USP1227-VALIDATION OF MICROBIAL RECOVERY FROM PHARMACOPEIAL ARTICLES美国药典微生物回收率验证
USP40 1226 药典的确认中英文对照

1226 VERIFICATION OF COMPENDIAL PROCEDURES药典方法的确认The intent of this general information chapter is to provide general information on the verification of compendial procedures that are being performed for the first time to yield acceptable results utilizing the personnel, equipment, and reagents available.此章节的意图是对药典方法的确认提供基本资料,使用人员,设备和试剂使第一次进行运用药典方法以产生可接受的结果。
This chapter is not intended for retroactive application to already successfully established laboratory procedures. The chapter Validation of Compendial Procedures <1225>provides general information on characteristics that should be considered for various test categories and on the documentation that should accompany analytical procedures submitted for inclusion in USP–NF.Verification consists of assessing selected analytical performance characteristics, such as those that are described in chapter <1225>to generate appropriate, relevant data rather than repeating the validation process.此章节并不旨在对已经成功建立的实验室方法进行回顾性运用。
USP微生物试验说明

美國藥典微生物試驗說明Essentials of USP Microbiological Testing研討會大綱I.基礎微生物學II.良好微生物實驗室規範III.藥典協同之改變IV.IV.滅菌滅菌滅菌與無菌保證與無菌保證V.微生物方法之確效微生物學研究微小生命形式的生物學分支研究微小生命形式的生物學分支。
典型的微小生命形式是單細胞的生命形式的微小生命形式是單細胞的生命形式,,也包括非常少也包括非常少量量的多細胞生物體 新陳代謝基本需求能量(如光、硫、一氧化碳或氨一氧化碳或氨))碳、氮、鈣、磷、硫、鎂、鉀和鈉 水(即使真菌也需要0.6 Aw 以上的游以上的游離離水)分類學生物環境多樣性嗜低溫菌嗜中溫菌嗜高溫菌生物環境多樣性嗜酸性菌嗜中性菌嗜鹼性菌依DNA在細胞中的情況可將細胞分為兩大類:真核細胞Eukaryotic cells原核細胞Prokaryotic cells原核生物Prokaryotic cells大部分細菌對人體無害如: 乳酸菌但, 也有微生物會致病細菌(分2界) 古細菌 真細菌原核生物革原核生物革蘭蘭氏分類氏分類法法Hans C.J. Gram –丹麥細菌學家(1853-1938).發現發現了了細胞學上的染色和染色技術細胞學上的染色和染色技術((革蘭氏染色染色),),),用於區分細菌的分用於區分細菌的分用於區分細菌的分類類學組群學組群。
脂多糖(相對耐熱)如果進入人體,能導致發燒、白血球減少、腹瀉、休克(熱原Pyrogen)(內毒素Endotoxin)革蘭氏陽性結構革蘭氏陰性結構什麼是芽孢?在惡劣惡劣的環境條件下產生的有再生能的環境條件下產生的有再生能的環境條件下產生的有再生能力力的細胞或細胞群的細胞或細胞群。
特徵特徵::–厚壁–耐惡劣惡劣環境環境–非常緩慢的代謝速非常緩慢的代謝速度度分4界原生動物 動物植物真菌真菌-特徵皆為化學異異營菌皆為化學具強大的酵素 (腐生或寄生腐生或寄生) ) ) 具強大的酵素pH5))酸性的生活環境((pH5 比細菌比細菌更更酸性的生活環境高鹽、、高糖環境中生長高鹽黴菌和蕈是多細胞酵母是單細胞新陳代謝Metabolism 依據分解代謝是否能夠使用O2為什麼了解微生物對新陳代謝的需求很重要?為的是想培養微生物生長更為的是防止微生物生長USP微生物相關務必遵守章節<51> 防腐效力/Antimicrobial Preservatives –Effectiveness耐受力力測試<55> 生物指示劑–耐受Biological Indicators –Resistance Performance Tests微生物計數數試驗非無菌產品的微生物檢驗::微生物計<61> 非無菌產品的微生物檢驗Microbiological Examination of Nonsterile Products:Microbial Enumeration Tests<62> 特定菌的微生物檢查/ Microbiological Proceduresfor Absence of Specified Microorganisms<71> 無菌試驗/ Sterility Tests<85> 細菌內毒素試驗Bacterial Endotoxins TestUSP微生物相關章節建議遵循<1035> 滅菌生物指示劑<1072> 消毒劑與殺菌劑<1111> 非無菌產品的微生物評估<1112> 水活性應用<1116> 清淨室和其他管制環境的微生物評估<1117> 微生物實驗室規範 <1207> 無菌產品包裝–完整性評估 <1209> 滅菌–化學和物理化學指示劑,及綜合指示劑<1211> 滅菌與無菌保證<1222> 最終滅菌藥品–參數放行 <1223> 微生物方法確效<1227> 藥物的微生物回收率確效 <1231> 製藥用水<2021> 微生物計數試驗–營養和膳食補充劑<2022> 特定菌的微生物檢查–營養和膳食補充劑<2023> 非無菌營養和膳食補充劑的微生物評估EP & USP & JP 藥典方法的國際協合International Harmonization of Pharmacopoeia時限–現行的EP 只維持到2008年12月31日–USP 將維持至2009年4月影響行適用性試驗並需重新進行–各藥廠將必需提早因應,並需重新進批產品來來測試其一致性.( Suitability Test),以至少3 批產品此協合後的方法不不僅在產品( non-sterile)的稀釋液此協合後的方法及預試驗,生長試驗上有改變,在部份培養基的組成有所改變,甚至新增一些檢驗項目與培養基,在有些品管菌株及接種量量都有改變品管菌株及接種微生物限量(USP <61> & EP 2.6.12) Microbial Enumeration TestTAMC= Total aerobic microbial count (on TSA, incl. fungi)TYMC= Total combined yeast/mould count (on Sab.4%-Dextrose Agar, incl. bacteria)特定微生物USP <62> & EP 2.6.13 Tests for Specific Microorganisms目前–Escherichia coli–Salmonella–Pseudomonasaeruginosa–Staphylococcusaureus 協調後–Escherichia coli–Salmonella–Pseudomonas aeruginosa–Staphylococcus aureus–Clostridia 產芽孢梭菌–Bile-tolerant Gram-negative bacteria 耐膽鹽革耐膽鹽革蘭蘭氏陰性菌–Candida albicans白色白色念念珠菌Test for Escherichia coli:Presence/Absence (Current USP Method)Test for Escherichia coli:Presence/Absence (Harmonized Method)<1112> 水活性應用水活性是水活性是什什麼?–產品中水的蒸氣壓P 與相同溫與相同溫度度下純水中的蒸氣壓Po 的比的比率率。
USP401225药典的验证中英文对照

VALIDATION OF COMPENDIAL PROCEDURES药典方法的验证Test procedures for assessment of the quality levels of pharmaceutical articles are subject to various requirements. According to Section 501 of the Federal Food, Drug, and Cosmetic Act, assays and specifications in monographs of the United States Pharmacopeia and the National Formulary constitute legal standards. The Current Good Manufacturing Practice regulations [21 CFR 211.194(a)] require that test methods, which are used for assessing compliance of pharmaceutical articles with established specifications, must meet proper standards of accuracy and reliability. Also, according to these regulations [21 CFR 211.194(a)(2)], users of analytical methods described in USP NF are not required to validate the accuracy and reliability of these methods, but merely verify their suitability under actual conditions of use. Recognizing the legal status of USP and NF standards, it is essential, therefore, that proposals for adoption of new or revised compendial analytical procedures be supported by sufficient laboratory data to document their validity.用于评估药品质量的检验方法需要满足不同的要求。
注射用盐酸吉西他滨无菌方法验证

注射用盐酸吉西他滨无菌方法验证目的对注射用盐酸吉西他滨的无菌检查方法进行探讨,建立其无菌检查方法。
方法按中国药典2010附录ⅪH无菌检查方法进行。
结果在验证条件下,该方法能消除对微生物生长的抑制作用。
结论可以采用此法对本品进行无菌检查。
标签:注射用盐酸吉西他滨;无菌方法;验证盐酸吉西他滨(gemcitabine hydrochloride)化学名2-脱氧-2,2-盐酸二氟脱氧胞苷(β-异构体),是一种阿糖胞苷类似物,由美国礼来公司研发的核苷类抗代谢抗癌药[1]。
