同位素丰度表
地球表层化学元素丰度

地球表层化学元素丰度一、丰度的概念:即为该元素在自然体中的丰富程度abundance of elements),是指一种化学元素在某个自然体中的重量占这个自然体总重量的相对份额(如百分数)。
丰度表示方法主要分为重量丰度、原子丰度和相对丰度。
二、定义:同位素在自然界中的丰度,又称天然存在比,指的是该同位素在这种元素的所有天然同位素中所占的比例。
丰度的大小一般以百分数表示;人造同位素的丰度为零。
周期表上所列的原子量实际上是各种同位素按丰度加权的平均值,这是因为各种同位素在自然界中往往分布的比较均匀,取平均值计算比较准确。
一种化学元素在某个自然体中的重量占这个自然体总重量的相对份额(如百分数),称为该元素在自然体中的丰度。
三、研究地球表层化学元素丰度的意义研究元素丰度是研究地球化学基础理论问题的重要素材之一。
宇宙天体是怎样起源的?地球又是如何形成的?地壳与地幔中的主要元素有什么不一样?生命体是怎么产生和演化的?这些研究都离不开地球化学体系中元素丰度分布特征和规律。
元素丰度是每一个地球化学体系的基本数据,可在同一或不同体系中用元素的含量值来进行比较,通过纵向(时间)、横向(空间)上的比较,了解元素动态情况,从而建立起元素集中、分散、迁移活动等一些地球化学概念。
从某种意义上来说,也就是在探索和了解丰度这一课题的过程中,逐渐建立起近代地球化学。
四、发现历史自从1889年F.W.克拉克发表元素在地壳中的平均含量的资料以来,人们已经积累了大量有关陨石、太阳、恒星、星云等各种天体中元素及其同位素分布的资料。
1937年,戈尔德施米特首次绘制出太阳系的元素丰度曲线。
1956年,修斯和尤里根据地球、陨石和太阳的资料绘制出更详细、更准确的元素丰度曲线。
1957年,伯比奇夫妇、福勒和霍伊尔就是以该丰度曲线为基础,提出他们的核合成假说的。
四十年代,人们只知道大多数恒星的化学组成与太阳相似,因而就认为分布在整个宇宙的元素丰度可能是一样的。
南京大学同位素地质学-10-K-Ar-Ar-Ar同位素年代学(含作业)

40 Ar 36 Ar
总
40 Ar 36 Ar
堇青石、辉石和电气石经常含有过剩40Ar,而角闪 石、长石、金云母、黑云母和方钠石中较少出现 过 剩 Ar 。 过 剩 40 ( York and MacIntyre, 1965; Livingston et al., 1967) 40Ar也曾经在金刚石中发现(Ozima et al., 。 1983)
(2)
两边除于36Ar,得:
40 36
Ar Ar
40 36
Ar Ar
i
e
40 K 36 Ar
et
1
y
x
该方程具有以下形式: y = b + xm
(4) (5)
把方程(4)中初始(40Ar/36Ar)i一项扩展为包括大 气Ar和继承Ar两项:
Ar是一个惰性气体元素,原子量为39.948,Ar 在地球大气中的含量为0.93%。根据Nier (1950) 的测定,地球大气中Ar的同位素丰度为:
40Ar 99.60%
38Ar 0.063%
36Ar 0.337% 因而大气40Ar/36Ar=295.5。
2、K-Ar法定年
放 射 性 的 40K 分支衰变为 40Ca和40Ar:
由于全岩抵御热扰动保存Ar的能力最差,因此 K-Ar定年中,只有当所有矿物相都太细而无法 分离时,才采用全岩样品。
一般而言,K-Ar年龄代表矿物/岩石冷却到Ar 扩散丢失微乎其微的温度以来所经历的时间。
另一方面,一些含K矿物中发现存在过剩的40Ar, 在K含量较低或较年轻的矿物中,过剩40Ar的存在 对K-Ar定年的影响最明显。
关于同位素测定

