第三章-化学反应速率理论
合集下载
无机化学第三章化学反应速率和化学平衡ppt课件

生成2mol NH3
ppt课件
5
反应进度 0=∑νBB B
对于化学计量方程式
dξ = dnB /νB
nB——B的物质的量 ξ的单位为mol νB——为B的化学计量数
改写为 dnB = νB dξ
开始时ξ0=0, 、 nB =0 反应进度为ξ时: nB =νBξ
ppt课件
6
反应进度
nB =νBξ
基元反应:反应物一步就直接转变为产物
如:
2NO2→ 2NO + O2
NO2 + CO →NO + CO2
非基元反应:反应物经过若干步(若干个
基元反应步骤)才转变为产物。
如 又如
2NO + 2H2 → N2 + 2H2O ① 2NO + H2→N2+ H2O2 ② H2O2 + H2 → 2H2O
ቤተ መጻሕፍቲ ባይዱ
0.3mol L1 s1
v NH3
cNH3
t
0.4 0.0mol L1 2 0s
0.2mol L1 s1
v N2 : v H2 : v NH3 1: 3 : 2
●瞬间速率:某瞬间(即t0)的反应速率
vNH3
dcB,则得:
v 1 dcB B dt
ppt课件
11
对于上述 N2 3H2 2NH3 化学计量方程式 :
v 1 d 1 dcB V dt B dt
1 dcN2 1 dcH2 1 dcNH3
1 dt
3 dt 2 dt
aA + bB
反应 2W+X Y+Z 哪种速率表达式是正确的?
ppt课件
5
反应进度 0=∑νBB B
对于化学计量方程式
dξ = dnB /νB
nB——B的物质的量 ξ的单位为mol νB——为B的化学计量数
改写为 dnB = νB dξ
开始时ξ0=0, 、 nB =0 反应进度为ξ时: nB =νBξ
ppt课件
6
反应进度
nB =νBξ
基元反应:反应物一步就直接转变为产物
如:
2NO2→ 2NO + O2
NO2 + CO →NO + CO2
非基元反应:反应物经过若干步(若干个
基元反应步骤)才转变为产物。
如 又如
2NO + 2H2 → N2 + 2H2O ① 2NO + H2→N2+ H2O2 ② H2O2 + H2 → 2H2O
ቤተ መጻሕፍቲ ባይዱ
0.3mol L1 s1
v NH3
cNH3
t
0.4 0.0mol L1 2 0s
0.2mol L1 s1
v N2 : v H2 : v NH3 1: 3 : 2
●瞬间速率:某瞬间(即t0)的反应速率
vNH3
dcB,则得:
v 1 dcB B dt
ppt课件
11
对于上述 N2 3H2 2NH3 化学计量方程式 :
v 1 d 1 dcB V dt B dt
1 dcN2 1 dcH2 1 dcNH3
1 dt
3 dt 2 dt
aA + bB
反应 2W+X Y+Z 哪种速率表达式是正确的?
第三章 化学反应速率
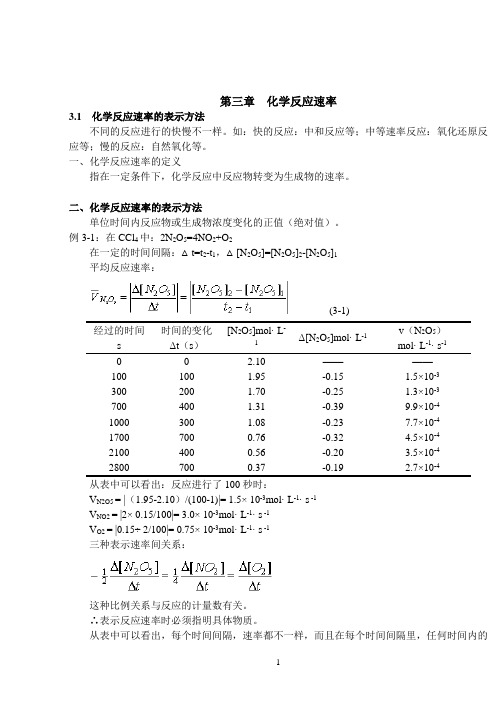
第三章化学反应速率3.1化学反应速率的表示方法不同的反应进行的快慢不一样。
如:快的反应:中和反应等;中等速率反应:氧化还原反应等;慢的反应:自然氧化等。
一、化学反应速率的定义指在一定条件下,化学反应中反应物转变为生成物的速率。
二、化学反应速率的表示方法单位时间内反应物或生成物浓度变化的正值(绝对值)。
例3-1:在CCl4中:2N2O5=4NO2+O2在一定的时间间隔:△t=t2-t1,△[N2O5]=[N2O5]2-[N2O5]1平均反应速率:(3-1)经过的时间s 时间的变化Δt(s)[N2O5]mol· L-1Δ[N2O5]mol· L-1v(N2O5)mol· L-1· s-10 0 2.10 ————100 100 1.95 -0.15 1.5×10-3300 200 1.70 -0.25 1.3×10-3700 400 1.31 -0.39 9.9×10-41000 300 1.08 -0.23 7.7×10-41700 700 0.76 -0.32 4.5×10-42100 400 0.56 -0.20 3.5×10-42800 700 0.37 -0.19 2.7×10-4从表中可以看出:反应进行了100秒时:V N2O5 = |(1.95-2.10)/(100-1)|= 1.5× 10-3mol· L-1· s -1V NO2 = |2× 0.15/100|= 3.0× 10-3mol· L-1· s -1V O2 = |0.15÷ 2/100|= 0.75× 10-3mol· L-1· s -1三种表示速率间关系:这种比例关系与反应的计量数有关。