属于新型抗嘧啶核苷酸代谢化疗药物,属细胞周期特异性抗代谢类药物,主要作用于DNA合成期的肿瘤细胞,即S期细胞。
在一定条件下,可以阻止G1期向S期的进展;具有抗瘤谱广、作用机制独特、毒性反应低、与其他化疗药物无交叉耐药且毒性反应无叠加等特点。
在过去的临床工作中,观察到盐酸吉西他滨是疗效较好而毒副反应较少的化疗药物之一[2-3]。
无菌检查、细菌内毒素、微生物限度检查是药品安全性检查的重要项目[4],本品为冻干粉针按照中国药典2010版附录ⅠB[5]的要求,要进行细菌内毒素检查。
1 材料与方法1.1 仪器HTY-2000A全封闭集菌仪和NKF集菌培养器(杭州高德泰林有限公司),LS-B50L立式压力蒸汽灭菌锅,SP120水夹套恒温培养箱,PYX-DHS-50X65-BS 隔水式电热恒温培养箱,MJX-160B霉菌培养箱。
AKL1500净化工作台。
1.2 试剂培养基:硫乙醇酸盐流体培养基,改良马丁培养基,营养琼脂培养基,玫瑰红钠琼脂培养基,0.1%无菌蛋白胨水溶液,0.9%无菌氯化钠溶液。
实验菌株:白色念珠菌[CMCC(F)98 001],黑曲霉[CMCC(F)98 003],铜绿假单胞菌[CMCC (B)10 104],金黄色葡萄球菌[CMCC(B)26 003],枯草芽孢杆菌[CMCC(B)63 501],生孢梭菌[CMCC(B)64 941],大肠埃希菌[CMCC(B)44 102]以上菌种均为江苏省康华医药科技实业中心。
USP 1223 VALIDATION OF ALTERNATIVE MICROBIOLOGICAL METHODS

1223VALIDATION OF ALTERNATIVE MICROBIOLOGICAL METHODSINTRODUCTIONThe purpose of this chapter is to provide guidance for validating methods for use as alternatives to the official compendial microbiological methods. For microbial recovery and identification, microbiological testing laboratories sometimes use alternative test methods to those described in the general chapters for a variety of reasons, such as economics, throughput, and convenience. Validation of these methods is required. Some guidance on validation of the use of alternate methods is provided in the Tests and Assays section in the General Notices and Requirements. This section also notes that in the event of a dispute, only the result obtained by the compendial test is conclusive.Validation studies of alternate microbiological methods should take a large degree of variability into account. When conducting microbiological testing by conventional plate count, for example, one frequently encounters a range of results that is broader (%RSD 15 to 35) than ranges in commonly used chemical assays (%RSD 1 to 3). Many conventional microbiological methods are subject to sampling error, dilution error, plating error, incubation error, and operator error.Validation of Compendial Procedures 1225defines characteristics such as accuracy, precision, specificity, detection limit, quantification limit, linearity, range, ruggedness, and robustness in their application to analytical methods. These definitions are less appropriate for alternate microbiological m ethod validation as ―at least equivalent to the compendial method‖ given the comparative nature of the question (see the Tests and Assays—Procedures section in General Notices and Requirements). The critical question is whether or not the alternate method will yield results equivalent to, or better than, the results generated by the conventional method.Other industry organizations have provided guidance for the validation of alternate microbiological methods.* The suitability of a new or modified method should be demonstrated in a comparison study between the USP compendial method and the alternate method. The characteristics defined in this chapter may be used to establish this comparison.TYPES OF MICROBIOLOGICAL TESTSIt is critical to the validation effort to identify the portion of the test addressed by an alternate technology. For example, there is a variety of technologies available to detect the presence of viable cells. These techniques may have application in a variety of tests (e.g., bioburden, sterility test) but may not, in fact, replace the critical aspects of the test entirely. For example, a sterility test by membrane filtration may be performed according to the compendial procedure up to the point of combining the processed filter with therecovery media, and after that the presence of viable cells might then be demonstrated by use of some of the available technologies. Validation of this application would, therefore, require validation of the recovery system employed rather than the entire test.There are three major types of determinations specific to microbiological tests. These include tests to determine whether microorganisms are present in a sample, tests to quantify the number of microorganisms (or to enumerate a specific subpopulation of the sample), and tests designed to identify microorganisms. This chapter does not address microbial identification.Qualitative Tests for the Presence or Absence of MicroorganismsThis type of test is characterized by the use of turbidity in a liquid growth medium as evidence of the presence of viable microorganisms in the test sample. The most common example of this test is the sterility test. Other examples of this type of testing are those tests designed to evaluate the presence or absence of a particular type of microorganism in a sample (e.g., coliforms in potable water and E. coli in oral dosage forms).Quantitative Tests for MicroorganismsThe plate count method is the most common example of this class of tests used to estimate the number of viable microorganisms present in a sample. The membrane filtration and Most Probable Number (MPN) multiple-tube methods are other examples of these tests. The latter was developed as a means to estimate the number of viable microorganisms present in a sample not amenable to direct plating or membrane filtration.General