同位素测定报告#12732.05“PMU”型铜粉批号#3/05-07由TAG GIREDMET抽样。
原子分率的测定使用了火花源质谱分析法。
应用了日本电子公司(日本)制造的JMS-01-BM2双聚焦质谱分析仪。
高分辨率质谱是在Ilford-Q板上摄取的。
Joyce Loebl(英国)的MDM6测微密度计和NOVA 4(美国)在线微型计算机被用于识别质谱线。
产生量估算由原版的MS&GC实验室软件计算得出。
同位素丰度测量的相对标准偏差为0.01-0.05。
稀有气体和超铀元素没有制进表格中,因为它们的浓度低于百万分之0.001的检测极限。
结果用原子百分比表示“PMU”型铜粉的化学成分证书批号#3/05-07净重 199,785kg装于14个箱子中的1392个玻璃安瓿实验室MS&GC Lab任何对于此样本的参考均要引用以上的名称和号码。
铜粉中杂质(镁、铝、钛、铁、镍、钼、钶、锑)的总含量不超过重量的0.002%。
铜粉的纯度级别为99.998%。
此数据由100%铜粉和杂质总量的差额计算得出。
杂质列表与TU 1793-001-56993504-2004相一致。
铜粉在放射性方面是安全的。
铜粉的总放射性不超过1.10-11 Ci/g。
样品由TAG Giredmet抽样。
抽样程序报告始于2005年5月16日。
箱子由TAG Giredmet “GAC-68”铅垂探测。
铜粉中杂质含量与检测技术列于报告#12732.05中(请翻页)。
杂质检测报告#12732.05球状铜粉批号#3/05-07样品由TAG GIREDMET抽样。
总杂质分析采用火花源质谱分析法。
应用了日本电子公司(日本)制造的JMS-01-BM2双聚焦质谱分析仪。
高分辨率质谱是在Ilford-Q板上摄取的。
Joyce Loebl(英国)的MDM6测微密度计和NOVA 4(美国)在线微型计算机被用于识别质谱线。
产生量估算由原版的MS&GC 实验室软件计算得出。
质谱知识总结

第四章:质谱法第一节经验1)在正离子模式下,样品主要以[M+H]+、[M+Na]+、[M+K]+准分子离子被检测;在负离子模式下,样品则大多以[M-H]-、[M+Cl]-准分子离子被检测。
2)正离子模式下,样品还会出现M—1(M-H),M—15(M-CH3), M—18(M-H2O),M—20(M—HF), M-31(M—OCH3)等的峰.分子离子峰应具有合理的质量丢失。
也即在比分子离子质量差在4—13,21—26,37-,50-53,65,66 是不可能的也是不合理的,否则,所判断的质量数最大的峰就不是分子离子峰,。
因为一个有机化合物分子不可能失去4~13个氢而不断键。
如果断键,失去的最小碎片应为CH3,它的质量是15个质量单位.3)分子离子峰应为奇电子离子,它的质量数应符合氮规则:在有机化合物中,凡含有偶数氮原子或不含氮原子的,相对分子质量一定为偶数,反之,凡今吸奇数氮原子的,相对分子质量一定是奇数,这就是氮规则。
运用氮规则将有利于分子离子峰的判断和分子式的推定,经元素分析确定某化合物的元素组成后,若最高质量的离子的质量与氮规则不符,则该离子一定不是分子离子。
如果某离子峰完全符合上述3项判断原则,那么这个离子峰可能是分子离子峰;如果3项原则中有一项不符合,这个离子峰就肯定不是分子离子峰.应该特别注意的是,有些化合物容易出现M-1峰或M+1峰。
基峰研究高质量端离子峰, 确定化合物中的取代基M-15(CH3); M-16(O, NH2M-17(OH, NH3); M-18(H2O);M-19(F); M-26(C2H2);M-27(HCN, C2H3); M-28(CO, C2HM-29(CHO, C2H5); M-30(NO);M-31(CH2OH, OCH3); M-32(S, CHM-35(Cl); M-42(CH2CO, CH M-43(CH3CO, C3H7); M-44(CO2, CS15 (。
质谱解谱教程 (1)

