶的稳定性5.酶的转换数是指(C)A.酶催化底物转化为产物的数量B.每个酶分子催化底物转化为产物的分子数C.每个酶分子每分钟催化底物转化为产物的分子数D.每摩尔酶催化底物转化为产物的摩尔数6.酶的固定化常用的固定方式不包括(D)A.吸附B.包埋C.连接D.将酶加工成固体7.通过酶工程生产的酶制剂中酶的化学本质是(B)A.蛋白质B.有机物C.RNAD. 核酸8.当前生产酶制剂所需的酶主要的来自(C)A.动物组织和器官B.植物组织和器官C.微生物D.基因工程二、名词解释酶的提取:指在一定的条件下,用适当的溶剂或溶液处理含酶原料,使酶充分溶解到溶剂或溶液中的过程。
酶分子修饰:通过各种方法使酶分子的结构发生某些改变,从而改变酶的某些特性和功能的技术过程称为酶分子修饰。
固定化酶:通过物理的或化学的手段,将酶束缚于水不溶的载体上,或将酶束缚在一定的空间内,限制酶分子的自由流动,但能使酶充分发挥催化作用;曾称其为水不溶酶或固相酶。
包埋法:将酶或含酶菌体包埋在各种多孔载体中,使酶固定化的方法称为包埋法。
核酶(ribozyme):具有催化功能的RNA分子,是生物催化剂。
又称核酸类酶、酶RNA、核酶类酶RNA。
多药耐药性( MDR):是指肿瘤细胞对一种化疗药物表现出耐药性,同时对其他许多结构不同、作用靶点亦不相同的化疗药物也表现出交叉耐药的现象,是通过化疗药物治疗肿瘤急需突破的一大难题。
常用核酸探针标记方法
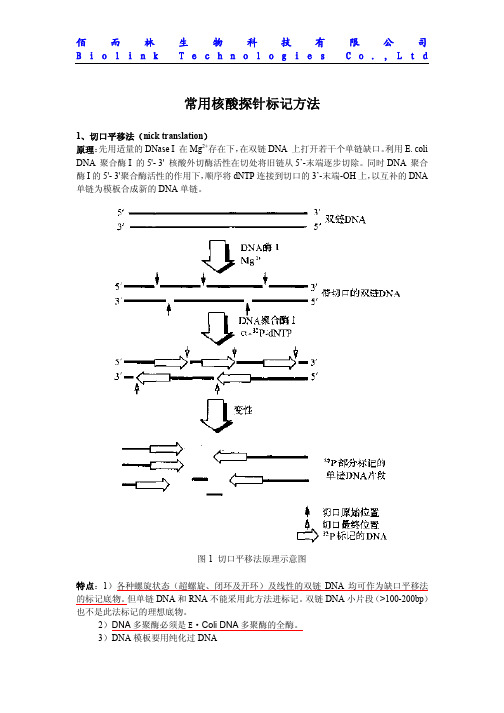
常用核酸探针标记方法1、切口平移法(nick translation)原理:先用适量的DNase I 在Mg2+存在下,在双链DNA 上打开若干个单链缺口。
利用E. coli DNA 聚合酶I 的5'- 3' 核酸外切酶活性在切处将旧链从5’-末端逐步切除。
同时DNA 聚合酶I的5'- 3'聚合酶活性的作用下,顺序将dNTP连接到切口的3’-末端-OH上,以互补的DNA 单链为模板合成新的DNA单链。
图1 切口平移法原理示意图特点:1)各种螺旋状态(超螺旋、闭环及开环)及线性的双链DNA均可作为缺口平移法的标记底物。
但单链DNA和RNA不能采用此方法进标记。
双链DNA小片段(>100-200bp)也不是此法标记的理想底物。
2)DNA多聚酶必须是E·Coli DNA多聚酶的全酶。
3)DNA模板要用纯化过DNA2、随机引物法(random priming)原理:将待标记的DNA探针片段变性后与随机引物一起杂交,然后以此杂交的寡核苷酸为引物,大肠杆菌DNA聚合酶I大片段(E·Coli DNA polymerase I Klenow Fragment)的催化下,合成与探针DNA互补的DNA链。
当反应液中含有标记的dNTP时,即形成标记的探针。
图2 随机引物标记法原理示意图特点:1)除了能进行双链DNA标记外,也可用于单链DNA和RNA探针的标记。
2)所得到的标记产物是新合成的DNA单链,而所加入的DNA片段本身并不能被标记。
3)新形成的标记DNA单链的长度与加入寡核苷酸引物的量成反比,因为加入的寡核苷酸数量越多,合成起点也越多,得到的片段的长度也越短。
按标准方法得到的标记产物长度一般为200-400bp。
3、末端标记与切口平移法和随机引物法不同,DNA末端标记法并不将DNA片段的全长进行标记,而是只将其一端(5’或3’端)进行部分标记。
其特点是可得到全长DNA片段,DNA片段并非均匀标记,标记活性不高。
医学分子生物学 7 分子生物学常用技术

5′
3′
*********G —OH
*********C T T A A — P
3′
5′
5′
3′
P A A T T C*********
OH— G*********
3′ 5′
EcoR I: dATP+ [α-32P]-dTTP
19
根据反应原理确定同位素反应底物名称
• DNA的切口平移标记法 • 随机引物标记法 • DNA的3′末端标记
