Impurity and correlation effects on transport in one-dimensional quantum wires
ASTM_B117-2007 盐雾试验规程

Designation:B117–07Standard Practice forOperating Salt Spray(Fog)Apparatus1This standard is issued under thefixed designation B117;the number immediately following the designation indicates the year of original adoption or,in the case of revision,the year of last revision.A number in parentheses indicates the year of last reapproval.A superscript epsilon(e)indicates an editorial change since the last revision or reapproval.This standard has been approved for use by agencies of the Department of Defense.1.Scope1.1This practice covers the apparatus,procedure,and conditions required to create and maintain the salt spray(fog) test environment.Suitable apparatus which may be used is described in Appendix X1.1.2This practice does not prescribe the type of test speci-men or exposure periods to be used for a specific product,nor the interpretation to be given to the results.1.3The values stated in SI units are to be regarded as the standard.The values given in parentheses are for information only.1.4This standard does not purport to address all of the safety concerns,if any,associated with its use.It is the responsibility of the user of this standard to establish appro-priate safety and health practices and determine the applica-bility of regulatory limitations prior to use.2.Referenced Documents2.1ASTM Standards:2B368Test Method for Copper-Accelerated Acetic Acid-Salt Spray(Fog)Testing(CASS Test)D609Practice for Preparation of Cold-Rolled Steel Panels for Testing Paint,Varnish,Conversion Coatings,and Related Coating ProductsD1193Specification for Reagent WaterD1654Test Method for Evaluation of Painted or Coated Specimens Subjected to Corrosive EnvironmentsE70Test Method for pH of Aqueous Solutions With the Glass ElectrodeE691Practice for Conducting an Interlaboratory Study to Determine the Precision of a Test MethodG85Practice for Modified Salt Spray(Fog)Testing3.Significance and Use3.1This practice provides a controlled corrosive environ-ment which has been utilized to produce relative corrosion resistance information for specimens of metals and coated metals exposed in a given test chamber.3.2Prediction of performance in natural environments has seldom been correlated with salt spray results when used as stand alone data.3.2.1Correlation and extrapolation of corrosion perfor-mance based on exposure to the test environment provided by this practice are not always predictable.3.2.2Correlation and extrapolation should be considered only in cases where appropriate corroborating long-term atmo-spheric exposures have been conducted.3.3The reproducibility of results in the salt spray exposure is highly dependent on the type of specimens tested and the evaluation criteria selected,as well as the control of the operating variables.In any testing program,sufficient repli-cates should be included to establish the variability of the results.Variability has been observed when similar specimens are tested in different fog chambers even though the testing conditions are nominally similar and within the ranges speci-fied in this practice.4.Apparatus4.1The apparatus required for salt spray(fog)exposure consists of a fog chamber,a salt solution reservoir,a supply of suitably conditioned compressed air,one or more atomizing nozzles,specimen supports,provision for heating the chamber, and necessary means of control.The size and detailed con-struction of the apparatus are optional,provided the conditions obtained meet the requirements of this practice.4.2Drops of solution which accumulate on the ceiling or cover of the chamber shall not be permitted to fall on the specimens being exposed.1This practice is under the jurisdiction of ASTM Committee G01on Corrosionof Metals and is the direct responsibility of Subcommittee G01.05on LaboratoryCorrosion Tests.Current edition approved March1,2007.Published March2007.Originallyapproved st previous edition approved in2003as B117–03.2For referenced ASTM standards,visit the ASTM website,,orcontact ASTM Customer Service at service@.For Annual Book of ASTMStandards volume information,refer to the standard’s Document Summary page onthe ASTM website.Copyright©ASTM International,100Barr Harbor Drive,PO Box C700,West Conshohocken,PA19428-2959,United States.4.3Drops of solution which fall from the specimens shall not be returned to the solution reservoir for respraying.4.4Material of construction shall be such that it will not affect the corrosiveness of the fog.4.5All water used for this practice shall conform to Type IV water in Specification D1193(except that for this practice limits for chlorides and sodium may be ignored).This does not apply to running tap water.All other water will be referred to as reagent grade.5.Test Specimens5.1The type and number of test specimens to be used,as well as the criteria for the evaluation of the test results,shall be defined in the specifications covering the material or product being exposed or shall be mutually agreed upon between the purchaser and the seller.6.Preparation of Test Specimens6.1Specimens shall be suitably cleaned.The cleaning method shall be optional depending on the nature of the surface and the contaminants.Care shall be taken that specimens are not recontaminated after cleaning by excessive or careless handling.6.2Specimens for the evaluation of paints and other organic coatings shall be prepared in accordance with applicable specification(s)for the material(s)being exposed,or as agreed upon between the purchaser and the supplier.Otherwise,the test specimens shall consist of steel meeting the requirements of Practice D609and shall be cleaned and prepared for coating in accordance with the applicable procedure of Practice D609.6.3Specimens coated with paints or nonmetallic coatings shall not be cleaned or handled excessively prior to test.6.4Whenever it is desired to determine the development of corrosion from an abraded area in the paint or organic coating, a scratch or scribed line shall be made through the coating with a sharp instrument so as to expose the underlying metal before testing.The conditions of making the scratch shall be as defined in Test Method D1654,unless otherwise agreed upon between the purchaser and the seller.6.5Unless otherwise specified,the cut edges of plated, coated,or duplex materials and areas containing identification marks or in contact with the racks or supports shall be protected with a suitable coating stable under the conditions of the practice.N OTE1—Should it be desirable to cut test specimens from parts or from preplated,painted,or otherwise coated steel sheet,the cut edges shall be protected by coating them with paint,wax,tape,or other effective media so that the development of a galvanic effect between such edges and the adjacent plated or otherwise coated metal surfaces,is prevented.7.Position of Specimens During Exposure7.1The position of the specimens in the salt spray chamber during the test shall be such that the following conditions are met:7.1.1Unless otherwise specified,the specimens shall be supported or suspended between15and30°from the vertical and preferably parallel to the principal direction offlow of fog through the chamber,based upon the dominant surface being tested.7.1.2The specimens shall not contact each other or any metallic material or any material capable of acting as a wick.7.1.3Each specimen shall be placed to permit unencum-bered exposure to the fog.7.1.4Salt solution from one specimen shall not drip on any other specimen.N OTE2—Suitable materials for the construction or coating of racks and supports are glass,rubber,plastic,or suitably coated wood.Bare metal shall not be used.Specimens shall preferably be supported from the bottom or the side.Slotted wooden strips are suitable for the support offlat panels.Suspension from glass hooks or waxed string may be used as long as the specified position of the specimens is obtained,if necessary by means of secondary support at the bottom of the specimens.8.Salt Solution8.1The salt solution shall be prepared by dissolving561 parts by mass of sodium chloride in95parts of water conforming to Type IV water in Specification D1193(except that for this practice limits for chlorides and sodium may be ignored).Careful attention should be given to the chemical content of the salt.The salt used shall be sodium chloride with not more than0.3%by mass of total impurities.Halides (Bromide,Fluoride,and Iodide)other than Chloride shall constitute less than0.1%by mass of the salt content.Copper content shall be less than0.3ppm by mass.Sodium chloride containing anti-caking agents shall not be used because such agents may act as corrosion inhibitors.See Table1for a listing of these impurity restrictions.Upon agreement between the purchaser and the seller,analysis may be required and limits TABLE1Maximum Allowable Limits for Impurity Levels inSodium Chloride A,B,CImpurity Description Allowable Amount Total Impurities#0.3% Halides(Bromide,Fluoride and Iodide)excluding Chloride<0.1% Copper<0.3ppmAnti-caking Agents noneA A common formula used to calculate the amount of salt required by mass to achieve a5%salt solution of a known mass of water is:0.0533Mass of Water5Mass of NaCl requiredThe mass of water is1g per1mL.To calculate the mass of salt required in grams to mix1L of a5%salt solution,multiply0.053by1000g(35.27oz,the mass of1L of water).This formula yields a result of53g(1.87oz)of NaCl required for each liter of water to achieve a5%salt solution by mass.The0.053multiplier for the sodium chloride used above is derived by the following:1000g~mass of a full L of water!divided by0.95~water is only95%of the total mixture by mass!yields1053gThis1053g is the total mass of the mixture of one L of water with a5%sodium chloride concentration.1053g minus the original weight of the L of water,1000g, yields53g for the weight of the sodium chloride.53g of total sodium chloride divided by the original1000g of water yields a0.053multiplier for the sodium chloride.As an example:to mix the equivalent of200L(52.83gal)of5%sodium chloride solution,mix10.6kg(23.37lb)of sodium chloride into200L(52.83gal)of water. 200L of water weighs200000g.200000g of water30.053(sodium chloride multiplier)=10600g of sodium chloride,or10.6kg.B In order to ensure that the proper salt concentration was achieved when mixing the solution,it is recommended that the solution be checked with either a salimeter hydrometer or specific gravity hydrometer.When using a salimeter hydrometer,the measurement should be between4and6%at25°C(77°F).When using a specific gravity hydrometer,the measurement should be between1.0255and1.0400at 25°C(77°F).C If the purity of the salt used is>99.9%,then the limits for halides can be ignored.This is due to the fact that the halides cannot be$0.1%with a salt purity of>99.9%.If the salt used is of lower purity,then test forhalides. --````````,`,,,`,``,`,`,`````-`-`,,`,,`,`,,`---established for elements or compounds not specified in the chemical composition given above.8.2The pH of the salt solution shall be such that when atomized at 35°C (95°F)the collected solution will be in the pH range from 6.5to 7.2(Note 3).Before the solution is atomized it shall be free of suspended solids (Note 4).The pH measurement shall be made at 25°C (77°F)using a suitable glass pH-sensing electrode,reference electrode,and pH meter system in accordance with Test Method E 70.N OTE 3—Temperature affects the pH of a salt solution prepared from water saturated with carbon dioxide at room temperature and pH adjust-ment may be made by the following three methods:(1)When the pH of a salt solution is adjusted at room temperature,and atomized at 35°C (95°F),the pH of the collected solution will be higher than the original solution due to the loss of carbon dioxide at the higher temperature.When the pH of the salt solution is adjusted at room temperature,it is therefore necessary to adjust it below 6.5so the collected solution after atomizing at 35°C (95°F)will meet the pH limits of 6.5to 7.2.Take about a 50-mL sample of the salt solution as prepared at room temperature,boil gently for 30s,cool,and determine the pH.When the pH of the salt solution is adjusted to 6.5to 7.2by this procedure,the pH of the atomized and collected solution at 35°C (95°F)will come within this range.(2)Heating the salt solution to boiling and cooling to 35°C (95°F)and maintaining it at 35°C (95°F)for approximately 48h before adjusting the pH produces a solution the pH of which does not materially change when atomized at 35°C (95°F).(3)Heating the water from which the salt solution is prepared to 35°C (95°F)or above,to expel carbon dioxide,and adjusting the pH of the salt solution within the limits of 6.5to 7.2produces a solution the pH of which does not materially change when atomized at 35°C (95°F).N OTE 4—The freshly prepared salt solution may be filtered or decanted before it is placed in the reservoir,or the end of the tube leading from the solution to the atomizer may be covered with a double layer of cheesecloth to prevent plugging of the nozzle.N OTE 5—The pH can be adjusted by additions of dilute ACS reagent grade hydrochloric acid or sodium hydroxide solutions.9.Air Supply9.1The compressed air supply to the Air Saturator Tower shall be free of grease,oil,and dirt before use by passing through well-maintained filters.