第四章:质谱法第一节: 概述1.1 发展历史1.1886年,E. Goldstein在低压放电实验中观察到正电荷粒子.2. 1898年,W. Wen发现正电荷粒子束在磁场中发生偏转.3.现代质谱学之父: J. J. Thomson(获1906年诺贝尔物理奖).4.1922年, F.W.Aston[英]因发明了质谱仪等成就获诺贝尔化学奖. 1942年, 第一台商品质谱仪.5.50年代起,有机质谱研究(有机物离子裂解机理, 运用质谱推断有机分子结构)6.各种离子源质谱, 联机技术的研究及其在生物大分子研究中的应用(CI, FD, FAB, ESI-MS等)1.2 特点:1.灵敏度高(几微克甚至更少的样品, 检出极限可达10-14克)2.是唯一可以确定分子式的方法.3.分析速度快(几秒)4.可同色谱联用.第二节: 基本原理2.1基本原理质谱是唯一可以确定分子式的方法。
而分子式对推测结构是至关重要的。
质谱法的灵敏度远远超过其它方法,测试样品的用量在不断降低,而且其分析速度快,还可同具有分离功能的色谱联用。
具有一定压力的气态有机分子,在离子源中通过一定能量(70ev)的电子轰击或离子分子反应等离子化方式,使样品分子失去一个电子产生正离子, 继而还可裂解为一系列的碎片离子,然后根据这些离子的质荷比(m/z e)的不同,用磁场或磁场与电场等电磁方法将这些正离子进行分离和鉴定。
由此可见质谱最简单形式的三项基本功能是:(1)气化挥发度范围很广的化合物;(2)使气态分子变为离子(除了在气化过程中不产生中性分子而直接产生离子的化合物);(3)根据质荷比(m/z e)将它们分开,并进行检测、记录。
由于多电荷离子产生的比例比单电荷离子要小得多,通常取z等于1,e为常数(1个电子的电荷),因而就表征了离子的质量。
这样,质谱就成为了产生并称量离子的装置。
由于各化合物所形成的离子的质量以及各种离子的相对强度都是各化合物所特有的,故可从质谱图形中确定分子量及其结构。
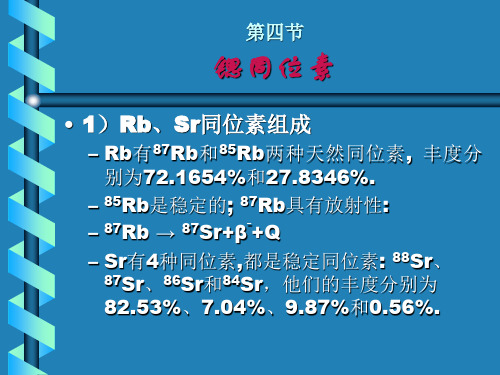
Sr同位素解析
2 、通过测定矿脉中脉石矿物液体包 裹体的 Rb-Sr 同位素组成确定成矿年 龄。
• 通过分析液体包裹体的Rb-Sr同位素组成有可能确定成 矿年龄。 • 为使测定的年龄值真正代表成矿事件,所分析的液体包 裹体必须都是原生的, • 石英最常见,化学纯度又高,其同位素交换不会影响液 体包裹体的Rb-Sr同位素组成,因而石英是一个比较理 想的测定对象。 • 据T. J. Shephard和D. P. F. Darbyshire(1981) 研究,Carrock Fell钨矿床中,早、晚两期石英中液 体包裹体的Rb-Sr同位素分析结果构成一条很好的等时 线。等时年龄为393士5Ma,与矿脉中白云母的K-Ar 年龄(387士6Ma)在误差范围内一致,因而393Ma可 以作为该矿床的成矿年龄。
3)地球锶同位素
• 地球的初始值可能与陨石相当。 • 对被任为起源于地幔的,又没有受到地壳 Sr 明显混入的玄武岩和大的辉长岩体的 分 析 , 得 到 上 地 幔 的 87Sr/86Sr=0.704±0.002
• 对900个年轻的玄武质和中性成分的火山 岩的 87Sr/86Sr 值统计,表明大多数岩石 落在了0.704±0.002的范围内。 • 但是随着环境的不同比值也有差别(见 图),原因可能是多方面的,如大陆物质 的混染,与海水的作用,或地幔的分异。
1
86
87
87
花岗岩:
• 与地幔有关的: • (87Sr/86Sr)0=0.704±0.002 • 地壳成因的: • (87Sr/86Sr)0=0.720±0.005
• 玄武岩类 • 大洋玄武岩:0.7012—0.7059 • 大陆玄武岩:0.703—0.712(可能是受到地 壳物质的影响。 • 岛弧安山岩 • 世界若干主要的岛弧地区的安山岩、英安岩和 玄武岩的锶同位素比值与大洋玄武岩是重叠的, 在0.7036—0.7066之间。 • 超基性岩 • 阿尔卑斯性超铁镁质岩类和火山岩中的超铁镁 质岩捕虏体被认为近似地代表原始地幔的组成。 • 大部分超铁镁质岩为:0.7012—0.7057 • 但是,地幔肯定是不均一的,因此有些超铁镁 质岩可能会高于这个范围。
硫同位素地球化学

硫同位素地球化学硫有四种稳定同位素:32S,33S,34S,36S,其大致丰度为95.02%,0.75%,4.21%,0.02%。
以S34S/32S来表示硫同位素的分馏。
硫同位素标准是CDT。
自然界硫同位素组成范围大,最重的硫酸盐的δ34S为95‰,最轻的硫化物为-65‰。
等亚稳定络合物,不同价态含硫原子团富集34S的能力不同。
硫化物和硫酸盐之间的氧化还原作用,地表条件下微生物的还原作用,以及硫酸盐和硫化物的溶解度的极大差异,是造成硫的轻、重同位素分馏的重要原因。
7.4.1硫同位素分馏硫同位素的分馏过程主要有:各种硫化合物(硫酸盐、硫化物)之间的同位素交换反应,是一种平衡的同位素分馏;硫化合物发生价态改变的单向化学反应,是一种不可逆的氧化还原反应,具有动力分馏的性质,它既可是无机环境改变引起,也可是生物细菌的有机作用,而且生物细菌的作用往往能引起大的动力分馏。
岩浆环境和250℃以上热液流体中的硫酸盐和溶解的硫化氢、火山喷气口的二氧化硫和硫化氢气体、热液流体中溶解的硫化氢和沉淀的硫化物等是同位素平衡交换的典型体系,平衡条件下硫的重同位素倾向于富集在具有较强硫键的化合物中,由高价到低价,δ34S依次降低,因此各种含硫原子团7.3表示了一些含硫化合物和H2S之间的同位素分馏曲线,硫化物—H2S达到平衡时各种硫化物富34S的顺序大致如下:辉钼矿>黄铁矿>闪锌矿(磁黄铁矿)>H2S>黄铜矿>(HS1-)>铜蓝>方铅矿>辰砂>辉铜矿(辉锑矿)>辉银矿>S2-。
实测数据和理论计算结果大致相符。
低很小。
硫化合物的无机氧化还原作用是一种非平衡的单向化学反应。
硫化物氧化为硫酸盐是一种动力分馏过程,但分馏不明显。
硫酸盐无机还原为硫化物制,它的同位素效应比较明显。
但硫酸盐的无机还原作用需要较高的活化能,低温下参与反应的物质数量很少,因而有实际意义的反应多发生在约250℃以上的热液体系和地壳深部环境,如热液流体中水溶性硫酸盐被还原成水溶硫化物,火山气体中SO2被H2S还原底火山作用条件下,反应是海水演化成为成矿热液的重要反应。
质谱

第四章 质谱(Mass Specrometry)早在1921年,就已经出现了质谱仪,那时UV 、IR 、NMR 、GC 及HPLC 等分析技术都还没有出现。
早期质谱仪的最大成功是同位素的发现及其相对丰度的测量。
所以,那时的质谱仪主要用来作同位素的研究,随着石油化工的发展,到了40~50年代,质谱的应用开始向石油化工转移,1951年Brown 发表了测定汽油烃类组成的质谱法。
1957年I. Holms 和F. Moreell 首次实现了色谱-质谱联用。
50年代末,Beyon 、Bienann 和Mclafferty 都提出:官能团对分子中化学键的断裂有引导作用,因此有机质谱得到迅速发展。
4.1 概述质谱用于结构分析有点像:»¨»»ò»é»é»(»»»»»»÷»»¬»»»»)(»»»»»»÷»»¬»»»»)»»»ó»»¨»即:»»ö«»¬»»»¬»é»»«»¬»»¬»(M)( M )+根据各种碎片离子的质荷比及其相对丰度,就可以进行结构分析。
4.1.1定义质谱——按照离子的质荷比的大小依次排列的谱图。
从本质上讲,质谱并非电磁波谱,而是物质的质量谱,因而质谱中并无透光率、吸光度、波长等概念。