• 极高的特异性
缺点 半寿期短, 故要随用随标 放射性污染
9
1. DNA探针放射性标记方法
(1) 切口平移标记法 (2) 随机引物标记法 (3) PCR标记法 (4) 5′-末端标记 (5) 3′-末端标记
10
(1) DNA的切口平移标记法
5′
3 ′ 双链DNA
3′
5′
DNase I,Mg2+
② 反应产物的长度与加入的寡核苷酸引物的量呈反比。
当需要较长片段探针,可适当减少随机引物的加入量。
③ 所得到的标记产物为新合成的DNA单链。当采用单链
DNA片段或RNA作为模板时,必须注意得到的标记探针并 不是其本身.
④ 反应条件要控制在pH值6.6,抑制Klenow DNA聚合酶
的3′-5′外切酶的活性。
第七章 分子生物学常用技术
1
第一节 核酸分子杂交
2
*核酸分子杂交:
具有一定互补序列的不同来源的核苷酸单 链在一定条件下按照碱基配对的原则形成
杂交双链的过程。
实质: 核酸分子的变性与复性过程
变性:将双链DNA分子解聚成为单链的过程 复性:使单链聚合成双链的过程,又称为退火. 特点:高度特异性和灵敏性 杂交的双方: (靶,target) --待测序列
基因诊断

第九章基因诊断基因诊断是通过检测基因的存在状态或缺陷对疾病作出诊断的方法。
基因诊断的主要技术:1、核酸分子杂交2、聚合酶链反应(PCR)3、基因芯片技术第一节核酸分子杂交技术核酸杂交(Nucleic acid hybridization)是指具有一定同源性的两条单链核酸在一定条件下,按碱基互补的原则重新配对形成双链的过程。
一、核酸杂交的基本原理DNA的变性和复性:在一定的条件(如适当的温度、有机溶剂存在等)下,DNA的双链可解开成为单链,这一过程称为DNA的变性(Denaturatioin)。
高温、低盐和有机溶剂促进DNA变性。
Tm值是反映DNA的热稳定性的一个参数,称为DNA的熔化温度,系指一半的双链DNA解离成为单链时的温度。
DNA的热稳定性或Tm值直接与其碱基组成特别是GC碱基对含量有关,GC碱基对含量越高,Tm值也越高。
DNA的杂交即复性(Renaturation)是变性的单链DNA在一定的条件下(低于Tm的温度下)与其互补序列退火形成双链的过程,因此杂交与Tm值相关。
影响杂交的主要因素:温度:一般在低于Tm约15至25度的温度下杂交速率最快。
盐浓度:钠离子增加杂交分子的稳定性,降低钠离子浓度强烈地影响Tm值和复性速率。
但当钠离子浓度超过0.4M时,对复性速度和Tm值影响不大。
甲酰胺:有机溶剂如甲酰胺能减少双链核酸的稳定性。
每增加1%的甲酰胺,DNA/DNA或DNA/RNA双链的Tm值减少0.72℃。
常用50%甲酰胺硫酸葡聚糖:使杂交速率增加,但有时可能增加杂交本底。
二、核酸探针的选择和标记核酸探针是指能与待检测的靶核酸序列互补杂交的某种已知核酸片段,它必须具有高度的特异性,并且带有某种适当的标记以便被检测。
(一)核酸探针的类型1、克隆的DNA片段,常用cDNA探针。
2、RNA探针(Riboprobe)RNA探针的优点是特异性高;杂交效率(灵敏度)更高。
适合于Northen杂交、原位杂交等。
《实验心理学》部分术语解释

实验心理学部分术语1.ABAB设计(ABAB design)也称为轮回设计(reversal design),是一种单被试实验设计。
其实验程序包括四个阶段:基线测试、实验处理条件下测试、返回基线测试段、实验处理条件下的再测等阶段。
2.听觉掩蔽(Auditory masking)就是指由于某一声音刺激的存在而使另一声音刺激的强度阈限提高的现象,这是一种常见的听觉现象。
3.被试间实验设计(Between-subjects experimental design)也叫做独立测量设计(independent-measures design)、独立组设计(independent groups design)或组间设计(between-groups design),是将互相独立但又相等的被试组分派在不同实验处理下进行测量,然后对各组测量结果进行比较的一种实验设计。