(Note 6)This air should be maintained at a sufficient pressure at the base of the Air Saturator Tower to meet the suggested pressures of Table 2at the top of the Air Saturator Tower.N OTE 6—The air supply may be freed from oil and dirt by passing it through a suitable oil/water extractor (that is commercially available)to stop any oil from reaching the Air Saturator Tower.Many oil/water extractors have an expiration indicator,proper preventive maintenance intervals should take these into account.9.2The compressed air supply to the atomizer nozzle or nozzles shall be conditioned by introducing it into the bottomof a tower fillwed with water.A common method of introduc-ing the air is through an air dispersion device (X1.4.1).The level of the water must be maintained automatically to ensure adequate humidification.It is common practice to maintain the temperature in this tower between 46and 49°C (114–121°F)to offset the cooling effect of expansion to atmospheric pressure during the atomization process.Table 2shows the temperature,at different pressures,that are commonly used to offset the cooling effect of expansion to atmospheric pressure.9.3Careful attention should be given to the relationship of tower temperature to pressure since this relationship can have a direct impact to maintaining proper collection rates (Note 7).It is preferable to saturate the air at temperatures well above the chamber temperature as insurance of a wet fog as listed in Table 2.N OTE 7—If the tower is run outside of these suggested temperature and pressure ranges to acheive proper collection rates as described in 10.2of this practice,other means of verifying the proper corrosion rate in the chamber should be investigated,such as the use of control specimens (panels of known performance in the test conducted).It is preferred that control panels be provided that bracket the expected test specimen performance.The controls allow for the normalization of test conditions during repeated running of the test and will also allow comparisons of test results from different repeats of the same test.(Refer to Appendix X3,Evaluation of Corrosive Conditions,for mass loss procedures).10.Conditions in the Salt Spray Chamber10.1Temperature —The exposure zone of the salt spray chamber shall be maintained at 3562°C (9563°F).Each set point and its tolerance represents an operational control point for equilibrium conditions at a single location in the cabinet which may not necessarily represent the uniformity of condi-tions throughout the cabinet.The temperature within the exposure zone of the closed cabinet shall be recorded (Note 8)at least once daily (except on Saturdays,Sundays,and holidays when the salt spray test is not interrupted for exposing,rearranging,or removing test specimens or to check and replenish the solution in the reservoir)N OTE 8—A suitable method to record the temperature is by a continu-ous recording device or by a thermometer which can be read from outside the closed cabinet.The recorded temperature must be obtained with the salt spray chamber closed to avoid a false low reading because of wet-bulb effect when the chamber is open.10.2Atomization and Quantity of Fog —Place at least two clean fog collectors per atomizer tower within the exposure zone so that no drops of solution will be collected from the test specimens or any other source.Position the collectors in the proximity of the test specimens,one nearest to any nozzle and the other farthest from all nozzles.A typical arrangement is shown in Fig.1.The fog shall be such that for each 80cm 2(12.4in.2)of horizontal collecting area,there will be collected from 1.0to 2.0mL of solution per hour based on an average run of at least 16h (Note 9).The sodium chloride concentration of the collected solution shall be 561mass %(Notes 9-11).The pH of the collected solution shall be 6.5to 7.2.The pH measurement shall be made as described in 8.2(Note 3).N OTE 9—Suitable collecting devices are glass or plastic funnels withTABLE 2Suggested Temperature and Pressure guideline for the top of the Air Saturator Tower for the operation of a test at 35°C(95°F)Air Pressure,kPaTemperature,°CAir Pressure,PSITemperature,°F83461211496471411711048161191244918121--````````,`,,,`,``,`,`,`````-`-`,,`,,`,`,,`---the stems inserted through stoppers into graduated cylinders,or crystal-lizing dishes.Funnels and dishes with a diameter of 10cm (3.94in.)have an area of about 80cm 2(12.4in.2).N OTE 10—A solution having a specific gravity of 1.0255to 1.0400at 25°C (77°F)will meet the concentration requirement.The sodium chloride concentration may also be determined using a suitable salinity meter (for example,utilizing a sodium ion-selective glass electrode)or colorimetrically as follows.Dilute 5mL of the collected solution to 100mL with distilled water and mix thoroughly;pipet a 10-mL aliquot into an evaporating dish or casserole;add 40mL of distilled water and 1mL of 1%potassium chromate solution (chloride-free)and titrate with 0.1N silver nitrate solution to the first appearance of a permanent red coloration.A solution that requires between 3.4and 5.1mL of 0.1N silver nitrate solution will meet the concentration requirements.N OTE 11—Salt solutions from 2to 6%will give the same results,though for uniformity the limits are set at 4to 6%.10.3The nozzle or nozzles shall be so directed or baffled that none of the spray can impinge directly on the test specimens.11.Continuity of Exposure11.1Unless otherwise specified in the specifications cover-ing the material or product being tested,the test shall be continuous for the duration of the entire test period.Continu-ous operation implies that the chamber be closed and the spray operating continuously except for the short daily interruptions necessary to inspect,rearrange,or remove test specimens,to check and replenish the solution in the reservoir,and to make necessary recordings as described in Section 10.Operations shall be so scheduled that these interruptions are held to a minimum.12.Period of Exposure12.1The period of exposure shall be as designated by the specifications covering the material or product being tested or as mutually agreed upon between the purchaser and the seller.N OTE 12—Recommended exposure periods are to be as agreed upon between the purchaser and the seller,but exposure periods of multiples of 24h are suggested.13.Cleaning of Tested Specimens13.1Unless otherwise specified in the specifications cover-ing the material or product being tested,specimens shall be treated as follows at the end of the test:13.1.1The specimens shall be carefully removed.13.2Specimens may be gently washed or dipped in clean running water not warmer than 38°C (100°F)to remove salt deposits from their surface,and then immediately dried.14.Evaluation of Results14.1A careful and immediate examination shall be made as required by the specifications covering the material or product being tested or by agreement between the purchaser and the seller.15.Records and Reports15.1The following information shall be recorded,unless otherwise prescribed in the specifications covering the material or product being tested:15.1.1Type of salt and water used in preparing the salt solution,15.1.2All readings of temperature within the exposure zone of the chamber,15.1.3Daily records of data obtained from each fog-collecting device including the following:15.1.3.1V olume of salt solution collected in millilitres per hour per 80cm 2(12.4in.2),15.1.3.2Concentration or specific gravity at 35°C (95°F)of solution collected,and15.1.3.3pH of collectedsolution.N OTE —This figure shows a typical fog collector arrangement for a single atomizer tower cabinet.The same fog collector arrangement is also applicable for multiple atomizer tower and horizontal (“T”type)atomizer tower cabinet constructions as well.FIG.1Arrangement of FogCollectors--````````,`,,,`,``,`,`,`````-`-`,,`,,`,`,,`---15.2Type of specimen and its dimensions,or number or description of part,15.3Method of cleaning specimens before and after testing, 15.4Method of supporting or suspending article in the salt spray chamber,15.5Description of protection used as required in6.5, 15.6Exposure period,15.7Interruptions in exposure,cause,and length of time, and15.8Results of all inspections.N OTE13—If any of the atomized salt solution which has not contacted the test specimens is returned to the reservoir,it is advisable to record the concentration or specific gravity of this solution also.16.Keywords16.1controlled corrosive environment;corrosive condi-tions;determining mass loss;salt spray(fog)exposureAPPENDIXES(Nonmandatory Information)X1.CONSTRUCTION OF APPARATUSX1.1CabinetsX1.1.1Standard salt spray cabinets are available fromseveral suppliers,but certain pertinent accessories are requiredbefore they will function according to this practice and provideconsistent control for duplication of results.X1.1.2The salt spray cabinet consists of the basic chamber,an air-saturator tower,a salt solution reservoir,atomizingnozzles,specimen supports,provisions for heating the cham-ber,and suitable controls for maintaining the desired tempera-ture.X1.1.3Accessories such as a suitable adjustable baffle orcentral fog tower,automatic level control for the salt reservoir,and automatic level control for the air-saturator tower arepertinent parts of the apparatus.X1.1.4The size and shape of the cabinet shall be such thatthe atomization and quantity of collected solution is within the limits of this practice.X1.1.5The chamber shall be made of suitably inert mate-rials such as plastic,glass,or stone,or constructed of metal and lined with impervious plastics,rubber,or epoxy-type materials or equivalent.X1.1.6All piping that contacts the salt solution or spray should be of inert materials such as plastic.Vent piping should be of sufficient size so that a minimum of back pressure exists and should be installed so that no solution is trapped.The exposed end of the vent pipe should be shielded from extreme air currents that may causefluctuation of pressure or vacuum in the cabinet.X1.2Temperature ControlX1.2.1The maintenance of temperature within the salt chamber can be accomplished by several methods.It is generally desirable to control the temperature of the surround-ings of the salt spray chamber and to maintain it as stable as possible.This may be accomplished by placing the apparatus in a constant-temperature room,but may also be achieved by surrounding the basic chamber of a jacket containing water or air at a controlled temperature.X1.2.2The use of immersion heaters in an internal salt solution reservoir or within the chamber is detrimental where heat losses are appreciable because of solution evaporation and radiant heat on the specimens.X1.3Spray NozzlesX1.3.1Satisfactory nozzles may be made of hard rubber, plastic,or other inert materials.The most commonly used type is made of plastic.Nozzles calibrated for air consumption and solution-atomized are available.The operating characteristics of a typical nozzle are given in Table X1.1.X1.3.2It can readily be seen that air consumption is relatively stable at the pressures normally used,but a marked reduction in solution sprayed occurs if the level of the solution is allowed to drop appreciably during the test.Thus,the level of the solution in the salt reservoir must be maintained automatically to ensure uniform fog delivery during the test.3 X1.3.3If the nozzle selected does not atomize the salt solution into uniform droplets,it will be necessary to direct the spray at a baffle or wall to pick up the larger drops and prevent them from impinging on the test specimens.Pending a com-plete understanding of air-pressure effects,and so forth,it is important that the nozzle selected shall produce the desired 3A suitable device for maintaining the level of liquid in either the saturator tower or reservoir of test solution may be designed by a local engineering group,or it may be purchased from manufacturers of test cabinets as an accessory.TABLE X1.1Operating Characteristics of Typical Spray Nozzle SiphonHeight,cmAir Flow,dm3/min Solution Consumption,cm3/hAir Pressure,kPa Air Pressure,kPa34691031383469103138 101926.531.5362100384045845256 201926.531.536636276037204320 301926.531.5360138030003710 401926.631.536078021242904SiphonHeight,in.Air Flow,L/minSolutionConsumption,mL/hAir Pressure,psi Air Pressure,psi51015205101520 41926.531.5362100384045845256 81926.531.536636276037204320 121926.531.5360138030003710 161926.631.536078021242904 --````````,`,,,`,``,`,`,`````-`-`,,`,,`,`,,`---condition when operated at the air pressure selected.Nozzles are not necessarily located at one end,but may be placed in the center and can also be directed vertically up through a suitable tower.X1.4Air for AtomizationX1.4.1The air used for atomization must be free of grease, oil,and dirt before use by passing through well-maintainedfilters.Room air may be compressed,heated,humidified,and washed in a water-sealed rotary pump if the temperature of the water is suitably controlled.Otherwise cleaned air may be introduced into the bottom of a towerfilled with water through a porous stone or multiple nozzles.The level of the water must be maintained automatically to ensure adequate humidification.A chamber operated in accordance with this method and Appendix X1will have a relative humidity between95and 98%.Since salt solutions from2to6%will give the same results(though for uniformity the limits are set at4to6%),it is preferable to saturate the air at temperatures well above the chamber temperature as insurance of a wet fog.Table X1.2 shows the temperatures,at different pressures,that are required to offset the cooling effect of expansion to atmospheric pressure.X1.4.2Experience has shown that most uniform spray chamber atmospheres are obtained by increasing the atomizing air temperature sufficiently to offset heat losses,except those that can be replaced otherwise at very low-temperature gradi-ents.X1.5Types of ConstructionX1.5.1A modern laboratory cabinet is shown in Fig.X1.1. Walk-in chambers are usually constructed with a sloping ceiling.Suitably located and directed spray nozzles avoid ceiling accumulation and drip.Nozzles may be located at the ceiling,or0.91m(3ft)from thefloor directed upward at30to 60°over a passageway.The number of nozzles depends on type and capacity and is related to the area of the test space.An11 to19L(3to5-gal)reservoir is required within the chamber, with the level controlled.The major features of a walk-in type cabinet,which differs significantly from the laboratory type, are illustrated in Fig.X1.2.Construction of a plastic nozzle, such as is furnished by several suppliers,is shown in Fig.X1.3.TABLE X1.2Temperature and Pressure Requirements forOperation of Test at95°FAir Pressure,kPa8396110124 Temperature,°C46474849Air Pressure,psi12141618 Temperature,°F114117119121 --````````,`,,,`,``,`,`,`````-`-`,,`,,`,`,,`---。