∴表示反应速率时必须指明具体物质。
大学无机化学课件03化学反应速率

应速率越大。 ③活化配合物分解成反应物的速率:速率越小,
反应速率越大。
26
反应进程势能图
1.ΔH = E正 – E逆
2. 微观可逆性原理
27
实验活化能
❖ 反应物分子须经过一个活化中间状态才能 转化为产物,该中间状态即活化分子。
❖ 由普通反应物分子转变为活化分子需吸收 的能量即活化能Ea。
❖ 根据活化能的大小,和反应能量变化曲线, 可以知道反应是吸热的还是放热的,以及 反应速率的相对快慢。
30
3.3 浓度对反应速率的影响
一、基元反应、简单反应与复合反应
➢ 基元反应(elementary reaction) :反应物分子一步 作用直接转化成产物的反应。由一个基元反应构 成的化学反应称为简单反应。 例:N2O4 = 2NO2 是基元反应,也是简单反应。
➢ 非基元反应:由两个或两个以上基元反应组成的 化学反应,又称复合反应(complex reaction) 、 总反应(overall reaction)。
2. 当具有足够能量的分子以适当的空间取向靠近 时,要进行化学键重排,能量重新分配,形成 一个过渡状态的活化配合物。
3. 过渡状态的活化配合物是一种不稳定状态。 可形成生成物,也可回到反应物。
23
3.2 反应速率理论简介
二、过渡态理论
如: A+ B-C [A…B…C] = A-B +C 反应物 活化配合物 产物
化学反应涉及两个基本问题: ①反应的方向和限度: 反应热力学
反应是否自发,自发的方向和程度 ②反应的速率和具体步骤: 反应动力学
研究途径和速率问题
3
研究反应速率的意义
反应速率
快 ,如爆炸反应,中和反应。 慢,金属的锈蚀,食品的腐烂,
反应速率越大。
26
反应进程势能图
1.ΔH = E正 – E逆
2. 微观可逆性原理
27
实验活化能
❖ 反应物分子须经过一个活化中间状态才能 转化为产物,该中间状态即活化分子。
❖ 由普通反应物分子转变为活化分子需吸收 的能量即活化能Ea。
❖ 根据活化能的大小,和反应能量变化曲线, 可以知道反应是吸热的还是放热的,以及 反应速率的相对快慢。
30
3.3 浓度对反应速率的影响
一、基元反应、简单反应与复合反应
➢ 基元反应(elementary reaction) :反应物分子一步 作用直接转化成产物的反应。由一个基元反应构 成的化学反应称为简单反应。 例:N2O4 = 2NO2 是基元反应,也是简单反应。
➢ 非基元反应:由两个或两个以上基元反应组成的 化学反应,又称复合反应(complex reaction) 、 总反应(overall reaction)。
2. 当具有足够能量的分子以适当的空间取向靠近 时,要进行化学键重排,能量重新分配,形成 一个过渡状态的活化配合物。
3. 过渡状态的活化配合物是一种不稳定状态。 可形成生成物,也可回到反应物。
23
3.2 反应速率理论简介
二、过渡态理论
如: A+ B-C [A…B…C] = A-B +C 反应物 活化配合物 产物
化学反应涉及两个基本问题: ①反应的方向和限度: 反应热力学
反应是否自发,自发的方向和程度 ②反应的速率和具体步骤: 反应动力学
研究途径和速率问题
3
研究反应速率的意义
反应速率
快 ,如爆炸反应,中和反应。 慢,金属的锈蚀,食品的腐烂,
第三章 化学反应反应速率

上页 下页 目录 返回
(1) 能量因素
4 反应物之间的接触状况对反应速 率的影响
(1) 气相或溶液中进行的化学反应不考虑接触状况。 (2) 固体参加,其接触面积和形状不可忽视。红热状态的 块状铁与水蒸气之间的反应进行得非常缓慢, 而同样
温度下铁粉的反应则要快得多: 3Fe (s) + 4 H2O(g) =
Fe3O4(s) + 4 H2(g) ↑
化为生成物的基元反应!