4.天花板效应(Ceiling effect)分数聚集在测量量表的高分端,使分数提高的可能性很少甚至没有。
这是一种全距效应。
5.混淆变量(Confounding variable)混淆变量(confounding variable),是指那些不拟研究但却与自变量一起发生系统性改变的变量,它使得被试心理或行为变化的原因变得无法确定。
6.知会同意表(Consent form)是研究者提供的书面声明,上面包含所有知会同意的项目和被试签名行。
研究实施之前,要将知会同意表提供给可能被试,以便他们了解在决定是否愿意参加研究之前需要知道的全部信息。
7.控制组(Control group)在一项研究中不接受处理或只接受安慰剂处理的被试组。
8.抵消平衡(Counterbalancing)在被试内设计中,一种为了使顺序效应和与时间有关的影响降到最小而将被试分成若干小组以便各小组以不同的顺序接受一系列实验处理的实验程序,这样做的目的是使在每一种可能的处理顺序上都有相等的被试按照各自顺序参加实验。
第十二章 印迹杂交技术
二、RNA印迹法(Northern blot)
操作:与Southern印迹极为相似,但不需要酶切可直 接变性转移; RNA经变性后再电泳(可用甲酰胺、甲醛等 使RNA变性,不能用碱,易使RNA降解); 电泳时不能加EB,影响RNA与膜结合; 严防RNase污染。 用途:检测RNA(定性或定量分析细胞内总RNA或 某一特定RNA,特别是分析mRNA的大小和含 量)——研究基因插入缺失等突变、基因表达(金 标准)。
一、核酸探针
——带有标记物且序列已知的核酸片段, 能与待测核酸中的特定序列特异杂交。
• 核酸探针是否合适是决定核酸杂交分析 能否成功的关键。
核酸杂交与分子探针
核酸探针应具有的条件
① 特异性高:只与待测核酸样品中的 互补序列杂交。 ② 带有标记物:标记物灵敏度高而稳 定,检测方便。
核酸探针的种类
常用核酸杂交分类
杂交方法
Southern印迹 Northern印迹 斑点杂交 菌落杂交和噬斑杂交
适用范围
检测经凝胶电泳分开的DNA 分子,需转印到膜上 检测经凝胶电泳分开的RNA 分子,需转移到膜上 检测未经分离的,固定在膜上的 DNA或RNA分子 检测固定在膜上的,经裂解后从 细菌和噬菌体中释放的 DNA分子 检测细胞或组织中的DNA或 RNA分子
2.非放射性标记物
优点:无环境污染,可长时间贮存。 • 生物素:
一种小分子水溶性维生素,连接在碱基上 和抗生物素蛋白(avidin)特异结合 抗生物素蛋白上连接有显色物质(酶、荧光素等) 。 • 酶促显色:
碱性磷酸酶(ALP或AKP):催化底物(5-溴-4-氯-4吲哚 磷酸,BCIP)和硝基蓝四氮唑(NBT),生成不溶性紫色 化合物二甲臜; 辣根过氧化物酶(HRP):催化底物二氨基联苯胺(DAB) 生成红棕色沉淀物;或催化底物四氨基联苯胺(TMB)生 成蓝色沉淀物。
核酸分子杂交
影响复性的因素:
❖ 单链核酸的起始浓度 ❖ 核酸链长度(分子量) ❖ 核酸分子的复杂性,即核酸分子中不同序列的总长度 ❖ 温度 ❖ 离子强度
hybridization single strand to double strand
DNA/DNA、DNA/RNA、RNA/RNA
影响核酸分子杂交的因素:
➢DNA酶Ⅰ的用量和E.coli DNA聚合酶的质量会影响产物 片段的大小。
➢DNA模板中的抑制物如琼脂糖会抑制酶的活性, 故应使 用仔细纯化后的DNA。
(2)随机引物法 (random priming)
随机引物:含有各种可能 排列顺序的寡核苷酸片段
的混合物。46=4096
DNA聚合酶ⅠKlenow片段: 5’→3’DNA聚合酶活性 弱3’→5’外切核酸酶活性 无5’→3’外切核酸酶活性
体+ ABC试剂
(2)显色反应 酶法:通过酶促反应使其底物形成有颜色的反应产物