219316024_鲜活虹鳟鱼的呼吸频率与肌肉品质的相关性

吴艺文,赵曼曼,尤孝鹏,等. 鲜活虹鳟鱼的呼吸频率与肌肉品质的相关性[J]. 食品工业科技,2023,44(12):29−36. doi:10.13386/j.issn1002-0306.2022080201WU Yiwen, ZHAO Manman, YOU Xiaopeng, et al. Correlation between Respiratory Rate and Muscle Quality of Fresh Rainbow Trout (Oncorhynchus mykiss )[J]. Science and Technology of Food Industry, 2023, 44(12): 29−36. (in Chinese with English abstract). doi:10.13386/j.issn1002-0306.2022080201· 研究与探讨 ·鲜活虹鳟鱼的呼吸频率与肌肉品质的相关性吴艺文1,2,赵曼曼1,2,尤孝鹏1,2,汪 兰2,石 柳2,丁安子2,吴文锦2,陈 胜2,孙卫青1, *,熊光权2,*(1.长江大学生命科学学院,湖北荆州 434025;2.湖北省农业科学院农产品加工与核农技术研究所,农业农村部农产品冷链物流技术重点实验室,湖北武汉 430064)摘 要:为探讨鲜活虹鳟鱼的呼吸频率与肌肉品质之间的相关性,研究了不同呼吸频率所对应的肌肉品质的变化。
测定了乳酸、糖原、丙二醛(Malondialdehyde ,MDA )以及核苷酸类化合物含量和加压失水率。
结果表明,随着呼吸频率的上升,糖原含量下降、丙二醛和核苷酸类化合物含量、加压失水率上升。
当呼吸频率从83升至95次/min ,乳酸含量由初始值1.46 mmol/g prot 显著上升至2.37 mmol/g prot (P <0.01)。
皮尔逊相关分析显示鲜活虹鳟鱼的呼吸频率与乳酸含量、MDA 值、加压失水率呈现出极显著相关性(P <0.01),与糖原含量、AMP 含量、IMP 含量以及Hx 和HxR 总含量等肌肉品质呈现出显著相关性(P <0.05),相关系数分别为0.987、0.951、0.939、−0.897、0.847、0.896、0.882。
电感耦合等离子体质谱法测定金属锂中6种元素

化学分析计量CHEMICAL ANALYSIS AND METERAGE第29卷,第4期2020年7月V ol. 29,No. 4Jul. 202035doi :10.3969/j.issn.1008–6145.2020.04.008电感耦合等离子体质谱法测定金属锂中6种元素郑亚蕾(中核建中核燃料元件有限公司,四川宜宾 644000)摘要 建立电感耦合等离子体质谱(ICP–MS )法同时测定金属锂中的钛、铬、锰、镍、钡和铅6种杂质元素。
样品用硝酸消解,在线加入混合内标元素,根据待测元素的质荷比不同以ICP–MS 技术进行分离,经质量分析器和检测系统,测试质谱的信号强度对元素进行定量分析。
6种杂质元素适用于He 模式下检测,样品酸度确定为0.15 mol /L 。
6种元素的质量浓度在1~100 μg /L 范围内与离子强度线性关系良好,相关系数均大于0.999,钛,铬,锰,镍,钡和铅元素检出限分别为0.059,0.065,0.048,0.074,0.062,0.071 μg /L ,测定结果的相对标准偏差为1.56%~3.94%,加标回收率为91.7%~109.4%。
该法操作简便、灵敏度高、准确度高,适用于金属锂中6种杂质元素的测定,提高了分析效率和检测水平。
关键词 电感耦合等离子体质谱法;检测;金属锂中图分类号:O657.6 文献标识码:A 文章编号:1008–6145(2020)04–0035–05Determination of six elements in lithium metal by inductively coupled plasma mass spectrometryZHENG Yalei( CNNC Jianzhong Nuclear Fuel Co., Ltd., Yibin 644000, China )Abstract An inductively coupled plasma mass spectrometry (ICP–MS) was established for the simultaneous determination of 6 impurity elements of titanium, chromium, manganese, nickel, barium, plumbum in lithium metal. Sample digestion with nitric acid and added internal standard elements online. ICP–MS separated the elements according to mass-to-charge ratio. Mass spectrometer and detection system detected signal intensity of mass spectrum to quantitative analysis for elements. Six kinds of impurity elements applied to He mode, and acidity of sample was 0.15 mol /L. The mass concentration of six elements had a good linear relationship with ion strength in the range of 1–100 μg /L, and correlation coefficient than 0.999, the detection limits of titanium, chromium, manganese, nickel, barium, plumbum were 0.059, 0.065, 0.048, 0.074, 0.062, 0.071 μg /L, respectively, the relative standard deviation of detection of results was 1.56%–3.94%, the recovery was 91.7%–109.4%. The method is simple, high sensitivity and high accuracy. It is suitable for determination of six impurity elements in lithium metal, which improve analysis efficiency and detection level.Keywords inductively coupled plasma mass spectrometry; determination; lithium metal金属锂是一种银白色、质软的轻金属,密度非常小,仅有0.534 g /cm³,被称为“高能金属”,广泛应用于电池材料、核电、军工、航空航天等行业。
制药行业专业英语词典

Abbe refractometer 阿贝折射计 absorbance 吸光度absorbance ratio 吸光度比值 absorption curve 吸收曲线 absorption spectrum 吸收光谱 accuracy 准确度acidbase indicator 酸碱指示剂acidbase titration 酸碱滴定 acidimetry 酸量法acidity 酸度acid 酸aciddye colorimetry酸性染料比色法 acid value 酸值acidinsoluble ash 酸不溶性灰分 action and use 作用与用途active constituent 活性成分 additive 添加剂additivity加和性adjusted retention time 调整保留时间 adsorption吸附affinity chromatography亲和色谱法 alkalinity 碱度alkaloid 生物碱alkyloxy determination 烷氧基测定alumina 氧化铝amino acid 氨基酸analysis error 分析误差analytical balance 分析天平analytical chemistry分析化学analysis of variance 方差分析analytical quality control(AQC)分析质量控制 angstrom,Å 埃anhydrous 无水的anhydrous basis,anhydrous substance 干燥品 antioxidant 抗氧剂apparatus error 仪器误差apparent viscosity 表观黏度appendix 附录application of sample 点样area normalization method 面积归一化法 argentimetry 银量法aromatic hydrocarbon 芳烃arsenic 砷arsenic stain 砷斑artificial neural network 人工神经网络artificial intelligence 人工智能ash 灰分assay含量测定asymmetrical stretching vibration 不对称伸缩振动 atmospheric pressure ionization(API)大气压离子化 atomic absorption spectrometry(AAS)原子吸收光谱法 attenuation衰减average 平均值average deviation 平均偏差back extraction 反萃取back titration 反滴定band absorption 谱带吸收bar graph 棒图baseline correction 基线校正 baseline drift 基线漂移base 碱baseline resolved peak 基线分离峰 batch,lot 批biotransformation 生物转化 bioequivalence 生物等效性 bioavailability 生物利用度blank test 空白试验blue shift 蓝移boiling range 沸程British Pharmacopeia (BP) 英国药典 bromate titration溴酸盐滴定法 bromocresol green 溴甲酚绿 bromocresol purple 溴甲酚紫 bromophenol blue 溴酚蓝 bromothymol blue 溴麝香草酚蓝 buffer action 缓冲作用buffer capacity 缓冲容量buffer solutions 缓冲溶液bulk drug,pharmaceutical product 原料药 buret 滴定管byproduct 副产物calibrate 校准calibration curve 校准曲线calomel electrode 甘汞电极capacity factor 容量因子capillary electrophoresis(CE)毛细管电泳 capillary gas chromatography 毛细管气相色谱法 capillary melting point determination 毛细管熔点测定 carrier gas 载气capsule 胶囊剂characteristics,description 性状characteristic spectrum 特征光谱chemical constituent 化学成分chemical drugs 化学药品check sample 对照试样check test 对照试验chelate compound 螯合物chemically bonded phase 化学键合相chemical equivalent 化学当量Chinese Pharmacopoeia (ChP) 中国药典Chinese patent medicine 中成药Chinese materia medica 中药学Chinese materia medica preparation 中药制剂Chinese Pharmaceutical Association (CPA)中国药学会 chiral stationary phase(CSP)手性固定相chiral separation手性分离chirality手性chiral carbon atom手性碳原子chiral molecule 手性分子chloride 氯化物chromatography 色谱法chromatogram色谱图chromatographic column 色谱柱chromatographic condition 色谱条件 chromatographic system 色谱系统chromatographic data processor 色谱数据处理机 chromatographic work station 色谱工作站cistrans isomerism顺反异构clarity澄清度clathrate, inclusion compound 包合物clearance 清除率clinical pharmacy 临床药学coefficient of distribution 分配系数coefficient of variation 变异系数coenzyme 辅酶color reaction 显色反应colorimetric analysis 比色分析column capacity 柱容量column dead volume 柱死体积column interstitial volume 柱隙体积column outlet pressure 柱出口压column temperature 柱温column pressure 柱压column volume 柱体积column overload 柱超载column switching柱切换committee of drug evaluation药品审评委员会 complex 络合物component,constituent 组分compound medicines 复方药concentration浓度controlled trial对照试验coordination compound 配位化合物 correlation coefficient 相关系数 comparative test 比较试验completeness of solution 溶液的澄清度 complexometric titration 络合滴定computeraided pharmaceutical analysis 计算机辅助药物分析 condensation reaction缩合反应condensation substance 缩合物confidence interval 置信区间confidence level 置信水平confidence limit 置信限confidence probability 置信概率congealing point 凝点content 含量,内含物content uniformity 装量差异contrast test 对照试验crude drug 生药crystal结晶crystal violet 结晶紫crystallinity 结晶性(结晶度)cyclodextrin inclusion compound 环糊精包合物cyanide 氰化物dead space 死体积deadstop titration永停滴定法dead time 死时间decomposition point 分解点deflection偏差deflection point 拐点degassing脱气deionized water 去离子水derivative spectrophotometry导数分光光度法detection 检查dextrose 右旋糖,葡萄糖diastereomer 非对映(异构)体diazotization titration method 重氮化滴定法2,6dichlorindophenol titration2,6二氯靛酚滴定法 differential thermal analysis(DTA) 差示热分析 differential scanning calorimetry(DSC)差示扫描热量法 differential pulse polarography示差脉冲极谱法 digestion 消化dilute 稀释diphasic titration 双相滴定direct potentiometry直接电位法dissociation constant 解离常数dissociation degree 解离度distribution chromatography 分配色谱distribution coefficient 分配系数dissolubility 溶解度dissolution溶出度disintegration 崩解时限distillation 蒸馏dose 剂量drug release 药物释放度drug quality management 药品质量管理drug control institutions 药检机构drug 药物drug metabolism enzyme 药物代谢酶drug quality control药品质量控制drug standard 药品标准dryness 干燥dual wavelength spectrophotometry 双波长分光光度法 duplicate test 重复试验excipient 赋形剂effective constituent 有效成分efficacy 效能,有效性effective plate number 有效板数efficiency of column 柱效electron transition 电子跃迁electrospray interface 电喷雾接口 electromigration injection电迁移进样elution洗脱eluting effect 洗脱效应elution curve 洗脱曲线elimination 消除emission spectrochemical analysis 发射光谱分析 end absorption末端吸收end point correction 终点校正end point error 终点误差enantiomer 对映(异构)体enzyme immunoassay(EIA)酶免疫分析 enzyme drug 酶类药物enzymatic reaction 酶促反应enzyme induction酶诱导enzyme inhibition 酶抑制epimer 差向异构体epimerization 差向异构化equilibrium constant 平衡常数 equivalence point 等当点equivalence potential等当点电位 equivalent weight 当量error in volumetric analysis 容量分析误差 extraction提取extract 提取物excite 激发excitation spectrum 激发光谱excitation wave length 激发波长 exclusion chromatography 排阻色谱法 expiration date 失效期external standard 外标准extrapolated method 外插法,外推法 expert system专家系统extraction gravimetry 提取重量法 extraction titration提取容量法factor 系数,因数feature 特征Fehling’s reaction费林反应fineness of the particles 颗粒细度finger print region指纹区finger print map 指纹图fixed phase 固定相flame ionization detector (FID)火焰离子化检测器flame emission spectrum 火焰发射光谱fluorimetry 荧光分析法fluorescamine 荧胺fluorescence immunoassay(FIA)荧光免疫分析fluorescence polarization immunoassay(FPIA)荧光偏振免疫分析 fluorescent agent 荧光剂fluorescence spectrophotometry 荧光分光光度法fluorescence detector 荧光检测器fluorescence efficiency 荧光效率fluorescence excitation spectrum 荧光激发光谱foreign odor 异臭foreign pigment 有色杂质formulary处方集freezing test 结冻试验functional group 官能团fused peaks, overlapped peaks 重叠峰gas chromatogram 气相色谱图gas chromatography (GC)气相色谱法gas chromatographflourier transform infrared spectrophotometer 气相 色谱傅里叶变换红外光谱联用仪glass electrode 玻璃电极gasliquid chromatography (GLC) 气液色谱法gas purifier 气体净化器gel chromatography凝胶色谱法general identification test 一般鉴别试验gradient elution 梯度洗脱Good Manufacturing Practice and Quality Control of Drug (GMP and QC of Drug)药品生产质量管理规范Good Manufacture Practices(GMP) 药品生产规范Good Laboratory Practice(GLP)实验室管理规范Good Clinical Practice(GCP)临床试验规范Good Supplying Practice(GSP)药品供应规范Gran’s plot 格兰作图法gravimetric analysis 重量分析法half peak width 半峰宽[halide]disk method,wafer method,pellet method 压片法headspace concentrating injector 顶空浓缩进样器heat conductivity 热导率heavy metal重金属height of an effective plate 有效板高度high resolution gas chromatography(HRGC)高分辨气相色谱法hghperformance liquid chromatography高效液相色谱法high performance thinlayer chromatography(HPTLC)高效薄层色谱 法hydroxyl value 羟值hyperchromic effect 深色效应hypsochromic effect 浅色效应hypothesis test 假设检验hydrophobicity 疏水性hydrophilicity亲水性hydrate 水合物hydrolysis 水解identification 鉴别immobile phase 固定相impurity 杂质Inactivation 失活index 索引indicator 指示剂indicator electrode 指示电极inhibitor抑制剂infrared absorption spectrum 红外吸收光谱 injecting septum 进样隔膜胶垫injection valve 进样阀integrator积分仪instrumental analysis仪器分析instrument error 仪器误差intermediate 中间体international unit(IU)国际单位internal standard substance 内标物质 iodometry 碘量法ion exchange chromatography 离子交换色谱法 ionic strength 离子强度ionize 电离ion pair chromatography 离子对色谱ion suppression 离子抑制 irreversible potential不可逆电位 isoelectric point 等电点isoosmotic solution 等渗溶液 immunoassay 免疫测定ionexchange cellulose离子交换纤维素 iodoform reaction 碘仿反应ion suppress 离子抑制iodide碘化物irreversible indicator不可逆指示剂 isosbestic point method等吸光点法课外阅读一 阿司匹林及其片剂的质量标准(USP )Aspirin O O H 3C HOOC 9H 8O 4 180.16Benzoic acid, 2(acetyloxy)Salicylic acid acetate [50782].>>Aspirin contains not less than 99.5 percent and not more than 100.5 percent of C 9H 8O 4, calculated on the dried basis. Packaging and storage Preserve in tight containers.USP Reference standards <11>-USP Aspirin RS .Identification -A : Heat it with water for several minutes, cool, and add 1 or 2 drops of ferric chloride TS: a violetred color is produced.B : Infrared Absorption <197K>Loss on drying <731>-Dry it over silica gel for 5 hours: it loses not more than 0.5% of its weight.Readily carbonizable substances <271>-Dissolve 500mg in 5 mL of sulfuric acid TS: the solution has no more color than Matching Fluid Q.Residue on ignition <281>: not more than 0.05%.Substances insoluble in sodium carbonate TS -A solution of 500mg in 10 mL of warm sodium carbonate TS is clear.Chloride <221>-Boil 1.5g with 75 mL of water for 5 minutes, cool, add sufficient water to restore the original volume, and filter. A 25mL portion of the filtrate shows no more chloride than corresponds to 0.10 mL of 0.020 N hydrochloric acid (0.014%).Sulfate Dissolve 6.0g in 37 mL of acetone, and add 3 mL of water. Titrate potentiometrically with 0.02 M lead perchlorate, prepared by dissolving 9.20 g of lead perchlorate in water to make 1000mL of solution, using a pH meter capable of a minimum reproducibility of ±0.1 mV (see pH <791>) equipped with an electrode system consisting of a leadspecific electrode and a silversilver chloride reference glasssleeved electrode containing a solution of tetraethylammonium perchlorate in glacial acetic acid (1 in 44)(see Titrimetry <541>): not more than 1.25mL of 0.02 M lead perchlorate is consumed (0.04%). [NOTE -After use, rinse the leadspecific electrode with water, drain the reference electrode, flush with water, rinse with methanol, and allow to dry.]Heavy metals -Dissolve 2 g in 25 mL of acetone, and add 1 mL of water. Add 1.2 mL of thioacetamideglycerin base TS and 2 mL of pH 3.5 Acetate Buffer (see Heavy Metals <231>), and allow to stand for 5 minutes: any color produced is not darker than that of a control made with 25 mL of acetone and 2 mL of Standard Lead Solution (see Heavy Metals <231>), treated in the same manner. The limit is 10μg per g.Limit of free salicylic acid-Dissolve 2.5g in sufficient alcohol to make 25.0 mL. To each of two matched colorcomparison tubes add 48 mL of water and 1 mL of a freshly prepared, diluted ferric ammonium sulfate solution (prepared by adding 1 mL of 1 N hydrochloric acid to 2 mL of ferric ammonium sulfate TS and diluting with water to 100 mL). Into one tube pipet 1 mL of a standard solution of salicylic acid in water, containing 0.10 mg of salicylic acid per mL. Into the second tube pipet 1 mL of the 1 in 10 solution of Aspirin. Mix the contents of each tube: after 30 seconds, the color in the second tube is not more intense than that in the tube containing the salicylic acid (0.1%).Organic volatile impurities, Method IV<467>: meets the