2.5.4 影响化学反应速率的因素
Influential factors on chemical
reaction rate
1 浓度对化学反应速率的影响
一、化学反应的速率方程
在温度恒定情况下, 增加反应物的浓度可以
增大反应速率。
用来表示反应速率 与反应物浓度之间定量 关系的方程式叫速率方 程又叫速率定律。
2N2O5 →4NO2+O2
dc( N2O 5 ) v dt
lim v
t 0
1.00
v=5.4×10-4 mol · dm-3 · s-1
c(N2O5)/mol· dm-3
0.80 0.60 0.40 0.20
v=2.7×10-4 mol · dm-3 · s-1
400
800
1200 1600 时间(s)
适合此历程的速率方程是什么?
a. kc2 (O3) c. kc(O3)2c(O2) b.kc(O3)c(O) d. kc2 (O3) c-1(O2)
2 温度对化学反应速率的影响
温度升高,大多数化学反应的速度加快 原因: ● 分子运动速率加快,反
应物分子间碰撞频率增大
● 活化分子百分数增大
(1). Van’t Hoff规则:
(1) 能量因素
4 反应物之间的接触状况对反应速 率的影响
(1) 气相或溶液中进行的化学反应不考虑接触状况。 (2) 固体参加,其接触面积和形状不可忽视。红热状态的 块状铁与水蒸气之间的反应进行得非常缓慢, 而同样
温度下铁粉的反应则要快得多: 3Fe (s) + 4 H2O(g) =
Fe3O4(s) + 4 H2(g) ↑
化为生成物的基元反应!
2.5.4 影响化学反应速率的因素
Influential factors on chemical
reaction rate
1 浓度对化学反应速率的影响
一、化学反应的速率方程
在温度恒定情况下, 增加反应物的浓度可以
增大反应速率。
用来表示反应速率 与反应物浓度之间定量 关系的方程式叫速率方 程又叫速率定律。
2N2O5 →4NO2+O2
dc( N2O 5 ) v dt
lim v
t 0
1.00
v=5.4×10-4 mol · dm-3 · s-1
c(N2O5)/mol· dm-3
0.80 0.60 0.40 0.20
v=2.7×10-4 mol · dm-3 · s-1
400
800
1200 1600 时间(s)
适合此历程的速率方程是什么?