碱性磷酸酶:
BCIP(5-溴-4氯 -3吲哚磷酸盐-4甲基胺蓝) NBT (四氮唑蓝)
辣根过氧化物酶:DAB(二氨基联苯胺)→红棕色
TMB(四甲基联苯胺)→蓝色
第三节 核酸分子杂交技术
➢ 膜上印迹杂交 ➢ 核酸原位杂交
将各种“探针”固定在基质 上,用于检测受检的各种标记样 品中与探针互补的核酸物质的变 化。
核酸杂交分类表
杂交方法
适用范围
Southern 印迹
检测经凝胶电泳分开的DNA分子,需转印到膜上
Northern 印迹
检测经凝胶电泳分开的RNA分子,需转印到膜上
斑点及狭缝印迹杂交 检测未经分离的、固定在膜上的DNA或RNA分子
制备探针
(1)缺口平移法
(推荐)比较基因组杂交原理
1 CGH的基本原理CGH是一种将消减杂交和FISH相结合,用于检测两个(或多个)基因组间相对DNA拷贝数变化(如扩增、增益(gains)、复制和丢失等),并将这些异常定位在染色体上,因此又称DNA拷贝数核型(DNA copy number karyotype)技术〔4〕。
与传统的原位杂交方法相反,该法不是将单一的、已定的DNA探针杂交在受检的分裂中期或间期的细胞上,而以被检组织的基因组DNA为杂交检测样本,正常组织的DNA样本为参照,分别用不同颜色的荧光标记,两者按1∶1混合,与正常淋巴细胞中期染色体进行杂交(即反向原位杂交),再通过检测两种颜色的荧光强度,根据颜色的比例来显示基因组的结构状况。
如果探针中某一染色体或染色体亚区呈现过度表达或低表达,则在正常染色体的相应区域内便呈现较强或较低的信号,表明该区域存在DNA序列的增益或丢失〔5〕。
2 CGH的基本方法随着CGH技术的广泛应用,其操作方法已逐步完善,经验表明,CGH技术的建立和常规应用,对每一步操作都必须细致。
CGH的主要步骤包括:(1)正常中期染色体的制备;(2)从肿瘤组织中分离高分子量的基因组DNA;(3)用不同的haptens标记正常和肿瘤DNA;(4)标记的DNA与正常中期染色体原位杂交;(5)荧光显微镜检查和数字化图像分析;(6)对中期染色体产生的绿红荧光密度比率相对应的拷贝数异常进行分析〔3~5〕。
2.1 正常中期染色体的制备正常中期染色体充当CGH杂交的靶,CGH分析的质量主要依赖中期染色体的特性,中期染色体的制备采用常规PHT刺激和氨甲喋呤同步化处理的外周血淋巴细胞培养技术,一次应制备大量片子,以保证整个实验都使用同一批片子。
此外,对制备的中期染色体玻片应选择重叠较少,形态保持较好,染色体较长的标本。
2.2 肿瘤标本的选择和基因组DNA的分离分离DNA的标本应尽可能的选择具有典型肿瘤组织学特征和比例高的恶性细胞,弃除坏死组织和炎症区域及周边正常细胞,因其中含有坏死细胞降解的DNA和一些正常细胞,可显著消弱拷贝数改变的分辨能力。
分子生物学常用的各种技术项目举例
环境。
溴化乙锭(EB)为致癌剂,操作时应戴手套,
尽量减少台面污染。
凝胶电泳原理-EB染色
核酸电泳的指示剂
• 核酸电泳常用的指示剂有两种:
•
⑴ 溴酚蓝
•
⑵ 二甲苯青,
溴酚蓝(bromophenol blue, Bb)呈蓝紫色;
二甲苯晴(xylene cyanol, Xc)呈蓝青色,它 携带的电荷量比溴酚蓝少,在凝胶中的的迁移 率比溴酚蓝慢。
分子生物学常用技术
3.1 凝胶电泳技术 3.2 分子杂交技术 3.3 P凝胶电泳技术
凝胶电泳(Gel electrophoresis)是分子克隆的中 心技术之一。
1. 琼脂糖凝胶用于分离200~1000bp的片段;
操作简单、快速,且分离范围广,分辨率高 2. 聚丙烯酰胺凝胶用于分离5~500bp的片段;