requirements.Assay-Place about 1.5g of Aspirin, accurately weighed, in a flask, add 50.0 mL of 0.5 N sodium hydroxide VS, and boil the mixture gently for 10 minutes. Add phenolphthalein TS, and titrate the excess sodium hydroxide with 0.5 N sulfuric acid VS. Perform a blank determination (see Residual Titrations under Titrimetry <541>). Each mL of 0.5 N sodium hydroxide is equivalent to 45.04 mg of C9H8O4.Aspirin Tablets>> Aspirin Tablets contain not less than 90.0 percent and not more than 110.0 percent of the labeled amount of C9H8O4. Tablets of larger than 81mg size contain no sweeteners or other flavors.N OTETablets that are entericcoated meet the requirements for Aspirin Delayedrelease Tablets.Packaging and storagePreserve in tight containers. Preserve flavored or sweetened Tablets of 81mg size or smaller in containers holding not more than 36 Tablets each.USP Reference standards <11>USP Aspirin RS. USP Salicylic Acid RS.IdentificationA: Crush 1 Tablet, boil it with 50 mL of water for 5 minutes, cool, and add 1 or 2 drops of ferric chloride TS: a violetred color is produced.B: Infrared absorption <197K>Prepare the test specimen as follows. Shake a quantity of finely powdered Tablets, equivalent to about 500 mg of aspirin, with 10 mL of alcohol for several minutes. Centrifuge the mixture. Pour off the clear supernatant liquid, and evaporate it to dryness. Dry the residue in vacuum at 60℃ for 1 hour.Dissolution<711>Medium: 0.5 M acetate buffer, prepared by mixing 2.99 g of sodium acetate trihydrate and 1.66 mL of glacial acetic acid with water to obtain 1000mL of solution having a pH of 4.50±0.05; 500 mL.Apparatus 1 : 50 rpm.Time: 30 minutes.Procedure-Determine the amount of C9H8O4 dissolved from ultraviolet absorbances at the wavelength of the isosbestic point of aspirin and salicylic acid at 265±2nm of filtered portions of the solution under test, suitably diluted with Dissolution Medium. if necessary, in comparison with a Standard solution having a known concentration of USP Aspirin RS in the samemedium. [N OTEPrepare the Standard solution at the time of use. An amount of alcohol not to exceed 1% of the total volume of the Standard solution may be used to bring the Reference Standard into solution prior to dilution with Dissolution Medium.] TolerancesNot less than 80% (Q) of the labeled C9H8O4 is dissolved in 30 minutes.Uniformity of dosage units <905>: meet the requirementsLimit of free salicylic acidMobile phase and Diluting SolutionPrepare as directed in the Assay.Standard solutionDissolve an accurately weighed quantity of USP Salicylic Acid RS in the Standard preparation prepared as directed in the Assay, to obtain a solution having a known concentration of about 0.015 mg of salicylic acid per mL.Test preparationUse the Stock solution prepared as directed for Assay preparation in the Assay.Chromatographic systemUse the Chromatographic system described in the Assay. Chromatograph the Standard solution, and record the peak responses as directed under Procedure in the Assay. The relative standard deviation of the salicylic acid peak responses is not more than 4.0%. In a suitable chromatogram, the resolution, R, between salicylic acid and aspirin is not less than 2.0.ProcedureProceed as directed for Procedure in the Assay. The relative retention times are about 0.7 for salicylic acid and 1.0 for aspirin. Calculate the percentage of salicylic acid (C7H6O3) in the portion of Tablets taken by the formula:2000(C/Q A)(r u/r s),in which C is the concentration, in mg per mL, of USP Salicylic Acid RS in the Standard solution, Q A is the quantity, in mg, of aspirin (C9H8O4) in the portion of Tablets taken, as determined in the Assay, and rυ and r s are the peak responses of the salicylic acid peaks obtained from the Test preparation and the Standard solution, respectively: not more than 3.0% is found. In the case that are coated, not more than 3.0% is found.AssayMobile phaseDissolve 2 g of sodium 1heptanesulfonate in a mixture of 850 mL of water and 150 mL of acetonitrile, and adjust with glacial acetic acid to a pH of 3.4.Diluting solutionPrepare a mixture of acetonitrile and formic acid (99:1).Standard preparationDissolve an accurately weighed quantity of USP Aspirin RS in Diluting solution to obtain a solution having a known concentration of about 0.5 mg per mL.Assay preparationWeigh and finely powder not less than 20 Tablets. Transfer an accurately weighed quantity of the powder, equivalent to about 100 mg of aspirin, to a suitable container. Add 20.0 mL of Diluting solution and about 10 glass beads. Shake vigorously for about 10 minutes, and centrifuge (stock solution). Quantitatively dilute an accurately measured volume of the Stock solution with 9 volumes of Diluting solution (Assay preparation). Retain the remaining portion of Stock solution for the test for Limit of salicylic acid.Chromatographic system (see Chromatography <621>)-The liquid chromatograph is equipped with a 280nm detector and a 4.0mm×30cm column containing packing L1. The flow rate is about 2 mL per minute. Chromatograph the Standardpreparation, and record the peak responses as directed under Procedure: the relative standard deviation is not more than 2.0%. In a suitable chromatogram, the tailing factor is not greater than 2.0.ProcedureSeparately inject equal volumes (about 10m L) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the quantity, in mg, of aspirin (C9H8O4) in the portion of Tablets taken by the formula:200C(rυ/r s),in which C is the concentration, in mg per mL, of USP Aspirin RS in the Standard preparation, and rυ and r s are the peak responses of the aspirin peaks obtained from the Assay preparation and the Standard preparation, respectively.课外阅读二 分析方法论证ANALYTICAL PERFORMANCE CHARACTERISTICSAccuracyDefinitionThe accuracy of an analytical method is the closeness of test results obtained by that method to the true value. The accuracy of an analytical method should be established across its range.DeterminationIn the case of the assay of a drug substance, accuracy may be determined by application of the analytical method to an analyte of known purity (e.g., a Reference Standard) or by comparison of the results of the method with those of a second, wellcharacterized method, the accuracy of which has been stated or defined.In the case of the assay of a drug in a formulated product, accuracy may be determined by application of the analytical method to synthetic mixtures of the drug product components to which known amounts of analyte have been added within the range of the method. If it is not possible to obtain samples of all drug product components, it may be acceptable either to add known quantities of the analyte to the drug product (i.e., “to spike”) or to compare results with those of a second, wellcharacterized method, the accuracy of which has been stated or defined.In the case of quantitative analysis of impurities, accuracy should be assessed on samples (of drug substance or drug product) spiked with known amounts of impurities. Where it is not possible to obtain samples of certain impurities or degradation products, results should be compared with those obtained by an independent method. In the absence of other information, it may be necessary to calculate the amount of an impurity based on comparison of its response to that of the drug substance; the ratio of the responses of equal amounts of the impurity and the drug substance (response factor) should be used if known.Accuracy is calculated as the percentage of recovery by the assay of the known added amount of analyte in the sample, or as the difference between the mean and the accepted true value, together with confidence intervals.The ICH documents recommend that accuracy should be assessed using a minimum of nine determinations over a minimum of three concentration levels, covering the specified range (i.e., three concentrations and three replicates of each concentration).PrecisionDefinitionThe precision of an analytical method is the degree of agreement among individual test results when the method is applied repeatedly to multiple samplings of a homogeneous sample. The precision of an analytical method is usually expressed as the standard deviation or relative standard deviation (coefficient of variation) of a series of measurements. Precision may be a measure of either the degree of reproducibility or of repeatability of the analytical method under normal operating conditions. In this context, reproducibility refers to the use of the analytical procedure in different laboratories, as in a collaborative study. Intermediate precision expresses withinlaboratory variation, as on different days, or with different analystsor equipment within the same laboratory. Repeatability refers to the use of the analytical procedure within a laboratory over a short period of time using the same analyst with the same equipment. For most purposes, repeatability is the criterion of concern in USP analytical procedures, repeatability is the criterion of concern in USP analytical procedures, although reproducibility between laboratories or intermediate precision may well be considered during the standardization of a procedure before it is submitted to the Pharmacopeia.DeterminationThe precision of an analytical method is determined by assaying a sufficient number of aliquots of a homogeneous sample to be able to calculate statistically valid estimates of standard deviation or relative standard deviation (coefficient of variation). Assays in this context are independent analyses of samples that have been carried through the complete analytical procedure from sample preparation to final test result.The ICH documents recommend that repeatability should be assessed using a minimum of nine determinations covering the specified range for the procedure (i.e., three concentrations and three replicates of each concentration or using a minimum of six deter minations at 100% of the test concentration).SpecificityDefinitionThe ICH documents define specificity as the ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, and matrix components. Lack of specificity of an individual analytical procedure may be compensated by other supporting analytical procedures. [N OTEOther reputable international authorities (IUPAC, AOAC) have preferred the term “selectivity”, reserving “specificity” for those procedures that are completely selective.] For the test or assay methods below, the above definition has the following implications:IDENTIFICA TION TESTS: ensure the identity of the analyte.PURITY TESTS: ensure that all the analytical procedures performed allow an accurate statement of the content of impurities of an analyte (e.g., related substances test, heavy metals limit, organic volatile impurity limit).ASSAYS: provide an exact result, which allows an accurate statement on the content or potency of the analyte in a sample.DeterminationIn the case of qualitative analyses (identification tests), the ability to select between compounds of closely related structure that are likely to be present should be demonstrated. This should be confirmed by obtaining positive results (perhaps by comparison to a known reference material) from samples containing the analyte, coupled with negative results from samples that do not contain the analyte and by confirming that a positive response is not obtained from materials structurally similar to or closely related to the analyte.In the case of analytical procedure for impurities, specificity may be established by spiking the drug substance or product with appropriate levels of impurities and demonstrating that these impurities are determined with appropriated accuracy and precision.In the case of the assay, demonstration of specificity requires that it can be shown that the procedure is unaffected by the presence of impurities or excipients. In practice, this can be done by spiking the drug substance or product with appropriatelevels of impurities or excipients and demonstrating that the assay result is unaffected by the presence of these extraneous materials.If impurity or degradation product standards are unavailable, specificity may be demonstrated by comparing the test results of samples containing impurities or degradation products to a second wellcharacterized procedure (e.g., a pharmacopeial or other validated procedure). These comparisons should include samples stored under relevant stress conditions (e.g., light, heat, humidity, acid/base hydrolysis, oxidation). In the case of the assay, the results should be compared; in the case of chromatographic impurity tests, the impurity profiles should be compared.The ICH documents state that when chromatographic procedures are used, representative chromatograms should be presented to demonstrate the degree of selectivity, and peaks should be appropriatedly labeled. Peak purity tests (e.g., using diode array or mass spectrometry) may be useful to show that the analyte chromatographic peak is not attributable to more than one component.Detection LimitDefinitionThe detection limit is a characteristic of limit tests. It is the lowest amount of analyte in a sample that can be detected, but not necessarily quantitated, under the stated experimental conditions. Thus, limit tests merely substantiate that the amount of analyte is above or below a certain level. The detection limit is usually expressed as the concentration of analyte (e.g., percentage. parts per billion) in the sample.DeterminationFor noninstrumental methods, the detection limit is generally determined by the analysis of samples with known concentrations of analyte and by establishing the minimum level at which the analyte can be reliably detected.For instrumental procedures, the same method may be used as for noninstrumental. In the case of methods submitted for consideration as official compendial methods, it is almost never necessary to determine the actual detection limit. Rather, the detection limit is shown to be sufficiently low by the analysis of samples with known concentration of analyte above and below the require detection vevel. For example, if it is required to detect an impurity at the level of 0.1%, it should be demonstrated that the procedure will reliably detect the impurity at that level.In the case of instrumental analytical procedures that exhibit back ground noise, the ICH documents describe a common approach, which is to compare measure signals from samples with known low concentrations at which the analyte can reliably be detected is established, Typically acceptable signaltonoise ratios are 2:1 or 3:1. Other approaches depend on the determination of the slope of the calibation curve and the standard deviation of responses. Whatever method is used, the detection limit should be subsequently validated by the analysis of a suitable number of samples known to be near, or prepared at, the detection limit.Quantitation LimitDefinitionThe quantitation limit is a characteristic of quantitative assays for low levels of compounds in sample matrices, such as impurities in bulk drug substances and degradation products in finished pharmaceuticals. It is the lowest amount of analyte in a sample that can be determined with acceptable precision and accuracy under the stated experimental conditions.。