a. kc2 (O3) c. kc(O3)2c(O2) b.kc(O3)c(O) d. kc2 (O3) c-1(O2)
2 温度对化学反应速率的影响
温度升高,大多数化学反应的速度加快 原因: ● 分子运动速率加快,反
应物分子间碰撞频率增大
● 活化分子百分数增大
(1). Van’t Hoff规则:
3 化学反应速率(大学化学)

2 半衰期 当反应物A 的转化率为50%时所需的反应时间称为 半衰期,用t1/ 2表示。与浓度无关。
对于一级反应,其半衰期为:
c c k ln kt 或 lg t c0 c0 2.30
【例】某药物的分解反应为一级反应,在体温37℃时, 反应速率常数为0.46h-1。若复用该药物0.16g,问该 药在胃中停留多长时间方可分解90%? 解:根据
催化剂改变反应速率的作用就是催化作用。 加快反应速率的催化剂称为正催化剂。
而减慢反应速率的则称为负催化剂或阻化剂。 若不特别说明,所提到的催化剂均指正催化剂。
2 催化剂的特点
(1)催化剂参与反
应,改变反应的历程, 降低反应的 活化能。
(2)只能对热力学上可能发生的 反应起作用 (3)催化剂不改变反应体系的热 力学状态,不影响化学平衡与反 应的反应热。 (4)催化剂具有一定的选择性 (5)某些杂质对催化剂的性能有 很大的影响
即等于反应式相应物质分子式前的系数比
υ(N2O5L ):υ(NO2):υ(O2)=2:4:1 开始浓度/(mol· -1) 2.10 0 0 100秒浓度/(mol· -1) 1.95 L 0.30 0.075
2N2O5 → 4NO2 + O2
一般的化学反应: aA + bB = gG + dD 化学反应速率为:
1 van’t Hoff规则
范特霍夫(Van’t Hoff)由实验总结出:反应体系的温度 每升高10K, 化学反应速率将增大到原来的2~4倍。
kT 10 kT n10 kT kT
n
式中γ 为温度系数 ,一般为2~4。kT , kT+10 , kT+n · 分别 10 10)K 时的反应速率常数。 是TK , (T+10)K 和 (T+n·
第三章-化学反应速率理论

过渡态能量高于反应物,若要发生反应,必须克服这个能垒。 这个过渡态势能又低于其它所有可能的中间态的势能,可将 其看作是各种中间态中的势阱,因而可以说活化络合物具有 最低势能的原子构型。
. surface
一
势 能 )面 (
1. 势能面概念的提出:
反应体系从始态经活化状态到终态,必然伴随着势能的 起伏变化,如果将这连续变化的势能标示出来,就构成了一 个如山峦起伏的势能面。如果能计算出势能面上越过某一能 垒的频率,即能计算出反应速率。
根据碰撞理论:
k PBT1/ 2eEc / RT
d ln k dT
1 2T
Ec RT 2
公 式 的
Ea
RT 2
1 2T
Ec RT 2
Ea
Ec
1 2
RT
比
EC——临界能
较
Ea——表观活化能(实验值)
由此式可知Ea与T确实有关。
Ea与Ec的异同
Ea=E活-E反——表观活化能是2个平均能量之差,是一个
正面碰撞 活化络合物 B-C键拉长
A-B成键 AB与C分离
设:x轴表示rAB,y轴表示rBC,z轴表示V, 则可得一势能面图形:
将势能面投影到一个平面图中——势能曲线 (RTP曲线)图,每条曲线是等势能线,线上数字 越大,势能越高。RTP曲线代表由A+B-C→A-B+C 的耗能最少的途径——反应坐标。
过渡态(T)能量 是所有其它中间 态能量最低的。
由R到达P点,需跨 越的最低势垒是T点
R——反应物(A+B-C)势能——势阱; T——活化络合物势能([A…B…C])——过渡态——势垒 ——鞍点; P——产物(A-B+C)势能——势阱; D——某种中间态势能(A…B…C)
. surface
一
势 能 )面 (
1. 势能面概念的提出:
反应体系从始态经活化状态到终态,必然伴随着势能的 起伏变化,如果将这连续变化的势能标示出来,就构成了一 个如山峦起伏的势能面。如果能计算出势能面上越过某一能 垒的频率,即能计算出反应速率。
根据碰撞理论:
k PBT1/ 2eEc / RT
d ln k dT
1 2T
Ec RT 2
公 式 的
Ea
RT 2
1 2T
Ec RT 2
Ea
Ec
1 2
RT
比
EC——临界能
较
Ea——表观活化能(实验值)
由此式可知Ea与T确实有关。
Ea与Ec的异同
Ea=E活-E反——表观活化能是2个平均能量之差,是一个