ConcernsValidation of a microbiological method is the process by which it is experimentally established that the performance characteristics of the method meet the requirements for the intended application, in comparison to the traditional method. For example, it may not be necessary to fully validate the equivalence of a new quantitative method for use in the antimicrobial efficacy test by comparative studies, as the critical comparison is between the new method of enumeration and the plate count method (the current method for enumeration). As quantitative tests, by their nature, yield numerical data, they allow for the use of parametric statistical techniques. In contrast, qualitative microbial assays, e.g., the sterility test in the example above, may require analysis by nonparametric statistical methods. The validation of analytical methods for chemical assays followswell-established parameters as described in Validation of Compendial Procedures 1225. Validation of microbiological methods shares some of the same concerns, although consideration must be given to the unique nature of microbiological assays (see Table 1).Table 1. Validation Parameters by Type of Microbiological TestVALIDATION OF QUALITATIVE TESTS FOR DEMONSTRATION OF VIABLEMICROORGANISMS IN A SAMPLESpecificityThe specificity of an alternate qualitative microbiological method is its ability to detect a range of microorganisms that may be present in the test article. This concern is adequately addressed by growth promotion of the media for qualitative methods that rely upon growth to demonstrate presence or absence of microorganisms. However, for those methods that do not require growth as an indicator of microbial presence, the specificity of the assay for microbes assures that extraneous matter in the test system does not interfere with the test.Limit of DetectionThe limit of detection is the lowest number of microorganisms in a sample that can be detected under the stated experimental conditions. A microbiological limit test determines the presence or absence of microorganisms, e.g., absence of Salmonella spp. in 10 g. Due to the nature of microbiology, the limit of detection refers to the number of organisms present in the original sample before any dilution or incubation steps; it does not refer to the number of organisms present at the point of assay.One method to demonstrate the limit of detection for a quantitative assay would be to evaluate the two methods (alternative and compendial) by inoculation with a low number of challenge microorganisms (not more than 5 cfu per unit) followed by a measurement of recovery. The level of inoculation should be adjusted until at least 50% of the samples show growth in the compendial test. It is necessary to repeat this determination several times, as the limit of detection of an assay is determined from a number of replicates (notless than 5). The ability of the two methods to detect the presence of low numbers of microorganisms can be demonstrated using the Chi square test. A second method to demonstrate equivalence between the two quantitative methods could be through the use of the Most Probable Number technique. In this method, a 5-tube design in a ten-fold dilution series could be used for both methods. These would then be challenged with equivalent inoculums (for example, a 10–1, 10–2, and 10–3 dilution from a stock suspension of approximately 50 cfu per mL to yield target inocula of 5, 0.5, and 0.05 cfu per tube) and the MPN of the original stock determined by each method. If the 95% confidence intervals overlapped, then the methods would be considered equivalent.RuggednessThe ruggedness of a qualitative microbiological method is the degree of precision of test results obtained by analysis of the same samples under a variety of normal test conditions, such as different analysts, instruments, reagent lots, and laboratories. Ruggedness can be defined as the intrinsic resistance to the influences exerted by operational and environmental variables on the results of the microbiological method. Ruggedness is a validation parameter best suited to determination by the supplier of the test method who has easy access to multiple instruments and batches of components.RobustnessThe robustness of a qualitative microbiological method is a measure of its capacity to remain unaffected by small but deliberate variations in method parameters, and provides an indication of its reliability during normal usage. Robustness is a validation parameter best suited to determination by the supplier of the test method. As there are no agreed upon standards for current methods, acceptance criteria are problematic and must be tailored to the specific technique. It is essential, however, that an estimate of the ruggedness of the alternate procedure be developed. The measure of robustness is not necessarily a comparison between the alternate method and the traditional, but rather a necessary component of validation of the alternate method so that the user knows the operating parameters of the method.VALIDATION OF