FDA仿制药研发指南(原文 明胶软胶囊)
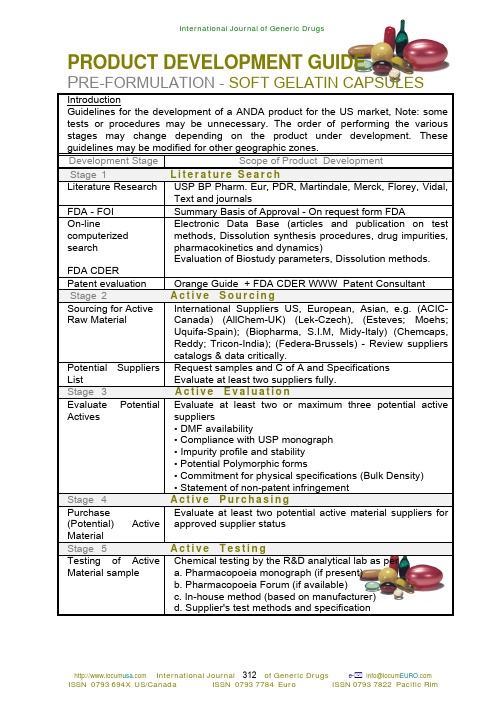
PRODUCT DEVELOPMENT GUIDEP RE-FORMULATION - SOFT GELATIN CAPSULES IntroductionGuidelines for the development of a ANDA product for the US market, Note: some tests or procedures may be unnecessary. The order of performing the various stages may change depending on the product under development. These guidelines may be modified for other geographic zones.Development Stage Scope of Product DevelopmentStage 1L i t e r a t u r e S e a r c hLiterature Research USP BP Pharm. Eur, PDR, Martindale, Merck, Florey, Vidal,Text and journalsFDA - FOI Summary Basis of Approval - On request form FDAOn-line computerized searchFDA CDER Electronic Data Base (articles and publication on test methods, Dissolution synthesis procedures, drug impurities, pharmacokinetics and dynamics)Evaluation of Biostudy parameters, Dissolution methods.Patent evaluation Orange Guide + FDA CDER WWW Patent Consultant Stage 2A c t i v e S o u r c i n gSourcing for Active Raw Material International Suppliers US, European, Asian, e.g. (ACIC-Canada) (AllChem-UK) (Lek-Czech), (Esteves; Moehs; Uquifa-Spain); (Biopharma, S.I.M, Midy-Italy) (Chemcaps, Reddy; Tricon-India); (Federa-Brussels) - Review suppliers catalogs & data critically.Potential Suppliers List Request samples and C of A and Specifications Evaluate at least two suppliers fully.Stage 3A c t i v e E v a l u a t i o nEvaluate Potential Actives Evaluate at least two or maximum three potential active suppliers• DMF availability• Compliance with USP monograph• Impurity profile and stability• Potential Polymorphic forms• Commitment for physical specifications (Bulk Density)• Statement of non-patent infringementStage 4A c t i v e P u r c h a s i n gPurchase (Potential) Active Material Evaluate at least two potential active material suppliers for approved supplier statusStage 5A c t i v e T e s t i n gTesting of Active Material sample Chemical testing by the R&D analytical lab as pera. Pharmacopoeia monograph (if present)b. Pharmacopoeia Forum (if available)c. In-house method (based on manufacturer)d. Supplier's test methods and specificationPRE-FORMULATION - SOFT GELATIN CAPSULES DevelopmentStageScope of Product Development Stage 6I n n o v a t o r's P r o d u c t P u r c h a s i n gDRUG PRODUCT Innovator Samples Purchase at least 3 different lots in smallest and largest pack size for each product strengthStage 7I n n o v a t o r's P r o d u c t T e s t i n gInnovator Testing Evaluate physical parameters:-Capsule size, capsule color / US approved dyes, coding forprinting, pack sizes containers materials, closure types;cotton and desiccants, blister packaging.Innovator Physical Testing Physical testingFill Weight & Uniformity; Shell disintegration & Dissolution Evaluation of capsule size and volume w.r.t. fill volumeEvaluation of Innovator formula ingredients Summary Formula in PDR; International PDRs (Italian, French, Swiss) and Innovators product's insert (obtain latest FOI -FDA)Perform actual analytical testing on innovator's product.Microscopic observation Particle/crystal information on:Particle size, crystal shape, habit, rugosity Viscosity agents used (PVP 30 / 90) Presence of vegetable oil (type) Presence of PEG 400Presence of polyethylene glycol solventEvaluation of Biostudy Review FDA CDER Home page for listing and Biostudy parametersDissolution profile USP monograph and FDA method - (where present)Dissolution; 12 unit Dissolution Profile.Stage 8B u l k A c t i v e T e s t i n gFIRST BATCH FROM APPROVED SUPPLIERFull Physical characterization Physical characterization of bulk batch• Polymorphism• B.E.T.• Particle size distribution (& method development)• Bulk and Tapped density (Need for size reduction of material)• Microscopic observationFULL CHEMICAL CHARACTERIZATI ON Chemical characterization • Assay• Stressed Analysis• Degradants (Expected)• Impurity profile• Optical rotation• Enantiomeric purity• O.V.I. TestingDEVELOPMENT BATCHES - SOFT GELATIN CAPSULES Development Stage Scope of Product DevelopmentStage 9E x c i p i e n t sEvaluate formulation with suitable excipients • Vegetable oil solubility / PEG 400 / PEG 600 / PG / PVP 30• Antioxidants (dl-alpha Tocopherol (Vit. E USP) / Propyl gallate)Stage 10C o n t a i n e r C l o s u r e S y s t e mEvaluation of suitableContainer-Closure System Choice of container-closure-liner system including:• material composition,• type of thermoplastic resin and resin pigments,• manufacturers and suppliers,• liners and seals used by closure manufacturer,• cotton and desiccants.• manufacturer's DMF numbers for all component parts• Letters of Access for regulatory authorities to view DMF dossiersStage 11M a n u f a c t u r i n g P r o c e s sEVALUATION SUITABLE MANUFACTURING PROCESSES Gelatin MassFill Preparation Encapsulation Drying stages • Gel mass blending (rpm & time)• Gel mass melting (temperature and vacuum)• Gel mass color addition (temperature and rpm & time)• Gel mass VISCOSITY and MOISTURE determination • Gel mass deaeration (holding time)• Fill mass (mixing rpm, time & propeller position)• Determination of Fill mass viscosity / SG / moisture• Determination of Bulk Uniformity Analysis• Wet shell weight / Fill weight• Seal thickness• Determination of Drying Parameters• Determination of rotary tumbler drying parameters & time • Determination of primary and secondary tray drying times • Determine Bareiss hardnessFill material Physical Properties of oil or paste fill • Viscosity - (Critical)• Fill moisture• SG (helps to control deaeration)FillingPhysical Properties of Filled Softgels • Weight Uniformity• Individual Fill Weight Limits (7.5%)• Content Uniformity • Average Fill Weight Limits (5.0%)• Disintegration• Dissolution profileFinal Formula Established Assessment of Final Master Formula and accelerated 1-3 month stability profile.Stage 12B u l k A c t i v e P u r c h a s e dActive material Bulk purchase Ordering of Active material for Process Qualification (PQ) and Pivotal Batch(es). On approval of final formula, order sufficient material for the PQ (2) and Pivotal Lots (sufficient for all strengths and batch sizes).NB: Never mix batch numbers in PQ and Pivotal Lots.FULL LABORATORY EVALUATION - SOFTGELS Development Scope of Product DevelopmentStage 13A n a l y t i c a l E v a l u a t i o nAnalytical testing of Softgels • Dissolution - in USP medium (Multipoint profiles) and other relevant media versus Innovator's product. Dissolution testing may not be possible where active strength is in micrograms (i.e. 0.25 or 0.5 mcg)• U of C-for low active concentrations. Refer to USP requirements for uniformity of content vs. uniformity of dosage units.• Validation of analytical package i.e. Assay; Dissolution ; Content Uniformity completed prior to Process QualificationPROCESS OPTIMIZATIONDevelopment Scope of Product Development Stage 14P r o c e s s O p t i m i z a t i o nGEL & FILL MATERIAL OPTIMIZATION [In suspension and paste fills size reduction of active material may be necessary i.e. to decrease active bulk densities from 0.6 -0.75g/cc to 0.35 - 0.45 g/cc]◊ Optimization of gel mass moisture (once per shell formula)◊ Optimization of gel mass viscosity (once per shell formula)◊ Deaeration of gel mass - (critical)◊ Optimization of antioxidant percentage.◊ Mixing process - rpm ; mixing time ; propeller position◊ Fill material uniformity◊ Ribbon Thickness adjustments for correct shell weights (once per shell formula)◊ Seal Thickness (once per shell formula NLT 0.006")1◊ Wet Shell Weight variation (± 8%)1◊ Fill Weight variation (± 2%)1◊ 1 Process capability performed.DRYING♦ Drying time temperature versus shell moisture (in rotarydryer)♦ Primary drying time versus shell moisture (in tray dryer)♦ Secondary drying time versus shell moisture (in traydryer)♦ Bareiss hardness versus drying times.♦ Softgel properties (Fill weights and Content Uniformity).♦ Evaluation of Fill weight Range Limits (Qualification)♦ Evaluation of stability results of optimized mfg. Process.♦ Printing Inspection LimitsPROCESS OPTIMIZATION REPORT (PO)Prepare PO Report.This Process Optimization Report forms part of the product Development ReportESTABLISHING AND INVITRO INVIVO CORRELATION DevelopmentScope of Product Development Stage 15A n a l y t i c a l E v a l u a t i o n IVIV Correlation(search literature)• Dissolution - in USP medium (Multipoint profiles) and other relevant media versus Innovator's product. Dissolutiontesting may not be possible where active strength is inmicrograms (i.e. 0.25 or 0.5 mcg)• Perform IVIV Bioavailability Study (where relevant)BSC System IVIVC 1 : 1 Correlation Dissolution settings Plasma parameters D evelopers are encouraged to develop IVIVC for IR dosageforms, where applicable to the BCS, (BiopharmaceuticalClassification System) in the expectation that the informationwill be useful in establishing appropriate dissolutionspecifications and thus permit certain post approvalformulation and manufacturing changes to be effected, -without additional bioequivalence studies.T he objective of developing an IVIVC is to establish apredictive mathematical model describing the relationshipbetween invitro dissolution settings and the actual invivodrug-plasma parameters found, (such as AUC, Cmax,Tmax).T he invitro dissolution settings are adjusted (via media, pHagitation) until a I : I correlation is achieved (Level A) or asingle dissolution point and a plasma parameter is shown tocorrelate (Level C).When more than one point correlates a multiple Level C isobtained - which may possibly be upgraded to a Level Awith additional development work.T his matching of dissolution settings with plasma levels, thatare derived from a specific IR formula and its correspondingmanufacturing process, is in fact simply an arbitrary set ofvalues that establish the so called 'predictive mathematicalmodel'.A n IVIVC should be evaluated to demonstrate thatpredictability of the invivo performance of the drug product(i.e. derived from the plasma parameters) from its in vitrodissolution characteristics (e.g. equipment settings / andmanufacturing changes) is maintained over the product'sdissolution profile.Not applicable to highly soluble materials Establish a Level A or C correlation without adjustingdissolution parameters and time scale.Not applicable to highly solublematerials• Adjust the dissolution parameters or time scale to achievea Level A or C correlation (adjust only if necessary)SCALE UPDevelopmentStageScope of Product DevelopmentStage16S c a l e-u pScale-up S cale-up lot prepared if larger batch size scale up problemsanticipated.S cale-up of gel massS cale-up of fill material (N2 atmosphere and final filtration)S cale-up of encapsulation procedure (seal thickness)S cale-up of primary and secondary drying (Time &moisture)S cale-up of sorting, sizing and printing.P rocess Qualification batch and Scale-up batch may beevaluated as a single batch.Scale-up Report T he preparation of a Scale-up Report. The Scale-up reportforms part of the overall Development Report PROCESS QUALIFICATIONDevelopmentStageScope of Product DevelopmentStage 17P r o c e s s Q u a l i f i c a t i o nT he process qualification batch is manufactured in order to detect any problems that may arise during the manufacture of production size batches, allowing a solution prior the manufacture of the pivotal demonstration batch.S cale-up to the pivotal batch size or 70% of the pivotal batch may be combined with qualifying the manufacturing process.A t this stage full manufacturing documentation is prepared alone standard procedures.PRODUCTION FACILITIES P rocess Qualification batch should be executed on a commercial production (or production type with same principle and operation) encapsulating machine in a production setting.The primary dryer and secondary tray or tunnel dryingequipment should be identical or similar.S ize of pivotal and marketing batch confirmed (NLT 100 000 net/ packed at target parameters or 10% of proposed market batch).Ideally for Soft Gelatin Capsules 120 000 - 150 000 or more units should be prepared for pivotal batch to allow for some level of qualification by testing and challenging both ends of the selected specification limits.BATCH DOCUMENTATION P reparation of Master Formula and Processing InstructionsDiscussion of formula, manufacturing process and control parameters with production personnel and QA StaffPROCESS QUALIFICATION - SOFTGELS Development Stage Scope of Product Development Stage 17 (Cont)P r o c e s s Q u a l i f i c a t i o nFINAL REVIEW and AUTHORIZATION Review of proposed formula, manufacturing process and control parameters with production personnel and QA Staff with authorization signatures (RD; QA-QC; RA; and Production)PROTOCOL PQ. protocol preparedKEY STEPS Critical manufacturing steps designated and sampling andtesting parameters