正面碰撞 活化络合物 B-C键拉长
A-B成键 AB与C分离
设:x轴表示rAB,y轴表示rBC,z轴表示V, 则可得一势能面图形:
将势能面投影到一个平面图中——势能曲线 (RTP曲线)图,每条曲线是等势能线,线上数字 越大,势能越高。RTP曲线代表由A+B-C→A-B+C 的耗能最少的途径——反应坐标。
过渡态(T)能量 是所有其它中间 态能量最低的。
由R到达P点,需跨 越的最低势垒是T点
R——反应物(A+B-C)势能——势阱; T——活化络合物势能([A…B…C])——过渡态——势垒 ——鞍点; P——产物(A-B+C)势能——势阱; D——某种中间态势能(A…B…C)
第3章 化学反应速率和化学平衡
4
例如:硫酸钡的沉淀反应,基本上是向 一个方向进行,其逆反应觉察不到:
Ba2++SO42- →BaSO4↓ 再如:氯酸钾的分解反应,在分解过程中 逆反应的条件还不具备,反应物就已耗尽:
2KClO3 ===M=△n=O=2== 2KCl+3O2 象这些反应实际上只能向一个方向进行 到底的反应称为不可逆反应。
化学反应速率和化学平衡是两类不同性质的问 题。前者属于化学动力学范畴,后者属于化学热力 学范畴,因此研究他们的方法有所不同。
2
第一节 化学平衡 (p44)
概述:化学平衡普遍存在于各类化 学反应中,它涉及到在给定条件下,一 种化学反应所能达到的最大限度。本节 主要讨论化学平衡的一般规律。
3
一、可逆反应与化学平衡
K = 0.197
最大转化率:反应达平衡后,反应物转 化为产物的百分数,也叫平衡转化率或理论 转化率。
28
三、平衡浓度和转化率的计算 转化率概念: 平衡时已转化了的某反应物的
量与转化前该反应物的量之比。即:
转化率
已转化了的某反应物的量 转化前该反应物的总量
100%
在恒容条件下可表示为:
转化率
某反应物的起始浓度 平衡浓度 某反应物的起始浓度
生成物产生 ([C]、[D]会增加),因此正反应 速率会逐渐降低,逆反应速率会逐渐增加。 直至某一時間t,正反应速率=逆反应速率, 反应达「化学平衡」。如图示:
7
反应速度→
V正 V正=V逆 (平衡)
V逆
0
达到平衡时间
时间→
化学平衡建立示意图 8
以 2NO2(g)→ N2O4(g) 进行实验所得数据說明之:
4 0.1600 5 0.0000
例如:硫酸钡的沉淀反应,基本上是向 一个方向进行,其逆反应觉察不到:
Ba2++SO42- →BaSO4↓ 再如:氯酸钾的分解反应,在分解过程中 逆反应的条件还不具备,反应物就已耗尽:
2KClO3 ===M=△n=O=2== 2KCl+3O2 象这些反应实际上只能向一个方向进行 到底的反应称为不可逆反应。
化学反应速率和化学平衡是两类不同性质的问 题。前者属于化学动力学范畴,后者属于化学热力 学范畴,因此研究他们的方法有所不同。
2
第一节 化学平衡 (p44)
概述:化学平衡普遍存在于各类化 学反应中,它涉及到在给定条件下,一 种化学反应所能达到的最大限度。本节 主要讨论化学平衡的一般规律。
3
一、可逆反应与化学平衡
K = 0.197
最大转化率:反应达平衡后,反应物转 化为产物的百分数,也叫平衡转化率或理论 转化率。
28
三、平衡浓度和转化率的计算 转化率概念: 平衡时已转化了的某反应物的
量与转化前该反应物的量之比。即:
转化率
已转化了的某反应物的量 转化前该反应物的总量
100%
在恒容条件下可表示为:
转化率
某反应物的起始浓度 平衡浓度 某反应物的起始浓度
生成物产生 ([C]、[D]会增加),因此正反应 速率会逐渐降低,逆反应速率会逐渐增加。 直至某一時間t,正反应速率=逆反应速率, 反应达「化学平衡」。如图示:
7
反应速度→
V正 V正=V逆 (平衡)
V逆
0
达到平衡时间
时间→
化学平衡建立示意图 8
以 2NO2(g)→ N2O4(g) 进行实验所得数据說明之:
4 0.1600 5 0.0000
有机化学 第三章化学反应速度
n(NH 3 ) 2.0mol - 0mol 1
4.0mol - 5.0mol - 1/ 2 7.0mol - 10mol - 3/ 2 2.0mol
( NH 3 )
n(N 2 )
Hale Waihona Puke .0mol (N 2 )n( H 2 )
(H 2 )
2.0mol
结论: ★对于同一反应方程式, 的值与选择何种物质 来求算无关。 ★反应式写法不同,或者说化学反应的基本单元 定义不同,反应进度也不同。 注意:
k t1 / 2 ln cO cO 2 ln 2
t1 2 0.693 k
注意: t1/2 与起始浓度C0无关。
例:反应2N2O5 (g)→4NO2(g) + O2(g)服从
速率方程式v = k c(N2O5)。设某温度时
k = l.68×10-2s-1,如在一个5.00L的容器
中放入2.50molN2O5,在该温度下反应
v = k c x(A) c y(B);
说明:x—反应物A的级数
y—反应物B的级数
总的反应级数:n = x + y
反应级数与反应分子数的比较
反应级数 速率方程 任何反应 整数或分数 不一定相等 反应分子数 反应机理 基元反应 整数(1,2,3) 相等
提出依据 使用范围 取值 与反应物系数和
基元反应与普通反应比较: 反应方程式:aA+bB→dD+eE
例: H2(g) + I2(g) = 2HI (g) H2(g) + 2I(g) = 2HI (g) 慢反应
控制速率步骤
•
一、简单反应与复合反应
第三章化学反应速率_大学化学
衡量化学反应快慢程度的物理量称为化学反应速率
。通常将它定义为:单位体积的反应系统中,反应进度 随时间的变化率,用符号表示。对于反应
aA + fF = gG + dD (3.1)
化学反应速率的定义式为:
=
1 d V dt
式中V为系统的体积, 按(2.8)式:
L · s 。 v 的单位常用mol·
加快2~4倍。
kT 10 K 24 kT
1.阿仑尼乌斯公式
阿仑尼乌斯根据实验,提出反应速率与温度的定量关
系:
k=Ae-Ea/RT
式中的A是指前因子;Ea叫做反应的活化能,单位是 kJ· mol-1。A与Ea都是反应的特性常数,当反应的温度区间 变化不大时,其值不随温度而改变,均可由实验求得。
活化能越高,活化分子数越少,反应速率越慢。活化能的大小取
决于反应物的本性,它是决定化学反应速率的内在因素。 活化能小于40kJ· mol-1反应,其反应速率非常大,反应可瞬间完 成;活化能大于400kJ· mol-1的反应,其反应速率非常小。前面提到的 汽车尾气NO治理反应活化能611kJ· mol-1,因此看不到该反应的进行。
Ea和A 是两个非常重要的动力学参量。由于Ea在指数
位置,所以它对k 的影响很大。 微分形式:
d ln k Ea 2 dt RT Ea Ea 积分形式 ln k2 ln A ln k1 ln A RT2 RT1 k2 Ea 1 1 ln ( ) k1 R T1 T2
例如的半衰期为8×108年,223Fr(钫)的半衰期为22 分钟,14C的半衰期为5730年等。某些放射性同位素的蜕变
可以作为估算古代化石、矿石、陨石以及地球年龄的基
础。如通常用于陨石和矿石年龄的估算,14C用于确定考古 学发现物和化石的年龄。 1947~1949年间美国科学家利比确立用14C确定地球年 代的概念与方法。为此他获得了1960年诺贝尔化学奖。
。通常将它定义为:单位体积的反应系统中,反应进度 随时间的变化率,用符号表示。对于反应
aA + fF = gG + dD (3.1)
化学反应速率的定义式为:
=
1 d V dt
式中V为系统的体积, 按(2.8)式:
L · s 。 v 的单位常用mol·
加快2~4倍。
kT 10 K 24 kT
1.阿仑尼乌斯公式
阿仑尼乌斯根据实验,提出反应速率与温度的定量关
系:
k=Ae-Ea/RT
式中的A是指前因子;Ea叫做反应的活化能,单位是 kJ· mol-1。A与Ea都是反应的特性常数,当反应的温度区间 变化不大时,其值不随温度而改变,均可由实验求得。
活化能越高,活化分子数越少,反应速率越慢。活化能的大小取
决于反应物的本性,它是决定化学反应速率的内在因素。 活化能小于40kJ· mol-1反应,其反应速率非常大,反应可瞬间完 成;活化能大于400kJ· mol-1的反应,其反应速率非常小。前面提到的 汽车尾气NO治理反应活化能611kJ· mol-1,因此看不到该反应的进行。
Ea和A 是两个非常重要的动力学参量。由于Ea在指数
位置,所以它对k 的影响很大。 微分形式:
d ln k Ea 2 dt RT Ea Ea 积分形式 ln k2 ln A ln k1 ln A RT2 RT1 k2 Ea 1 1 ln ( ) k1 R T1 T2
例如的半衰期为8×108年,223Fr(钫)的半衰期为22 分钟,14C的半衰期为5730年等。某些放射性同位素的蜕变