QUANTITATIVE ESTIMATION OF VIABLE MICROORGANISMS IN ASAMPLEAs colony-forming units follow a Poisson distribution, the use of statistical tools appropriate to the Poisson rather than those used to analyze normal distributions is encouraged. If the user is more comfortable using tools geared towards normally distributed data, the use of a data transformation is frequently useful. Two techniques are available and convenient for microbiological data. Raw counts can be transformed to normally distributed data either by taking the log10 unit value for that count, or by takingthe square root of count +1. The latter transformation is especially helpful if the data contain zero counts.AccuracyThe accuracy of this type of microbiological method is the closeness of the test results obtained by the alternate test method to the value obtained by the traditional method. It should be demonstrated across the operational range of the test. Accuracy is usually expressed as the percentage of recovery of microorganisms by the assay method. Accuracy in a quantitative microbiological test may be shown by preparing a suspension of microorganisms at the upper end of the range of the test, that has been serially diluted down to the lower end of the range of the test. The operational range of the alternate method should overlap that of the traditional method. For example, if the alternate method is meant to replace the traditional plate count method for viable counts, then a reasonable range might be from 100 to 106 cfu per mL. At least 5 suspensions across the range of the test should be analyzed for each challenge organism. The alternate method should provide an estimate of viable microorganisms not less than 70% of the estimate provided by the traditional method, or the new method should be shown to recover at least as many organisms as the traditional method by appropriate statistical analysis, an example being an ANOVA analysis of the log10 unit transforms of the data points. Note that the possibility exists that an alternate method may recover an apparent higher number of microorganisms if it is not dependent on the growth of the microorganisms to form colonies or develop turbidity. This is determined in the Specificity evaluation.PrecisionThe precision of a quantitative microbiological method is the degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of suspensions of laboratory microorganisms across the range of the test. The precision of a microbiological method is usually expressed as the standard deviation or relative standard deviation (coefficient of variation). However, other appropriate measures may be applied. One method to demonstrate precision uses a suspension of microorganisms at the upper end of the range of the test that has been serially diluted down to the lower end of the range of the test. At least 5 suspensions across the range of the test should be analyzed. For each suspension at least 10 replicates should be assayed in order to be able to calculate statistically significant estimates of the standard deviation or relative standard deviation (coefficient of variation). Generally, a RSD in the 15% to 35% range would be acceptable. Irrespective of the specific results, the alternate method should have a coefficient of variation that is not larger than that of the traditional method. For example, a plate count method might have the RSD ranges as shown in the following table.Table 2. Expected RSD as a Function of cfu per PlateThe specificity of a quantitative microbiological method is its ability to detect a panel of microorganisms suitable to demonstrate that the method is fit for its intended purpose. This is demonstrated using the organisms appropriate for the purpose of the alternate method. It is important to challenge the alternate technology in a manner that would encourage false positive results (specific to that alternate technology) to demonstrate the suitability of the alternate method in comparison to the traditional method. This is especially important with those alternate methods that do not require growth for microbial enumeration (for example, any that do not require enrichment or can enumerate microorganisms into the range of 1–50 cells).Limit of QuantificationThe limit of quantification is the lowest number of microorganisms that can be accurately counted. As it is not possible to obtain a reliable sample containing a known number of microorganisms, it is essential that the limit of quantification of an assay is determined from a number of replicates (n > 5) at each of at least 5 different points across the operational range of the assay. The limit of quantification should not be a number greater than that of the traditional method. Note that this may have an inherent limit due to the nature of bacterial enumeration and the Poisson distribution of bacterial counts (see Validation of Microbial Recovery from Pharmacopeial Articles 1227). Therefore, the alternate method need only demonstrate that it is at least as sensitive as the traditional method to similar lower limits.LinearityThe linearity of a quantitative microbiological test is its ability to produce results that are proportional to the concentration of microorganisms present in the sample within a given range. The linearity should be determined over the range of the test. A method to determine this would be to select at least 5 concentrations of each standard challenge microorganism and conduct at least 5 replicate readings of each concentration. An appropriate measure would be to calculate the square of the correlation coefficient, r2, from a linear regression analysis of the data generated above. While the correlation coefficient does not provide an estimate of linearity, it is a convenient and commonly applied measure to approximate the relationship. The alternate method should not have an r2 value less than 0.95.Limit of DetectionSee Limit of Detection under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.RangeThe operational range of a quantitative microbiological method is the interval between the upper and lower levels of microorganisms that have been demonstrated to be determined with precision, accuracy, and linearity.RuggednessSee Ruggedness under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.RobustnessSee Robustness under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.* PDA Technical Report No. 33. The Evaluation, Validation and Implementation of New Microbiological Testing Methods. PDA Journal of Pharmaceutical Science & Technology.54 Supplement TR#33 (3) 2000 and Official Methods Programs of AOAC International.。
微生物检测美国药典
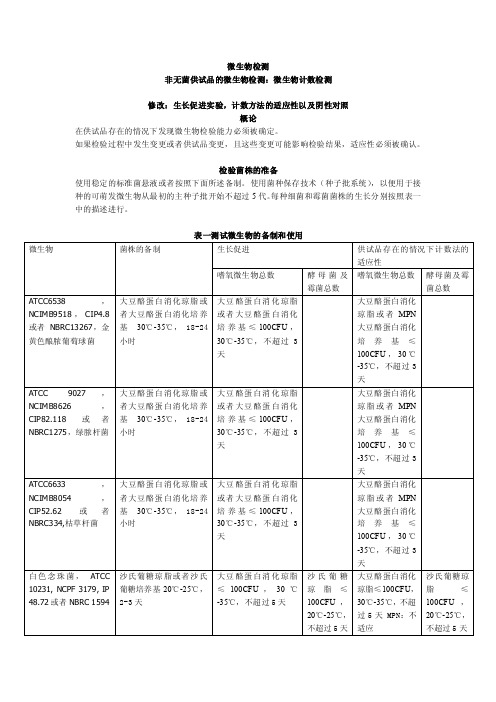
微生物检测非无菌供试品的微生物检测:微生物计数检测修改:生长促进实验,计数方法的适应性以及阴性对照概论在供试品存在的情况下发现微生物检验能力必须被确定。
如果检验过程中发生变更或者供试品变更,且这些变更可能影响检验结果,适应性必须被确认。
检验菌株的准备使用稳定的标准菌悬液或者按照下面所述备制。
使用菌种保存技术(种子批系统),以便用于接种的可萌发微生物从最初的主种子批开始不超过5代。
每种细菌和霉菌菌株的生长分别按照表一中的描述进行。
表一测试微生物的备制和使用使用pH7.0的缓冲氯化钠蛋白胨溶液或者pH7.2的磷酸盐缓冲溶液备制测试菌悬液;备制黑曲霉孢子悬浮液时,0.05%的聚山梨脂80可以被添加到缓冲液中。
测试菌悬液应在两小时内使用,或者在2-8℃的条件下24小时内使用。
也可以通过制备并稀释枯草芽胞杆菌营养细胞的新鲜悬液进行替代,制备稳定的胞子悬液,在接种测试中使用适当体积的胞子菌悬液,稳定的胞子悬液在2-8℃保存,保存期是经过验证的。
阴性对照为了确定检测条件,用选择好的稀释液代替测试备制来进行阴性对照。
必须没有微生物的生长。
当按照供试品检测中的描述进行检验时,也需要进行阴性对照。
如果阴性对照不合格需要进行调查。
培养基的生长促进检测每个批次的已经备制好的培养基以及通过脱水培养基或者描述的配料备制的每个批次的培养基。
在部分/盘大豆酪蛋白消化肉汤培养基以及大豆酪蛋白消化琼脂上接种少量(不超过100CFU)微生物,按表一中所示,每一种微生物应使用单独一部分/一盘培养基。
在沙氏葡萄糖琼脂上接种少量(不超过100CFU)微生物,按表一中所示,每一种微生物应使用单独的一盘培养基。
按照表一中所述的条件进行接种。
固体培养基的生长通过一个不大于2的系数的调节必须不能与标准接种体计算得到的数值有区别。
新鲜备制的接种体微生物的生长应与上一批通过检测的培养基的生长情形相同。
如果肉眼能清晰看到的微生物的生长与上一批通过检测的培养基的生长情形相同的话,液体培养基是适合的。
美国药典2011-5-3

171 VITAMIN B12 ACTIVITY ASSAY
Chemical Tests and Assays
Identification Tests
18BASES 191 IDENTIFICATION TESTS-GENERAL 194 IDENTIFICATION-TETRACYCLINES 197 SPECTROPHOTOMETRIC IDENTIFICATION TESTS 201 THIN-LAYER CHROMATOGRAPHIC IDENTIFICATION TEST
651 CONGEALING TEMPERATURE 661 CONTAINERS 671 CONTAINERS-PERMEATION 691 COTTON 695 CRYSTALLINITY 698 DELIVERABLE VOLUME 699 DENSITY OF SOLIDS 701 DISINTEGRATION 711 DISSOLUTION 721 DISTILLING RANGE 724 DRUG RELEASE
)尚未收入的新药和新制剂。 美国药典最新版为USP34-NF29,2010 年12 月出版,2011年5月1日生效。
Introduction
Front Matter
General Notices General Chapters Dietary Supplements Chapters Reagents
Chart 9 Chart 10 Chart 11 Chart 12 Chart 13
General Tests and Assays
Physical Tests and Determinations
601 AEROSOLS, NASAL SPRAYS, METEREDDOSE INHALERS,AND DRY POWDER INHALERS 616 BULK DENSITY AND TAPPED DENSITY 621 CHROMATOGRAPHY 631 COLOR AND ACHROMICITY 641 COMPLETENESS OF SOLUTION 643 TOTAL ORGANIC CARBON 645 WATER CONDUCTIVITY
化妆品微生物标准检验方

如有均质器,则采用无菌样品袋,将上述水溶性膏、霜、粉剂等,称10g样品加入90mL灭菌生理盐水,均质1min~2min;疏水性膏、霜及眉笔、口红等,称10g样品,加10mL灭菌液体石蜡,10mL吐温80,70mL灭菌生理盐水,均质3min~5min。
菌落总数
1范围
本标准规定了化妆品中菌落总数的检验方法。
本标准适用于化妆品菌落总数的测定。
2术语和定义
下列术语和定义适用于本标准:
菌落总数aerobic bacterial count化妆品检样经过处理,在一定条件下培养后(如培养基成分、培养温度、培养时间、pH值、需氧性质等),1g(1mL)检样中所含菌落的总数。所得结果只包括一群本方法规定的
条件下生长的嗜中温的需氧性和兼性厌氧菌落总数。测定菌落总数便于判明样品被细菌污染
10-1 10-2 10-3两稀释度
菌数之比
菌落总数
(CFU/mL或CFU/g)
报告方式
(CFU/mL或CFU/g)
1 1365 164 20─16400 16000或1.6×104 2 2760 295 46 1.6 38000 38000或3.8×104 3 2890 271 60 2.2 27100 27000或2.7×104 4不可计4650 513─513000 510000或5.1×105 5 27 11 5─270 270或2.7×102 6不可计305 12─30500 31000或3.1×104 7 0 0 0─<1×10
3仪器
3.1恒温水浴箱或隔水式恒温箱:44℃±0.5℃。
3.2温度计。
3.3显微镜。
3.4载玻片。
3.5接种环。
3.6电磁炉。
3.7三角瓶,250mL。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1227VALIDATION OF MICROBIAL RECOVERY FROM PHARMACOPEIAL ARTICLESThis chapter provides guidelines for the validation of methods for the estimation of the number of viable microorganisms, for the detection of indicators or objectionable microorganisms, for the validation of microbiological methods used in antimicrobial effectiveness testing, and for the sterility testing of Pharmacopeial articles. It is generally understood that if a product possesses antimicrobial properties because of the presence of a specific preservative or because of its formulation, this antimicrobial property must be neutralized to recover viable microorganisms. This neutralization may be achieved by the use of a specific neutralizer, by dilution, by a combination of washing and dilution, or by any combination of these methods.The tests under Antimicrobial Effectiveness Testing 51, Sterility Tests 71, and Microbial Limit Tests 61require the validation of recovery methods. To ensure that the results of the tests are credible, neutralization of antimicrobial properties of the test solution is required before estimating the number of viable microorganisms.INFLUENTIAL FACTORSSeveral factors affect the measurement of a test solution's antimicrobial activity, and these must be considered in the validation design. They include the nature of the microorganisms used as challenge organisms, the preparation of the inoculum of challenge organisms, the specific conditions of the test, and the conditions of recovery. These factors also affect the validation of recovery methods for aqueous or nonaqueous products, irrespective of their antimicrobial properties; thus, all test methods should be validated with these factors in mind.The nature of the challenge microorganism exerts a strong effect upon the response to the antimicrobial agent, and so upon the neutralization required for recovery. Represented among these organisms in compendial tests are Gram-positive bacteria, Gram-negative bacteria, yeasts, and molds. Each organism to be used in the test must be included in the validation.The preparation of the inoculum of challenge microorganisms also affects the testing of products having antimicrobial properties. The growth and preparation of the challenge organism determines the physiological state of the cell. This state has a direct influence on the results of any test of antimicrobial efficacy. Microbial tests do not use individual cells; rather, populations of cells are harvested for study. The data generated from these studies are less variable if the cell populations are homogeneous. Liquid cultures or confluent growths on solid medium are best suited for reproducible culture preparation. The conditions of organism preparation and storage must be standardized for the neutralizer evaluation and should reflect the conditions of the antimicrobial assay.The specific conditions of the test, including buffers used, water, light conditions, and temperature, must be reproduced in the validation study. All test conditions also should be standardized and performed in the validation study exactly as performed in the test.The conditions of microbial recovery are among the most crucial in accurately estimating the number of microorganisms present in a test solution. The first consideration is the recovery medium used to support the growth of survivors. This concern is discussed in detail below. The second consideration is the incubation conditions. Optimal conditions for growth must be present to ensure complete growth and reproducible results.METHODS OF NEUTRALIZING ANTIMICROBIAL PROPERTIESThree common methods are used to neutralize antimicrobial properties of a product: (1) chemical inhibition, (2) dilution, and (3) filtration and washing.Chemical InhibitionTable 1 shows known neutralizers for a variety of chemical antimicrobial agents and the reported toxicity of some chemical neutralizers to specific microorganisms. However, despite potential toxicity, the convenience and quick action of chemical inhibitors encourage their use. Chemical inhibition of bactericides is the preferred method for the antimicrobial efficacy test. The potential of chemical inhibitors should be considered in the membrane filtration and the direct transfer sterility tests. Antibiotics may not be susceptible to neutralization by chemical means, but rather by enzymatic treatment (e.g., penicillinase). These enzymes may be used where required.Table 1. Some Common Neutralizers for Chemical BiocidesDilutionA second approach to neutralizing antimicrobial properties of a product is by dilution, because the concentration of a chemical bactericide exerts a large effect on its potency. The relationship between concentration and antimicrobial effect differs among bactericidal agents but is constant for a particular antimicrobial agent. This relationship is exponential in nature, with the general formula:C t = k,in which C is the concentration; t is the time required to kill a standard inoculum; k is a constant; and the concentration exponent, , is the slope of the plot of log t versus log C. Antimicrobial agents with high values are rapidly neutralized by dilution, whereas those with low values are not good candidates for neutralization by dilution.Membrane FiltrationAn approach that is often used, especially in sterility testing, is neutralization by membrane filtration. This approach relies upon the physical retention of the microorganism on the membrane filter, with the antimicrobial agent passing through the filter into the filtrate. The filter is then incubated for recovery of viable microorganisms. However, filtration alone may not remove sufficient quantities of the bactericidal agent toallow growth of surviving microorganisms. Adherence of residual antimicrobial agents to the filter membrane may cause growth inhibition. Filtration through a low-binding filter material, such as polyvinylidene difluoride, helps to minimize this growth inhibition. Additionally, the preservative may be diluted or flushed from the filter by rinsing with a benign fluid, such as diluting Fluid A(see Diluting and Rinsing Fluids for Membrane Filtration under Sterility Tests 71for diluting fluid compositions). Chemical neutralizers in the rinsing fluid can ensure that any antimicrobial residue on the membrane does not interfere with the recovery of viable microorganisms.VALIDATION OF NEUTRALIZATION METHODS—RECOVERY COMPARISONSA validated method for neutralizing the antimicrobial properties of a product must meet two criteria: neutralizer efficacy and neutralizer toxicity. The validation study documents that the neutralization method employed is effective in inhibiting the antimicrobial properties of the product (neutralizer efficacy) without impairing the recovery of viable microorganisms (neutralizer toxicity). Validation protocols may meet these two criteria by comparing recovery results for treatment groups.The first is the test group, in which the product is subjected to the neutralization method, then a low level of challenge microorganism [less than 100 colony-forming units (cfu)] is inoculated for recovery. The second is the peptone control group, in which the neutralization method is used with peptone, or diluting Fluid A (see Sterility Tests 71), as the test solution. The third is the viability group, in which the actual inoculum is used without exposure to the neutralization scheme. Similar recovery between the test group and the peptone group demonstrates adequate neutralizer efficacy; similar recovery between the peptone group and the viability group demostrates adequate neutralizer toxicity.In principle, the protocol must show that recovery of a low inoculum (less than 100 cfu) is not inhibited by the test sample and the neutralization method. Validation protocols may meet these two criteria by comparing recovery among three distinct test groups: (1) neutralized product with inoculum, (2) challenge inoculum control in buffered solution, and(3) inoculum in the absence of product or neutralizer. This can be established by directly comparing the result in the treated solution (1) to the inoculum (3) above. If the growth on the treated solution is not comparable to the growth on the inoculum group, it should be determined whether the neutralization method itself is toxic to the microorganisms. Recovery on Agar MediumIn the tests under Antimicrobial Effectiveness Testing 51and Microbial Limit Tests 61, the number of viable challenge microorganisms in the product is estimated at various time intervals by calculating the concentration of cfu per mL by the plate count method. A design for validating neutralization would incorporate the treatment groups as described under Validation of Neutralization Methods—Recovery Comparisons. At least three independent replicates of the experiment should be performed, and each should demonstrate that the average number of cfu recovered from the challenge product is not less than 70% of that recovered from the inoculum control.If a greater number of replicates is required in the validation study, the comparisons may be evaluated by transforming the numbers of cfu to their logarithmic values and analyzing the data statistically by the Student t test (pairwise comparisons) or by analysis of variance (ANOVA) (for comparing all groups). If ANOVA is used, and significant differences among the populations are determined, a test such as Dunnett's test may be used, with the peptone group used as the control group.Recovery by Membrane FiltrationThis validation follows the procedure described for Validation Test under Sterility Tests 71, with the exception of plating on solid medium to quantitate recovery. Three 100-mL rinses are assumed, but the volume and number of rinses are subject to validation. Each validation run should be performed independently at least three times.In the test solution group, the product is filtered through the membrane filter, followed by two 100-mL portions of diluting-neutralizing fluid. After the second rinse has been filtered, a final 100-mL portion containing less than 100 cfu of the specific challengemicroorganism is passed through the filter. This filter is then placed on the appropriate agar recovery medium and incubated for recovery.The inoculum is directly plated onto the solid medium. It is possible that filtration will lead to reduced recovery of the challenge microorganism, either through inherent toxicity of the membrane or by adherence of the microrganism to the filtration vessel walls. A control group can be used to evaluate this component of membrane filtration validation. Diluting Fluid A is used as the dilution medium without exposing the filter to the product. After addition of the low-level inoculum to the final rinse, the filter is plated as above. Technique-specific loss of microorganisms can be estimated by comparing the recovery in the diluting Fluid A group to the inoculum count.It is assumed in this discussion that the test sample can be filtered. If it is necessary to solubilize the test sample, the effects of the solubilization method on viable microorganisms must be determined. This situation can occur when testing ointments, suspensions, or other articles.The method can be considered validated if the recovery rate in the three independent replicates is similar for the test solution and the diluting Fluid A control.Recovery in Liquid MediumIt is assumed in Direct Inoculation of the Culture Medium in the section Test for Sterility of the Product to be Examined under Sterility Tests 71that the recovery medium will allow for growth of all surviving microorganisms. The broth in that test must serve both to neutralize any antimicrobial properties of the test solution and to support the growth of the microorganisms. The treatment groups described under Validation of Neutralization Methods—Recovery Comparisons above can be used for validation of the recovery method, with the proportions of product and recovery medium varied to achieve adequate neutralization. The method can be considered validated if all groups show copious growth within 7 days for all microorganisms.RECOVERY OF INJURED MICROORGANISMSThe validation studies described above use challenge microorganisms that have never been exposed to antimicrobial agents, and thus are not identical to organisms seen in antimicrobial effectiveness testing or when a sterility test is performed on a preserved product. If the use of alternative media is desired, the recovery of injured microorganisms should be addressed in the validation study. This may be done by directly comparing the recovery of each challenge microorganism on the preferred medium and on the alternative medium, after exposure to the product. This exposure should include at least two time periods showing survival of less than 100 cfu per mL, unless the rate of kill of the antimicrobial agent is such that no recovery is possible even if the microorganism is plated within minutes of exposure. This comparison should be performed at least three times. The alternative medium is validated if the recovery seen on that medium is no less than that seen on the preferred medium, within an error of 0.5 log units.ESTIMATING THE NUMBER OF COLONY-FORMING UNITSThe accuracy of any estimate of viable cfu is affected by the number plated. As the number of viable cells plated increases, crowding effects decrease the accuracy of the count, reducing the estimate. As the number decreases, random error plays an increasing role in the estimate.The accepted range for countable colonies on a standard agar plate is between 25 and 250 for most bacteria and Candida albicans.This range was established in the food industry for counting coliform bacteria in milk. This range is acceptable for compendial organisms, except for fungi. It is not optimal for counting all environmental isolates. The recommended counting range for Aspergillus niger is between 8 and 80 cfu per plate. The use of membrane filtration to recover challenge microorganisms, or the use of environmental isolates as challenge microorganisms in antimicrobial effectiveness testing, requires validation of the countable range. This validation may be performed by statistical comparison of estimated cfu from successive pairs in a dilution series. Prepare a suspension so that plating will provide approximately 1000 cfu per plate, and then dilute twofold to a theoretical concentration of approximately 1 cfu per plate. Plate all dilutions in the series in duplicate, and incubate for recovery under the conditions of the AntimicrobialEffectiveness Testing 51. Compare the estimates of cfu per mL from paired tubes in the dilution series by the formula:in which L cfu is the number of colonies on the plate with the lower count (greater dilution), and H cfu is the number of colonies on the plate with the higher count (lesser dilution). The estimates of the cfu per mL provided by L cfu and H cfu should agree within the limits of the formula with a critical value of 1.96. The upper limit of plate counts is then defined as the number (H cfu) that reproducibly passes this test. This study should be independently repeated a sufficient number of times to establish an upper limit of cfu for the particular plating conditions.There is a lower limit at which the ability of the antimicrobial effectiveness test to recover microorganisms becomes untenable. If the first plating is performed with 1 mL of a 10–1 dilution, cfu in the range of 1 to 10 per mL would not be seen. On this dilution plating, only the lower number of cfu may be reduced to 3, allowing as few survivors as 30 cfu per mL to be reported.Lower counting thresholds for the greatest dilution plating in series must be justified. Numbers of colonies on a plate follow the Poisson distribution, so the variance of the mean value equals the mean value of counts. Therefore, as the mean number of cfu per plate becomes lower, the percentage error of the estimate increases (see Table 2). Three cfu per plate at the 10–1 dilution provide an estimate of 30 cfu per mL, with an error of 58% of the estimate.Table 2. Error as a Percentage of Mean for Plate Counts。