specified.OPERATING CONDITIONS Presence of production and control personnel during PQ manufacturePQ REPORT Upon completion prepare Process Qualification Report. ThisP-Q report forms part of the overall Development Report PIVOTAL BATCHDevelopment Scope of Product DevelopmentStage 18P i v o t a l P r o d u c t i o nPRODUCTION FACILITIES Pivotal batch MUST be compressed in a production Encapsulating machine (or production type with same principle and operation)BATCH DOCUMENTATION Preparation of FINAL Master Formula and Processing InstructionsREVIEW and AUTHORIZATION Review of FINAL formula, manufacturing process and control parameters with production personnel and QA Staff. Pivotal authorization signatures (RD; QA-QC; RA; and Production) attached.OPERATING CONDITIONS Operation of production and control personnel during Pivotal manufacture, aided by development team.REPORT The preparation of a Pivotal Report. This pivotal reportforms partof the overall Development Report. BIOEQUIVALENT STUDYStage Scope of Product DevelopmentStage19B I O S T U D Y E v a l u a t i o nBIOSTUDY Fasted Perform Fasted / Food Effect Biostudy on Pivotal LotSamplesBIOSTUDY [Food Effect]Perform Food Effect Biostudy on Pivotal Lot Samples (See food effect guidelines, where appropriate)HIGHEST DOSAGE Biostudy generally performed on highest strength of product One or two studies Fasted AND Food Effect Study may be requiredWAIVER CONDITIONS For multiple strength products Invitro dissolution testing conducted in three different pH media on lower dosage formsSIMILARITYTESTINGPerform Similarity Test [F2 Test] on dissolution results.PRE-SUBMISSION AUDITING - SOFTGELSDevelopment Stage Scope of Product DevelopmentStage20A N D A P r e-S u b m i s s i o n A u d i t i n gDevelopmentReportAudit all raw data supporting Development ReportANDA RegulatoryFileAudit Plant and Laboratory Documentation as per ANDA SOPs Review SOP System and Update levelcGMP Review cGMP of Manufacturing ProcessesBiostudy Report Evaluate and develop a IVIV correlation (Level A wherepossible.)Validation Protocol Product Process Validation Protocol complete and signed A NDA SUBMISSION - SOFTGELSDevelopment Stage Scope of Product DevelopmentStage21A N D A S u b m i s s i o nANDA Submission Submit ANDA structured carefully as per Feb. 1999Guidelines(9 Copies -as per Color system)(1 Field Copy)V ALIDATION BATCHESDevelopmentStageScope of Product DevelopmentStage 22P r o c e s s V a l i d a t i o nProtocol Process Validation Protocol for 3 consecutive marketing lots Execute validation Process Validation of 3 consecutive marketing lotsReport Process Validation ReportSimilarity Show intra-batch similarityBio-Validation Similarity Show inter-batch similarity between Biobatch (Pivotal) and the Commercial Validation LotsCOMMERCIAL RE-VALIDATION DUE TO MAJOR CHANGE DevelopmentStageScope of Product DevelopmentStage 23P r o c e s s R e-v a l i d a t i o nFormula Change Revalidate procedure with new formula process orequipment withProcess Change a different operating principleEquipment ChangeMinor change Follow SUPAC Rules Level I II or III。
HPLC法测定醋酸曲普瑞林微球的载药量及包封率

H P L C法测定醋酸曲普瑞林微球的载药量及包封率刘智慧,赵冰,王燕清,徐朋,戈芸芸,苏日佳•(丽珠医药集团股份有限公司,广东珠海519090 )摘要:目的建立H P L C法测定醋酸曲普瑞林微球的栽药量和包封率的方法。
方法色谱柱为P h e n o m e n e x K in etex®E V O C,8 (150 m m x4.6 m m,5 g m);流动相为0.05 m o l.L/1磷酸溶液(用三乙胺调节p H值至3.5)-乙腈(78:22);柱温35 流速为1.0 m L.m iiT1;检测波长为220 n m。
结果空白溶剂、空白微球及供试品溶液中其他杂质色谱峰对主峰曲普瑞林的测定无干扰,曲普瑞林与前后相邻已知杂质峰分离度大于丨.5;曲普瑞林浓度范围在44.5~133.6 p g.m L—线性良好,相关系数为0.999 9 (n=5);定量限为0.16 g g.m L—1;50%浓度回收率为100.1%, 100%浓度回收率为100.5%, 150%浓度回收率为100.5%, ftSZ)为 0.5%;重复性/«£»为0.8%。
结论高效液相色谱方法准确、可靠、专属性强、耐用,可用于醋酸曲普瑞林缓释微球中药物含量的测定和包封率的测定,为制剂的质量控制提供方法。
关键词:醋酸曲普瑞林;微球;H P L C;栽药量;包封率中图分类号:R917 文献标志码:A文章编号:1674-229X(2021) 05-0370-04D o i:10.12048/j.is s n.1674-229X.2021.05.010HPLC Determination of Drug Loading and Encapsulated Efficiency of Triptorelin Acetate MicrospheresLIU Zhihui,ZHAO Bing,WANG Yanqing,XU Peng,GE Yunyun,SU Rijia*(L it)z o n Pharmaceutical Group Co. ,L td., Zhuhai, Guangdong 519090.China)A B S T R A C T:O B J E C T I V E H P L C m e t h o d to established a H P L C m e t h o d for the d e t ermination of d r u g loading a n d e n c a p sulated efficiency of triptorelin acetate microspheres. M E T H O D S A P h e n o m e n e x K i n e t e x® E V O C,8( 150 m m x4.6 m m,5(x m)c o l u m n w a s a d o p t e d with a m o b i l e p h a s e of 0.05m o l*L~l p h o s p h o r i c acid solution(adjust p H to 3.5 with triethylamine )-acetonitrile( 78:22) .T h e flow rate w a s 1.0 m L* m i n''.T h e detection w a v e l e n g t h w a s set at 220n m a n d the c o l u m n t e m p e r a t u r e w a s35Tl. R E S U L T S T h e bl a n k solvent, b l a n k m i c r o s p h e r e s a n d other impurities in s a m p l e s did not interfere with the determination of triptorelin.The resolutionb e t w e e n triptoreline a n d adjacent impurity p e a k s w e r e m o r e t han 1.5.T h e s t andardcurve of triptorelin w a s at g o o d linear in the r a n g e of44.5-133.6^Jig*mL '.T h e correlation coefficient w a s reach 0.999 9(^=5).T h e quantitation limit w a s 0.16 |X g*m L_l.T h e recovery rate of concentration of 50%w a s100.1%,a n d the recovery rate of concentration of 100%w a s100.5%.T h e recovery rate of concentration of 150%w a s100.5%.A n d RSD w a s0.5%, the RSD of repeatability w a s0.8%.C O N C L U S I O N T h e m e t h o d of H P L C is accurate, reliable, specific a n d durable. It c a n b e u s e d to determ i n a t e the d r u g loading a n d e n c a p s u l a t e d efficiency of triptorelin acetate m i c r o s p h e r e s a n d for quality control of m i c r o s p h e r e s .K E Y W O R D S:triptorelin acetate;m i c r o s p h e r e s;H P L C;d r u g l oading;enc a p s u l a t e d efficiency醋酸曲普瑞林为人工合成的强效促性腺激素 释放激素(gonadotropin-releasing hormone,GnRH)。
ASTM B117盐雾测试标准

Designation:B 117–03Standard Practice forOperating Salt Spray (Fog)Apparatus 1This standard is issued under the fixed designation B 117;the number immediately following the designation indicates the year of original adoption or,in the case of revision,the year of last revision.A number in parentheses indicates the year of last reapproval.A superscript epsilon (e )indicates an editorial change since the last revision or reapproval.This standard has been approved for use by agencies of the Department of Defense.1.Scope1.1This practice covers the apparatus,procedure,and conditions required to create and maintain the salt spray (fog)test environment.Suitable apparatus which may be used is described in Appendix X1.1.2This practice does not prescribe the type of test speci-men or exposure periods to be used for a specific product,nor the interpretation to be given to the results.1.3The values stated in SI units are to be regarded as standard.The inch-pound units in parentheses are provided for information and may be approximate.1.4This standard does not purport to address all of the safety concerns,if any,associated with its use.It is the responsibility of the user of this standard to establish appro-priate safety and health practices and determine the applica-bility of regulatory limitations prior to use.2.Referenced Documents 2.1ASTM Standards:B 368Method for Copper-Accelerated Acetic Acid-Salt Spray (Fog)Testing (CASS Test)2D 609Practice for Preparation of Cold-Rolled Steel Panels for Testing Paint,Varnish,Conversion Coatings,and Related Coating Products 3D 1193Specification for Reagent Water 4D 1654Test Method for Evaluation of Painted or Coated Specimens Subjected to Corrosive Environments 3E 70Test Method for pH of Aqueous Solutions with the Glass Electrode 5E 691Practice for Conducting an Interlaboratory Study to Determine the Precision of a Test Method 6G 85Practice for Modified Salt Spray (Fog)Testing 73.Significance and Use3.1This practice provides a controlled corrosive environ-ment which has been utilized to produce relative corrosion resistance information for specimens of metals and coated metals exposed in a given test chamber.3.2Prediction of performance in natural environments has seldom been correlated with salt spray results when used as stand alone data.3.2.1Correlation and extrapolation of corrosion perfor-mance based on exposure to the test environment provided by this practice are not always predictable.3.2.2Correlation and extrapolation should be considered only in cases where appropriate corroborating long-term atmo-spheric exposures have been conducted.3.3The reproducibility of results in the salt spray exposure is highly dependent on the type of specimens tested and the evaluation criteria selected,as well as the control of the operating variables.In any testing program,sufficient repli-cates should be included to establish the variability of the results.Variability has been observed when similar specimens are tested in different fog chambers even though the testing conditions are nominally similar and within the ranges speci-fied in this practice.4.Apparatus4.1The apparatus required for salt spray (fog)exposure consists of a fog chamber,a salt solution reservoir,a supply of suitably conditioned compressed air,one or more atomizing nozzles,specimen supports,provision for heating the chamber,and necessary means of control.The size and detailed con-struction of the apparatus are optional,provided the conditions obtained meet the requirements of this practice.4.2Drops of solution which accumulate on the ceiling or cover of the chamber shall not be permitted to fall on the specimens being exposed.1This practice is under the jurisdiction of ASTM Committee G01on Corrosion of Metals and is the direct responsibility of Subcommittee G01.05on Laboratory Corrosion Tests.Current edition approved October 1,2003.Published October 2003.Originally approved in st previous edition approved in 2002as B 117–02.2Annual Book of ASTM Standards ,V ol 02.05.3Annual Book of ASTM Standards ,V ol 06.01.4Annual Book of ASTM Standards ,V ol 11.01.5Annual Book of ASTM Standards ,V ol 15.05.6Annual Book of ASTM Standards ,V ol 14.02.7Annual Book of ASTM Standards ,V ol 03.02.1Copyright ©ASTM International,100Barr Harbor Drive,PO Box C700,West Conshohocken,PA 19428-2959,UnitedStates.4.3Drops of solution which fall from the specimens shall not be returned to the solution reservoir for respraying.4.4Material of construction shall be such that it will not affect the corrosiveness of the fog.4.5All water used for this practice shall conform to Type IV water in Specification D1193(except that for this practice limits for chlorides and sodium may be ignored).This does not apply to running tap water.All other water will be referred to as reagent grade.5.Test Specimens5.1The type and number of test specimens to be used,as well as the criteria for the evaluation of the test results,shall be defined in the specifications covering the material or product being exposed or shall be mutually agreed upon between the purchaser and the seller.6.Preparation of Test Specimens6.1Specimens shall be suitably cleaned.The cleaning method shall be optional depending on the nature of the surface and the contaminants.Care shall be taken that specimens are not recontaminated after cleaning by excessive or careless handling.6.2Specimens for the evaluation of paints and other organic coatings shall be prepared in accordance with applicable specification(s)for the material(s)being exposed,or as agreed upon between the purchaser and the supplier.Otherwise,the test specimens shall consist of steel meeting the requirements of Practice D609and shall be cleaned and prepared for coating in accordance with the applicable procedure of Practice D609.6.3Specimens coated with paints or nonmetallic coatings shall not be cleaned or handled excessively prior to test.6.4Whenever it is desired to determine the development of corrosion from an abraded area in the paint or organic coating, a scratch or scribed line shall be made through the coating with a sharp instrument so as to expose the underlying metal before testing.The conditions of making the scratch shall be as defined in Test Method D1654,unless otherwise agreed upon between the purchaser and the seller.6.5Unless otherwise specified,the cut edges of plated, coated,or duplex materials and areas containing identification marks or in contact with the racks or supports shall be protected with a suitable coating stable under the conditions of the practice.N OTE1—Should it be desirable to cut test specimens from parts or from preplated,painted,or otherwise coated steel sheet,the cut edges shall be protected by coating them with paint,wax,tape,or other effective media so that the development of a galvanic effect between such edges and the adjacent plated or otherwise coated metal surfaces,is prevented.7.Position of Specimens During Exposure7.1The position of the specimens in the salt spray chamber during the test shall be such that the following conditions are met:7.1.1Unless otherwise specified,the specimens shall be supported or suspended between15and30°from the vertical and preferably parallel to the principal direction offlow of fog through the chamber,based upon the dominant surface being tested.7.1.2The specimens shall not contact each other or any metallic material or any material capable of acting as a wick.7.1.3Each specimen shall be placed to permit unencum-bered exposure to the fog.7.1.4Salt solution from one specimen shall not drip on any other specimen.N OTE2—Suitable materials for the construction or coating of racks and supports are glass,rubber,plastic,or suitably coated wood.Bare metal shall not be used.Specimens shall preferably be supported from the bottom or the side.Slotted wooden strips are suitable for the support offlat panels.Suspension from glass hooks or waxed string may be used as long as the specified position of the specimens is obtained,if necessary by means of secondary support at the bottom of the specimens.8.Salt Solution8.1The salt solution shall be prepared by dissolving561 parts by mass of sodium chloride in95parts of water conforming to Type IV water in Specification D1193(except that for this practice limits for chlorides and sodium may be ignored).Careful attention should be given to the chemical content of the salt.The salt used shall be sodium chloride with not more than0.3%by mass of total impurities.Halides (Bromide,Fluoride,and Iodide)other than Chloride shall constitute less than0.1%by mass of the salt content.Copper content shall be less than0.3ppm by mass.Sodium chloride containing anti-caking agents shall not be used because such agents may act as corrosion inhibitors.See Table1for a listing of these impurity restrictions.Upon agreement between the purchaser and the seller,analysis may be required and limits established for elements or compounds not specified in the chemical composition given above.TABLE1Maximum Allowable Limits for Impurity Levels inSodium Chloride A,BImpurity Description Allowable Amount Total Impurities#0.3% Halides(Bromide,Fluoride and Iodide)excluding Chloride#0.1% Copper<0.3ppmAnti-caking Agents0.0%A A common formula used to calculate the amount of salt required by mass to achieve a5%salt solution of a known mass of water is:.053X Mass of Water5Mass of NaCl requiredThe mass of water is1g per1mL.To calculate the mass of salt required in grams to mix1L of a5%salt solution,multiply.053by1000g(35.27oz.,the mass of 1L of water).This formula yields a result of53g(1.87oz.)of NaCl required for each liter of water to achieve a5%salt solution by mass.The0.053multiplier for the sodium chloride used above is derived by the following:1000g~mass of a full L of water!divided by0.95~water is only95%of the total mixture by mass!yields1053gThis1053g is the total mass of the mixture of one L of water with a5%sodium chloride concentration.1053g minus the original weight of the L of water,1000g, yields53g for the weight of the sodium chloride.53g of total sodium chloride divided by the original1000g of water yields a0.053multiplier for the sodium chloride.As an example:to mix the equivalent of200L(52.83gal)of5%sodium chloride solution,mix10.6kg(23.37lb)of sodium chloride into200L(52.83gal)of water. 200L of water weighs200,000g.200,000g of water x.053(sodium chloride multiplier)=10,600g of sodium chloride,or10.6kg.B In order to ensure that the proper salt concentration was achieved when mixing the solution,it is recommended that the solution be checked with either a salimeter hydrometer or specific gravity hydrometer.When using a salimeter hydrometer,the measurement should be between4and6%at25°C(77°F).When using a specific gravity hydrometer,the measurement should be between1.0255and1.0400at 25°C(77°F).8.2The pH of the salt solution shall be such that when atomized at35°C(95°F)the collected solution will be in the pH range from6.5to7.2(Note3).Before the solution is atomized it shall be free of suspended solids(Note4).The pH measurement shall be made at25°C(77°F)using a suitable glass pH-sensing electrode,reference electrode,and pH meter system in accordance with Test Method E70.N OTE3—Temperature affects the pH of a salt solution prepared from water saturated with carbon dioxide at room temperature and pH adjust-ment may be made by the following three methods:(1)When the pH of a salt solution is adjusted at room temperature,and atomized at35°C(95°F),the pH of the collected solution will be higher than the original solution due to the loss of carbon dioxide at the higher temperature.When the pH of the salt solution is adjusted at room temperature,it is therefore necessary to adjust it below6.5so the collected solution after atomizing at35°C(95°F)will meet the pH limits of6.5to 7.2.Take about a50-mL sample of the salt solution as prepared at room temperature,boil gently for30s,cool,and determine the pH.When the pH of the salt solution is adjusted to6.5to7.2by this procedure,the pH of the atomized and collected solution at35°C(95°F)will come within this range.(2)Heating the salt solution to boiling and cooling to35°C(95°F)and maintaining it at35°C(95°F)for approximately48h before adjusting the pH produces a solution the pH of which does not materially change when atomized at35°C(95°F).(3)Heating the water from which the salt solution is prepared to35°C (95°F)or above,to expel carbon dioxide,and adjusting the pH of the salt solution within the limits of6.5to7.2produces a solution the pH of which does not materially change when atomized at35°C(95°F).N OTE4—The freshly prepared salt solution may befiltered or decanted before it is placed in the reservoir,or the end of the tube leading from the solution to the atomizer may be covered with a double layer of cheesecloth to prevent plugging of the nozzle.N OTE5—The pH can be adjusted by additions of dilute ACS reagent grade hydrochloric acid or sodium hydroxide solutions.9.Air Supply9.1The compressed air supply to the Air Saturator Tower shall be free of grease,oil,and dirt before use by passing through well-maintainedfilters.(Note6)This air should be maintained at a sufficient pressure at the base of the Air Saturator Tower to meet the suggested pressures of Table2at the top of the Air Saturator Tower.N OTE6—The air supply may be freed from oil and dirt by passing it through a suitable oil/water extractor(that is commercially available)to stop any oil from reaching the Air Saturator Tower.Many oil/water extractors have an expiration indicator,proper preventive maintenance intervals should take these into account.9.2The compressed air supply to the atomizer nozzle or nozzles shall be conditioned by introducing it into the bottom of a towerfillwed with water.A common method of introduc-ing the air is through an air dispersion device(X1.4.1).The level of the water must be maintained automatically to ensure adequate humidification.It is common practice to maintain the temperature in this tower between46and49°C(114–121°F)to offset the cooling effect of expansion to atmospheric pressure during the atomization process.Table2in9.3of this practice shows the temperature,at different pressures,that are com-monly used to offset the cooling effect of expansion to atmospheric pressure.9.3Careful attention should be given to the relationship of tower temperature to pressure since this relationship can have a direct impact to maintaining proper collection rates(Note7). It is preferable to saturate the air at temperatures well above the chamber temperature as insurance of a wet fog as listed in Table2.N OTE7—If the tower is run outside of these suggested temperature and pressure ranges to acheive proper collection rates as described in10.2of this practice,other means of verifying the proper corrosion rate in the chamber should be investigated,such as the use of control specimens (panels of known performance in the test conducted).It is preferred that control panels be provided that bracket the expected test specimen performance.The controls allow for the normalization of test conditions during repeated running of the test and will also allow comparisons of test results from different repeats of the same test.(Refer to Appendix X3, Evaluation of Corrosive Conditions,for mass loss procedures).10.Conditions in the Salt Spray Chamber10.1Temperature—The exposure zone of the salt spray chamber shall be maintained at35+1.1−1.7°C (95+2−3°F).Each set point and its tolerance represents an operational control point for equilibrium conditions at a single location in the cabinet which may not necessarily represent the uniformity of conditions throughout the cabinet.The tempera-ture within the exposure zone of the closed cabinet shall be recorded(Note8)at least twice a day at least7h apart(except on Saturdays,Sundays,and holidays when the salt spray test is not interrupted for exposing,rearranging,or removing test specimens or to check and replenish the solution in the reservoir)N OTE8—A suitable method to record the temperature is by a continu-ous recording device or by a thermometer which can be read from outside the closed cabinet.The recorded temperature must be obtained with the salt spray chamber closed to avoid a false low reading because of wet-bulb effect when the chamber is open.10.2Atomization and Quantity of Fog—Place at least two clean fog collectors per atomizer tower within the exposure zone so that no drops of solution will be collected from the test specimens or any other source.Position the collectors in the proximity of the test specimens,one nearest to any nozzle and the other farthest from all nozzles.A typical arrangement is shown in Fig.1.The fog shall be such that for each80 cm2(12.4in.2)of horizontal collecting area,there will be collected from1.0to2.0mL of solution per hour based on an average run of at least16h(Note9).The sodium chloride concentration of the collected solution shall be561mass% (Notes9-11).The pH of the collected solution shall be6.5to 7.2.The pH measurement shall be made as described in8.2 (Note3).N OTE9—Suitable collecting devices are glass or plastic funnels with TABLE2Suggested Temperature and Pressure guideline for the top of the Air Saturator Tower for the operation of a test at35°C(95°F)Air Pressure,kPa Temperature,°C Air Pressure,PSI Temperature,°F 83461211496471411711048161191244918121the stems inserted through stoppers into graduated cylinders,or crystal-lizing dishes.Funnels and dishes with a diameter of 10cm (3.94in.)have an area of about 80cm 2(12.4in.2).N OTE 10—A solution having a specific gravity of 1.0255to 1.0400at 25°C (77°F)will meet the concentration requirement.The sodium chloride concentration may also be determined using a suitable salinity meter (for example,utilizing a sodium ion-selective glass electrode)or colorimetrically as follows.Dilute 5mL of the collected solution to 100mL with distilled water and mix thoroughly;pipet a 10-mL aliquot into an evaporating dish or casserole;add 40mL of distilled water and 1mL of 1%potassium chromate solution (chloride-free)and titrate with 0.1N silver nitrate solution to the first appearance of a permanent red coloration.A solution that requires between 3.4and 5.1mL of 0.1N silver nitrate solution will meet the concentration requirements.N OTE 11—Salt solutions from 2to 6%will give the same results,though for uniformity the limits are set at 4to 6%.10.3The nozzle or nozzles shall be so directed or baffled that none of the spray can impinge directly on the test specimens.11.Continuity of Exposure11.1Unless otherwise specified in the specifications cover-ing the material or product being tested,the test shall be continuous for the duration of the entire test period.Continu-ous operation implies that the chamber be closed and the spray operating continuously except for the short daily interruptions necessary to inspect,rearrange,or remove test specimens,to check and replenish the solution in the reservoir,and to make necessary recordings as described in Section 10.Operations shall be so scheduled that these interruptions are held to a minimum.12.Period of Exposure12.1The period of exposure shall be as designated by the specifications covering the material or product being tested or as mutually agreed upon between the purchaser and the seller.N OTE 12—Recommended exposure periods are to be as agreed upon between the purchaser and the seller,but exposure periods of multiples of 24h are suggested.13.Cleaning of Tested Specimens13.1Unless otherwise specified in the specifications cover-ing the material or product being tested,specimens shall be treated as follows at the end of the test:13.1.1The specimens shall be carefully removed.13.2Specimens may be gently washed or dipped in clean running water not warmer than 38°C (100°F)to remove salt deposits from their surface,and then immediately dried.14.Evaluation of Results14.1A careful and immediate examination shall be made as required by the specifications covering the material or product being tested or by agreement between the purchaser and the seller.15.Records and Reports15.1The following information shall be recorded,unless otherwise prescribed in the specifications covering the material or product being tested:15.1.1Type of salt and water used in preparing the salt solution,15.1.2All readings of temperature within the exposure zone of the chamber,15.1.3Daily records of data obtained from each fog-collecting device including the following:15.1.3.1V olume of salt solution collected in millilitres per hour per 80cm 2(12.4in.2),15.1.3.2Concentration or specific gravity at 35°C (95°F)of solution collected,and15.1.3.3pH of collectedsolution.N OTE —This figure shows a typical fog collector arrangement for a single atomizer tower cabinet.The same fog collector arrangement is also applicable for multiple atomizer tower and horizontal (“T”type)atomizer tower cabinet constructions as well.FIG.1Arrangement of FogCollectors15.2Type of specimen and its dimensions,or number or description of part,15.3Method of cleaning specimens before and after testing, 15.4Method of supporting or suspending article in the salt spray chamber,15.5Description of protection used as required in6.5, 15.6Exposure period,15.7Interruptions in exposure,cause,and length of time, and15.8Results of all inspections.N OTE13—If any of the atomized salt solution which has not contacted the test specimens is returned to the reservoir,it is advisable to record the concentration or specific gravity of this solution also.16.Keywords16.1controlled corrosive environment;corrosive condi-tions;determining mass loss;salt spray(fog)exposureAPPENDIXES(Nonmandatory Information)X1.CONSTRUCTION OF APPARATUSX1.1CabinetsX1.1.1Standard salt spray cabinets are available from several suppliers,but certain pertinent accessories are required before they will function according to this practice and provide consistent control for duplication of results.X1.1.2The salt spray cabinet consists of the basic chamber, an air-saturator tower,a salt solution reservoir,atomizing nozzles,specimen supports,provisions for heating the cham-ber,and suitable controls for maintaining the desired tempera-ture.X1.1.3Accessories such as a suitable adjustable baffle or central fog tower,automatic level control for the salt reservoir, and automatic level control for the air-saturator tower are pertinent parts of the apparatus.X1.1.4The size and shape of the cabinet shall be such that the atomization and quantity of collected solution is within the limits of this practice.X1.1.5The chamber shall be made of suitably inert mate-rials such as plastic,glass,or stone,or constructed of metal and lined with impervious plastics,rubber,or epoxy-type materials or equivalent.X1.1.6All piping that contacts the salt solution or spray should be of inert materials such as plastic.Vent piping should be of sufficient size so that a minimum of back pressure exists and should be installed so that no solution is trapped.The exposed end of the vent pipe should be shielded from extreme air currents that may causefluctuation of pressure or vacuum in the cabinet.X1.2Temperature ControlX1.2.1The maintenance of temperature within the salt chamber can be accomplished by several methods.It is generally desirable to control the temperature of the surround-ings of the salt spray chamber and to maintain it as stable as possible.This may be accomplished by placing the apparatus in a constant-temperature room,but may also be achieved by surrounding the basic chamber of a jacket containing water or air at a controlled temperature.X1.2.2The use of immersion heaters in an internal salt solution reservoir or within the chamber is detrimental where heat losses are appreciable because of solution evaporation and radiant heat on the specimens.X1.3Spray NozzlesX1.3.1Satisfactory nozzles may be made of hard rubber, plastic,or other inert materials.The most commonly used type is made of plastic.Nozzles calibrated for air consumption and solution-atomized are available.The operating characteristics of a typical nozzle are given in Table X1.1.X1.3.2It can readily be seen that air consumption is relatively stable at the pressures normally used,but a marked reduction in solution sprayed occurs if the level of the solution is allowed to drop appreciably during the test.Thus,the level of the solution in the salt reservoir must be maintained automatically to ensure uniform fog delivery during the test.8 X1.3.3If the nozzle selected does not atomize the salt solution into uniform droplets,it will be necessary to direct the spray at a baffle or wall to pick up the larger drops and prevent them from impinging on the test specimens.Pending a com-plete understanding of air-pressure effects,and so forth,it is important that the nozzle selected shall produce the desired 8A suitable device for maintaining the level of liquid in either the saturator tower or reservoir of test solution may be designed by a local engineering group,or may be purchased from manufacturers of test cabinets as an accessory.TABLE X1.1Operating Characteristics of Typical Spray Nozzle SiphonHeight,cmAir Flow,dm3/min Solution Consumption,cm3/hAir Pressure,kPa Air Pressure,kPa34691031383469103138 101926.531.5362100384045845256 201926.531.536636276037204320 301926.531.5360138030003710 401926.631.536078021242904SiphonHeight,in.Air Flow,L/minSolutionConsumption,mL/hAir Pressure,psi Air Pressure,psi51015205101520 41926.531.5362100384045845256 81926.531.536636276037204320 121926.531.5360138030003710 161926.631.536078021242904condition when operated at the air pressure selected.Nozzles are not necessarily located at one end,but may be placed in the center and can also be directed vertically up through a suitable tower.X1.4Air for AtomizationX1.4.1The air used for atomization must be free of grease, oil,and dirt before use by passing through well-maintainedfilters.Room air may be compressed,heated,humidified,and washed in a water-sealed rotary pump if the temperature of the water is suitably controlled.Otherwise cleaned air may be introduced into the bottom of a towerfilled with water through a porous stone or multiple nozzles.The level of the water must be maintained automatically to ensure adequate humidification.A chamber operated in accordance with this method and Appendix X1will have a relative humidity between95and 98%.Since salt solutions from2to6%will give the same results(though for uniformity the limits are set at4to6%),it is preferable to saturate the air at temperatures well above the chamber temperature as insurance of a wet fog.Table X1.2 shows the temperatures,at different pressures,that are required to offset the cooling effect of expansion to atmospheric pressure.X1.4.2Experience has shown that most uniform spray chamber atmospheres are obtained by increasing the atomizing air temperature sufficiently to offset heat losses,except those that can be replaced otherwise at very low-temperature gradi-ents.X1.5Types of ConstructionX1.5.1A modern laboratory cabinet is shown in Fig.X1.1. Walk-in chambers are usually constructed with a sloping ceiling.Suitably located and directed spray nozzles avoid ceiling accumulation and drip.Nozzles may be located at the ceiling,or0.91m(3ft)from thefloor directed upward at30to 60°over a passageway.The number of nozzles depends on type and capacity and is related to the area of the test space.An11 to19L(3to5-gal)reservoir is required within the chamber, with the level controlled.The major features of a walk-in type cabinet,which differs significantly from the laboratory type, are illustrated in Fig.X1.2.Construction of a plastic nozzle, such as is furnished by several suppliers,is shown in Fig.X1.3.TABLE X1.2Temperature and Pressure Requirements forOperation of Test at95°FAir Pressure,kPa8396110124 Temperature,°C46474849Air Pressure,psi12141618 Temperature,°F114117119121。
correlation 标准流程

correlation 标准流程英文回答:Correlation.Correlation is a statistical measure that expresses the extent to which two variables are linearly related. It is a value between -1 and 1, where -1 indicates a perfect negative correlation, 0 indicates no correlation, and 1 indicates a perfect positive correlation.The correlation coefficient is calculated by dividing the covariance of the two variables by the product of their standard deviations. The covariance is a measure of how much the two variables vary together, and the standard deviation is a measure of how much each variable varies on its own.Correlation is a useful tool for understanding the relationship between two variables. It can be used toidentify trends, make predictions, and test hypotheses. However, it is important to note that correlation does not imply causation. Just because two variables are correlated does not mean that one causes the other.Types of Correlation.There are three main types of correlation:Positive correlation: This type of correlation occurs when two variables increase or decrease together. For example, the number of hours you study for a test and your score on the test are positively correlated.Negative correlation: This type of correlation occurs when one variable increases and the other variable decreases. For example, the amount of money you spend ongas and your car's gas mileage are negatively correlated.No correlation: This type of correlation occurs when there is no relationship between two variables. For example, the number of times you flip a coin and the number of headsyou get are not correlated.Strength of Correlation.The strength of a correlation is determined by the absolute value of the correlation coefficient. The closer the correlation coefficient is to 1 or -1, the stronger the correlation. A correlation coefficient of 0 indicates that there is no correlation between the two variables.Significance of Correlation.The significance of a correlation is determined by the p-value. The p-value is the probability of obtaining a correlation coefficient as large as or larger than the one that was observed, assuming that there is no correlation between the two variables. A p-value less than 0.05 is considered to be statistically significant.Correlation Analysis.Correlation analysis is a statistical technique that isused to identify and measure the relationship between two or more variables. Correlation analysis can be used to:Identify trends.Make predictions.Test hypotheses.Control for confounding variables.Correlation analysis is a valuable tool for understanding the relationships between variables. However, it is important to note that correlation does not imply causation.中文回答:相关性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
We study transport through a one-dimensional quantum wire of correlated fermions connected to semi-infinite leads. The wire contains either a single impurity or two barriers, the latter allowing for resonant tunneling. In the leads the fermions are assumed to be non-interacting. The wire is described by a microscopic lattice model. Using the functional renormalization group we calculate the linear conductance for wires of mesoscopic length and for all relevant temperature scales. For a single impurity, either strong or weak, we find power-law behavior as a function of temperature. In addition, we can describe the complete crossover from the weak- to the strong-impurity limit. For two barriers, depending on the parameters of the enclosed quantum dot, we find temperature regimes in which the conductance follows power-laws with “universal” exponents as well as non-universal behavior. Our approach leads to a comprehensive picture of resonant tunneling. We compare our results with those of alternative approaches.
1
arXiv:cond-mat/0411310v2 [cond-mat.mes-hall] 17 Jan 2005
Max-Planck-Institut f¨ ur Festk¨ orperforschung, Heisenbergstr. 1, D-70569 Stuttgart, Germany 2 Institut f¨ ur Theoretische Physik, Universit¨ at G¨ ottingen, Friedrich-Hund-Platz 1, D-37077 G¨ ottingen, Germany
I.
INTRODUCTION源自The interplay of static impurities and correlations in one-dimensional (1d) Fermi systems leads to a variety of surprising effects. A detailed understanding of the physics involved is interesting from the basic-science point of view as well as from the perspective of a possible application of 1d quantum wires in future nanoelectronics. In a 1d metallic system, correlations have a strong effect on the low-energy properties. Quite differently from the conventional Fermi-liquid behavior and even in the homogeneous case, physical properties at low energy scales follow power-laws, described by the Tomonaga-Luttinger liquid (TLL) phenomenology.1 For spin-rotational invariant and spinless models the exponents can be expressed in terms of a single number, the interaction-dependent TLL parameter K . A single static impurity in a TLL with repulsive interaction (0 < K < 1) leads to a dramatic modification of the low-energy physics as is obvious from lowest order perturbation theory in the strength of the impurity.2–5 More elaborate methods show that for vanishing energy scale s the conductance of such a system vanishes following a power-law.6–10 The scaling exponent 2αB is independent of the bare impurity strength and determined by the exponent αB = 1/K − 1 (for spinless fermions) of the one-particle spectral weight of a TLL close to an open boundary.6 For weak impurities the correction to the impurity free conductance scales as s2(K −1) , which holds as long as the correction stays small. Assuming an infinite system the asymptotic low-energy properties have been investigated intensively within an integrable field theoretical model — the local sine-Gordon model (LSGM).6–8,10 For this model, with fixed K the conductance as a function of temperature follows one-parameter scaling for different strengths of the impurity. Similar results were obtained for a TLL connected by arbitrarily “smooth” (spatially adiabatic) contacts to semi-infinite Fermi-liquid leads.11–16 Also transport through a double barrier enclosing a
Impurity and correlation effects on transport in one-dimensional quantum wires
T. Enss,1 V. Meden,2 S. Andergassen,1 X. Barnab´ e-Th´ eriault,2, ∗ W. Metzner,1 and K. Sch¨ onhammer2
quantum dot has been studied over the last few years using field theoretical models.6,17–27 Tuning the dot energies by a gate voltage Vg resonant tunneling can be achieved. For appropriately chosen dot parameters (dot size, barrier height) and as a function of temperature T the conductance G shows different temperature regimes with distinctive power-law scaling and exponents which can be expressed in terms of K .17,19–21,24–27 For the double-barrier problem no exactly solvable generic model is known and applying different approximate analytical17,19–21,24,25,27 and numerical26 methods has not provided a consistent picture. To study the effect of the interplay of correlations and impurities on transport on all relevant energy scales we use the functional renormalization group (fRG) method. The fRG can be applied directly to microscopic models. We consider the lattice model of spinless fermions with nearest-neighbor hopping t and nearest-neighbor interaction U . The impurities are modeled by either locally raising site energies or reducing hopping matrix elements across bonds. The fRG was recently introduced28 as a new powerful tool for studying interacting Fermi systems. It provides a systematic way of resumming competing instabilities29 and goes beyond simple perturbation theory even in problems which are not plagued by infrared divergences.30 The fRG procedure we use starts from an exact hierarchy of differential flow equations for the oneparticle irreducible vertex functions,31–33 as e.g. the selfenergy and the effective two-particle interaction. It is derived by replacing the free propagator by a propagator depending on an infrared cutoff Λ and taking the derivative of the generating functional with respect to Λ. At T = 0 we introduced earlier two truncation schemes which led to a manageable number of coupled equations.34–36 The flow of the self-energy, which in particular encodes the renormalized impurity potential, is fully taken into account, while the two-particle vertex is parametrized by a single flowing coupling constant.36 The bare two-particle interaction U is taken as a small parameter, but the impurities can have arbitrary strength and