1. 放射性核素的标记 (1)切口平移法:该技术由Kelly等于1970年创立。是目前最常用 的一种脱氧核糖核酸的探针标记法。是利用大肠杆菌DNA聚合酶Ⅰ 的多种酶促活性将标记的dNTP掺入到新形成的DNA链中去,从而 合成高比活的均匀标记的DNA探针。线状、超螺旋及带缺口的双链 DNA匀可作为模板。首先,极微量的DNaseⅠ在Mg2+的存在下,在 DNA链上随机形成单链切口。利用大肠杆菌DNA聚合酶的5`→3′核 酸外切酶活性在切口处将从旧链5`—末端逐步切除。同时,在DNA 聚合酶Ⅰ的5`→3′聚合酶活性的催化下,
基因探针根据标记方法不同可粗分为放射性探针和非放射性探 针两大类,根据探针的核酸性质不同又可分为DNA探针,RNA探针, cDNA探针等几类,DNA探针还有单链和双链之分。 1.基因组DNA探针: DNA探针是最常用的核酸探针,指长度在几百 碱基对以上的双链DNA或单链DNA探针。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Random priming
DNA聚合酶要进行DNA聚合反应时一定要有引子 (primer),因为它只能以引子所提供的3’-OH为起始点进行延伸(extension) 的工作,而不能就地重新合成新的DNA (de novo synthesis)。
一般实验常以人工合成的寡核苷酸为引子进行DNA 聚合反应,这时固然可以根据已知序列设计核酸引子,但也可以采用逢机引子(random primers)。
所谓逢机引子即其序列为逢机序列,不经特别设计,目前应用最广者是以自动化机器所合成的、六到八个核苷酸长的寡核苷酸混合物;反应进行中若有任何一条逢机引子结合到模版DNA,即可引导DNA聚合反应的进行。
以random priming方法进行核酸标定,是以逢机引子引导Klenow fragment(large fragment of E. coli DNA polymerase I) 进行DNA聚合反应,并在反应过程中添加 [α-32P]dNTP 或DIG-11-dUTP等标定物质。
而为了避免random priming也发生于载体部份的DNA,最好不要直接以质体DNA为模版进行标定反应,而应预先将目标DNA自载体切除出來。
仪器用具:微量离心机;沸水浴及冰浴
药品试剂:
模版DNA (pBlueGus 的Bam HI-Hin dIII DNA 片段,将由助教提供)
0.2 M EDTA, pH 8.0
4 M LiCl
Random priming kit (Roche):
Hexanucleotide mix (random primer)
dNTP mixture (1 mM dATP, 1 mM dCTP, 1 mM dGTP, 0.65 mM dTTP, 0.35
mM DIG-11-dUTP, pH 7.5)
Klenow enzyme (2 U/μL)
绝对酒精
70%酒精
TE (10 mM Tris-Cl, pH 8.0; 1 mM EDTA)
方法步骤:
◆以下反应条件与步骤主要参照random priming kit 制造厂商所提供的产品说明书。
1) 取100 ng 模版DNA,加入适量去离子水,把体积补足为15 μL。
2) 于沸水中加热10 min。
◆请记得以夹子固定微量离心管的管盖部份,以免离心管在加热过程中盖子爆开、进水!
3) 加热后,迅速把微量离心管移至冰浴中,静置数分钟。
4) 离心数秒后,依序加入下列三种试剂:
2 μL hexanucleotide mix
2 μL dNTP mixture
1 μL Klenow enzyme
5) 以指头轻弹离心管,以便混合均匀。
6) 短暂离心后,置于37℃恒温水槽中作用2 h。
◆反应时间至少须要1 h,最长可反应过夜。
7) 作用完毕的后,加入2 μL 的0.2 M EDTA,以便终止DNA 聚合反应。
8) 加入2.5 μL 4 M LiCl及75 μL纯酒精,混合均匀,置 -20℃过夜。
隔天:
1) 于13,000 rpm,4℃离心15 min。
2) 倒出上清液,加入50 μL 的70%酒精,再离心5 min。
3) 倒出上清液并风干DNA。
4) 最后以50 μL TE 溶解DNA。