可以作为估算古代化石、矿石、陨石以及地球年龄的基
础。如通常用于陨石和矿石年龄的估算,14C用于确定考古 学发现物和化石的年龄。 1947~1949年间美国科学家利比确立用14C确定地球年 代的概念与方法。为此他获得了1960年诺贝尔化学奖。
第三章化学反应速率和化学平衡精品PPT课件
2、结论 相同条件下,Ea越小,活化分子百分数越 大,单位时间内有效碰撞越多,反应越快。 反之,Ea越大,反应越慢。
二、过渡态理论(transition state theory)简介 以量子化学对反应过程中的能量变化的
研究为依据,认为从反应物种到生成物种之 间存在一个过渡态
快
A + B-C
慢
[A···B···C] 活化络合物
结论: 1、反应物分子必须具有足够的能量,才能 越过能峰(能垒)变为产物分子。 2、反应的活化能Ea越大,能峰越高,能越 过能峰的分子数越少反应速率越小; 反之,Ea越小,反应速率越快。
§3-3 影响化学反应速率的因素 一、基元反应(elementary reaction) 基元反应:反应物分子一步作用直接转 化成产物的反应。 简单反应:由一个基元反应构成的化学 反应称为简单反应。 例:N2O4 = 2NO2 是简单反应,也是基元反应。
定义:时间间隔Δt趋于无限小(Δt→0)时的平均速率 的极限。
lim
c limA
dc A
A
t △t→0
dt
t0
作图法求瞬时速率 从瞬时速率的定义,可以归纳出:
(1)做浓度--时间曲线图;(2)在指定时间的曲线 位置上做切线;(3)求出切线的斜率(用做图法, 量出线段长, 求出比值)。
对于反应 aA + bB = gG + hH 某时刻的瞬时 速率之间, 乃有如此的关系:
第三章 化学反应速率和化学平衡
§3-1 化学反应速率及其表示法 §3-2 反应速率理论简介 §3-3 影响化学反应速率的因素 §3-4 化学平衡 §3-5 化学平衡的移动
§3-1 化学反应速率及其表示法
热力学解决化学反应的可能性和进行的程度问 题,但自发过程是否一定进行得很快? 例:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Ea
Ec
1 2
RT
100kJ mol1
A的计算
将
Ec
Ea
1 2
RT 代入碰撞理论k的表达式:
1
k
1
1
PBT 2eEc / RT e2
P
L 1000
DA2B
8eRT
M
2
eEa / RT
根据Arrhenious公式: k AeEa / RT ,与理论比较可得:
1
A
P
L 1000
DA2B
第三章 化学反应速率理论
底讨 是论 讨一 论个 基化 元学 反反 应应 的的 速速 率率 。,
归 根 结
1. 基元反应速率理论是Arrhenius理论:该理论指出反
应速率与活化分子的数目成比例,提出活化态和活化能概念, 为后来的理论发展奠定了基础。
2. 分子碰撞理论: 1918年Lewis在研究HI的合成和分解反应
根据气体分子运动论:
1
Z
AB
L2 1000
DA2 B
8RT
M
2
AB
ZA-B——A与B的碰撞次数(总数);
DAB——分子碰撞时刻,A、B二分子中心的最小间距:
DAB=(dA+dB)/2;
µM——A、B二分子形成中间物[A…B]的折合质量:[g·分子-1];
[A]和[B]——A、B二种气体的体积摩尔浓度; T——温度(K);
宏观量、统计值。
Ec——发生反应的一个最小的临界能值,与价电子的 相互作用有关,碰撞理论无法计算出Ec理论值。当T不太 高,Ea不太大时,在数值上:Ea≈Ec
例如,当T=300 K时,
1 RT 1 8.314300 1247.1 J mol1 1.25 kJ mol1 22
若Ea=100 kJ·mol-1,则
q e Ec / RT
双分子反应速率:
r d A ZAB q
dt
L
1
L 1000
DA2B
8 RT
M
2
A B e Ec
/ RT
(mol l1
s1)
k AB
所以:
1
k
L 1000
DA2B
8RT
M
2
eEc / RT
(mol-1
l s1)
BT
1
e2 Ec
/ RT
8eRT
M
2
PB eT 1/2
因此,可知指前因子A也与T有关。 另:因为碰撞理论无法计算P的理论值,因而也无法计算出A 的理论值。
theory of absolute rate process, AFT.
根据碰撞理论:
k PBT1/ 2eEc / RT
d ln k dT
1 2T
Ec RT 2
公 式 的
Ea
RT 2
1 2T
Ec RT 2
Ea
Ec
1 2
RT
比
EC——临界能
较
Ea——表观活化能(实验值)
由此式可知Ea与T确实有关。
Ea与Ec的异同
Ea=E活-E反——表观活化能是2个平均能量之差,是一个
验 的
ln k C 1 lnT Ec
2
RT
验 证
ln k C Ec
T
RT
以 ln k ~ 1 作图应为一直线。 TT
而实验结果与此相符。该结果与
Arrhenious理论结果相符。
从
k
1/ M
2
和EC的关系来验证
2. 用相同温度下的不同反应进行研究:
k式中的P、DAB、μM、k、EC皆因不同的反应物而不同,应 选某一反应物与一系列的物质分别实验,取其k,此时每个浓度
1
B
L 1000
DA2B
8R
M
2
B与T无关,与A、B本性有关。或
1
k
DA2B
8kBT
2
eEc / RT
(molec-1
cm3
s1)
BT
1
e2 Ec / RT
k
L
k
mol1 dm 3 s 1 1000 molec1 cm3 s 1
μ的单位为g·mol-1。
上述理论推导与实验结果有许多不符的情况,原因在于
的P和DAB相似,可认为是常数。
1
k
PL 1000
DA2B
8RT
M
2
e Ec
/ RT
ln
k
1/ M
2
C Ec RT
以
k
1/ M
2
~
Ec作图可得一直线,
说明E确与分子的质量有关。
. Arrhenious
四
Ea与Ec的关系
与
根据Arrhenious公式: Ea RT 2d ln k / dT
节
➢ A和B为刚性硬球,直径分别为dA和dB;
➢ 两球相碰后即连接为中间物[A…B];
分
➢ 分解为产物C和D; ➢ 这种碰撞并不是每次都能导致反应发生,只有那些碰撞
子
后体系内部能量达到或超过某一数值时,才能发生反应
碰
生成产物。
撞
理
ZAB——总的碰撞次数; q——有效碰撞分数;
论
εC——临界能(阈能,threshold energy)。
r P ZABq L
1
k PBT e2 Ec / RT
从碰撞理论无法推出P值,只能从实验得到:
P A实验 A理论
A为Arrhennius公式的指前因子。对于简单分子的反应,P≈1。
从k与T的关系验证。
三
1. 用同一反应、不同温度为研究对象,按碰撞理论:
.
实
k PBT1/ 2eEc / RT
M
mA mB mA mB
R——气体常数;
L——Avogadreo常数。
q——有效碰撞的分数。根据Boltzmann分布定律,体系中内 能超过某一定值εC的分子占总分子个数的百分数——q应为:
q ec / kBT
kB—为Boltzmann常数。
由于: Lc Ec, LkB R
所以,q就是Boltzmann因子
时测出了Arrhenius理论中A的数值,首次给予A定量解释,并 为解释理论与实验结果的偏离而引入了几率因素P的概念。该 理论的核心是分子间发生反应的首要条件是分子间发生碰撞, 而只有碰撞时形成的少数具有能量大于或等于某一数值的活化 络合物,才能导致反应的发生,从理论上解决了分子间碰撞频 率的计算。但未能从理论上计算出活化能和几率因素,还需实 验才能求得。
二
其两个假设:
.
理
两个分子一经相碰即得一个复杂分子。实际情况是:两
论
个分子碰撞时能量虽然达到要求,但由于碰撞瞬间的取向不
的 修
合适,这样的碰分子的内部能量有一个自由度的振动能超过Ec。
项
修正方法:
认为两个分子相碰后只有部分分子产生复杂分子,其占分
子总数的百分数为P——几率因素——空间因素(P<1)
3. 绝对反应速率理论:1935年,爱伦和鲍兰义(Eyring
and Polanyi)创立并发展。将基元反应过程中势能变化转化成势 能面上的一个想象点的运动,计算出反应速率。利用统计力学 中配分函数,计算出各类反应的A的绝对值。
一. 简单理论的推导
第
一个气相中进行的双分子反应:A+B→C+D
一
假设: