Thermal decomposition (pyrolysis) of urea in an open reaction vessel
羧基封端的聚苯硫醚的合成及热学性能研究

2021 年2 月Journal of Chemical Engineering of Chinese Universities Feb. 2021文章编号:1003-9015(2021)01-0051-06羧基封端的聚苯硫醚的合成及热学性能研究尹红1, 柳琳1, 袁慎峰1, 陈志荣1, 李沃源2, 邓杭军2(1. 浙江省化工高效制造技术重点实验室, 浙江大学化学工程与生物工程学院, 浙江杭州310027;2. 浙江新和成特种材料有限公司, 浙江上虞312369)摘要:为了研究羧基封端聚苯硫醚(PPS)结构对其反应活性和热学性能的影响,合成3种羧基封端PPS并通过红外(FTIR)、端基含量测定、裂解气质联用(py-GC/MS)、高温凝胶渗透色谱(HTGPC)、热重(TGA)、差示扫描量热(DSC)等方法,获得改性样品的端基转化率、裂解机理、反应活性和熔点等参数。
结果表明,羧基对PPS原有巯基端基的置换率约为50%。
裂解过程中改性样品发生更多的环化、重排和次级反应,生成含氧标志性裂解产物。
改性PPS的反应活性提高、热稳定性略有下降,起始分解温度和最高分解速率温度均有所降低,熔点升高,结晶度增大。
关键词:聚苯硫醚;端基改性;羧基;热性能中图分类号:TQ 324.8 文献标志码:A DOI:10.3969/j.issn.1003-9015.2021.01.006Synthesis and thermal properties of carboxyl-terminated polyphenylene sulfideYIN Hong1, LIU Lin1, YUAN Shen-feng1, CHEN Zhi-rong1, LI Wo-yuan2, DENG Hang-jun2(1. Zhejiang Provincial Key Laboratory of Advanced Chemical Engineering Manufacture Technology,College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China;2. Zhejiang NHU Special Materials Co. Ltd., Shangyu 312369, China)Abstract:Three kinds of carboxyl-terminated polyphenylene sulfide (PPS) were synthesized for studying the influence of their structure on the reactivity and thermal property. The end group conversion, pyrolysis mechanism, reactivity, and melting point were obtained through FTIR, end group content determination, pyrolysis-GC/MS, HTGPC, TGA and DSC, respectively. The results show that the carboxyl group replaced the original sulfhydryl group effectively with the substitution rate of about 50%. More cyclization, rearrangement and secondary reactions occured during the pyrolysis of carboxyl terminated products, which resulted in the formation of characteristic pyrolysis products containing oxygen. The thermal stability of modified PPS decreased slightly while the reactivity increased. Both the initial decomposition temperature and the maximum decomposition rate temperature decreased, but the melting point and crystallinity increased.Key words:polyphenylene sulfide; end-group modification; carboxyl; thermal properties1前 言聚苯硫醚(polyphenylene sulfide,PPS)作为热塑性结晶聚合物[1],具有优异的热稳定性、耐化学腐蚀性和机械性能[2],PPS注塑和纤维产品可以用于电子封装材料和特种除尘过滤布领域[3],PPS还可作为新兴复合材料的基质,用于锂电池[4]、轻量化汽车、航空材料[5-7]和污水中有机物处理[8]等领域。
油酸甲酯和硬脂酸甲酯热解特性及动力学研究

油酸甲酯和硬脂酸甲酯热解特性及动力学研究王学春;方建华;李亮;王九;吴江【摘要】通过热重分析仪对油酸甲酯和硬脂酸甲酯的热解特性进行了研究,发现油酸甲酯相变硬脂酸甲酯具有较差的热安定性。
在升温速率分别为10、15、20、30℃/min 条件下,利用 TG-DTG 曲线分析了它们的基本热解特性。
结果表明,随着升温速率的增加,它们的起始热分解温度、最大失重速率、最大失重速率峰值温度以及其他热分解参数均呈增大趋势。
同时,用多元线性回归法求得相应的反应级数、活化能和指前因子,发现油酸甲酯和硬脂酸甲酯的热解反应机理函数不同,且反应活化能和指前因子之间表现出较好的动力学补偿效应。
%The differences in pyrolysis process of methyl stearate and methyl oleate were investigated under nitrogen atmosphere in a thermogravimetric analyzer at heating rate of 10 ℃/min from room temperature to 600 ℃. The results show that the methyl oleate has relatively lower stability of thermal decomposition compared with methyl stearate. At the same time, the pyrolysis and kinetic characteristics of methyl oleate and methyl stearate were further studied at heating rates of 10, 15, 20 and 30 ℃/min, respectively. The results indicate that the initial decomposition temperature, peak temperature of DTG curve, the maximum weight loss rate and other pyrolysis parameters all increase. In addition, the corresponding activation energy, pre-exponential factor and reaction order were calculated by Multiple-Linear Regression method. It’s found that the pyrolysis reaction mechanism functions of two kinds of fatty acid methyl ester are different,and the pyrolysisactivation energy and pre-exponential factor present a good kinetic compensation effect.【期刊名称】《当代化工》【年(卷),期】2015(000)001【总页数】5页(P4-8)【关键词】油酸甲酯;硬脂酸甲酯;热重分析;热解;动力学【作者】王学春;方建华;李亮;王九;吴江【作者单位】后勤工程学院军事油料应用与管理工程系,重庆 401311;后勤工程学院军事油料应用与管理工程系,重庆 401311;后勤工程学院军事油料应用与管理工程系,重庆 401311;后勤工程学院军事油料应用与管理工程系,重庆401311;后勤工程学院军事油料应用与管理工程系,重庆 401311【正文语种】中文【中图分类】TQ050.4+6随着能源消耗量日益增加以及矿物燃料的日趋枯竭,迫切要求新型石油替代能源的快速发展,其中,生物柴油作为一种新型能源,具有清洁、储量大、可再生和环境友好,具有与石化柴油相近的燃料特性,已受到世界各国的普遍关注[1-5]。
氯化铽气溶胶的高温热解机理

氯化铽气溶胶的高温热解机理薛首峰;吴文远;边雪【摘要】以TbCl3气溶胶为前驱体,在微纳米尺度研究了TbCl3在空气氛围中的高温热解机理,用XRD和FESEM分别表征了热解产物的物相组成和微观形貌.基于实验结果和热力学分析,讨论了TbCl3气溶胶热解生成TbO2的化学反应机理,以及促进TbCl3热解转变为氧化铽的动力学原因.研究结果表明:TbCl3气溶胶在空气氛围中热解可生成具有化学计量比形式的氧化物TbO2,当热解温度高于800℃时,热解产物为非化学计量比形式的Tb7 O12,没有发现TbOCl物相;在气溶胶热解产物中可以观察到更多热解中间产物,从而对TbCl3的性质和热解过程的认识更充分.【期刊名称】《东北大学学报(自然科学版)》【年(卷),期】2018(039)010【总页数】5页(P1423-1427)【关键词】TbCl3;气溶胶;热解;机理;Tb(OH)2Cl;TbO2【作者】薛首峰;吴文远;边雪【作者单位】东北大学冶金学院, 辽宁沈阳 110819;东北大学冶金学院, 辽宁沈阳 110819;东北大学冶金学院, 辽宁沈阳 110819【正文语种】中文【中图分类】TF845.6稀土元素由于其特殊的物理化学性质引起了广泛关注[1],研究表明稀土元素铽在CO催化氧化、发光材料和水解制氢等多个领域表现出优异性能[2-4],因此对于铽的研究有着重要的理论及现实意义.无水氯化铽是熔盐电解制备金属铽的主要原料[5],由于具有较强的吸水性,氯化铽通常以水合物的形式存在[6],因此国内外学者曾使用不同方法对水合氯化铽的脱水过程进行了相关研究[7-9],然而关于氯化铽在空气氛围中的高温热解机理尚未见报道.气溶胶热解法被广泛用于制备粉体材料[10-11],其特点是在超声波的作用下将溶液雾化为粒径约小于5 μm的气溶胶,由载气带入高温炉发生热解反应,制备出分散性好的、形貌规则的超细粉体[12].为了研究氯化铽在微纳米尺度范围的热解行为,本文选择以氯化铽气溶胶为前驱体,在600~1 100 ℃的范围内研究了氯化铽的高温热解反应过程,结合热力学理论分析初步探讨了氯化铽的高温热解机理.1 实验材料和方法氧化铽(Tb4O7)的质量分数为99.99%,江苏国盛稀土有限公司生产;双氧水,分析纯,成都市科龙化工试剂厂生产;乙醇,分析纯,国家药品化学试剂有限责任公司生产.实验中所用氯化铽(TbCl3·6H2O)通过氧化铽与盐酸反应后重结晶制得.准确称取一定量的氯化铽,将其溶解到200 mL的蒸馏水中得到的溶液浓度约为0.05 mol/L.将配制好的溶液移入雾化器,将雾化器与温度恒定的高温管式炉相连,调整载气流速约为10 L/min,打开雾化器即可进行实验.待实验结束后在管式炉的末端收集热解产物.高温炉温度由程序控温仪精确控制.用XRD(PANalytical X’ Pert Powder)分析产物物相,用场发射扫描电镜(FESEM, Hitachi S-4800)表征产物形貌.2 结果与讨论2.1 热解产物的物相组成图1为TbCl3气溶胶在600~1 100 ℃热解产物的XRD结果.600 ℃时TbCl3气溶胶所得热解产物的衍射花样(图1a)中2θ角为29.6°,34.3°,49.4°和58.6°的衍射峰与PDF卡片75-0209相一致,说明有TbO2生成.但2θ角为14.2°,16.9°,24.2°和37.6°的衍射峰在ICSD晶体学数据库没有找到与其相匹配的含铽无机化合物的PDF卡片,说明在TbCl3气溶胶热解时有其他中间物相TbX形成.700 ℃时热解产物的主要物相为TbO2,次要物相为TbX(图1b). 800 ℃时热解产物中出现了两种不同形式的氧化铽即TbO2和Tb7O12(图1c);热解温度为900~1 100 ℃时,热解产物为Tb7O12和微量的TbX物相(图1d).图1 TbCl3气溶胶分别在不同温度热解产物的XRDFig.1 XRD patterns of pyrolysis products of TbCl3 aerosol at different temperatures(a)—600 ℃;(b)—700 ℃; (c)—800 ℃; (d)—900~1 100 ℃.通常认为TbCl3热解时首先发生水解反应,生成TbOCl,有趣的是TbCl3气溶胶的热解产物中并没有发现TbOCl物相.随着热解温度的升高,热解产物中氧化铽的含量逐渐增多,与此同时,热解过程中形成的中间物相TbX的衍射峰强度逐渐减小,说明中间物相TbX随着热解温度的升高逐渐转变为氧化铽.图2为使用FESEM配置的EDXS对600 ℃时TbCl3气溶胶热解产物的元素分析结果,在产物中共检测到了C,Tb,O和Cl四种元素.其中C是由于导电胶所致.Cl与Tb的原子比约为1.13,接近于1;O与Tb的原子比约为3.95,接近于4.XRD分析表明热解产物中没有TbCl3和TbOCl,热解产物中的O含量较高说明热解中间产物TbX中含有O元素.在600 ~1 100 ℃范围内TbCl3气溶胶的热解产物中始终存在中间产物TbX.由于镧系收缩效应,元素性质非常相似,其中Pr元素与Tb元素的物理化学性质极其相似,形成的稳定氧化物均以非化学计量比形式存在.PrCl3气溶胶热解机理研究表明[13]:在600 ℃时热解产物主要是Pr(OH)2Cl和PrOCl,随着温度的升高,热解产物中PrOCl和PrO2的含量逐渐增加,温度高于900 ℃时PrOCl 转化为PrO2的速度增大,热解产物中PrO2的含量明显增多,但是PrCl3气溶胶在1 100 ℃ 的热解产物中只有PrO2和Pr(OH)2Cl两种物相存在,而没有发现PrOCl相.TbCl3气溶胶热解呈现出相似的规律,即中间产物TbX在600~1 100 ℃范围内始终存在,且TbCl3气溶胶在1 100 ℃的热解产物中也只含有Tb7O12和中间产物TbX;由于镧系收缩效应的存在,镧系原子半径大小相差不大,因此镧系元素可形成晶体结构相同的化合物.据此推断,TbCl3气溶胶热解时形成的中间产物可能是Tb(OH)2Cl.图2 TbCl3气溶胶在600 ℃热解产物的EDXSFig.2 EDXS of pyrolysis product of TbCl3 aerosolat 600 ℃2.2 TbCl3气溶胶的热解机理稀土氯化物热解过程中易发生水解反应,生成氯氧化物,其在更高的温度将被氧化为稀土氧化物.然而TbCl3气溶胶热解产物中没有发现TbOCl物相.因此,有必要对TbCl3气溶胶的高温热解机理进行探讨.根据化学反应的物质守恒定理及TbCl3气溶胶在600~1 100 ℃时热解产物的物相组成,TbCl3气溶胶可能发生的热解反应如式(1)~式(11)所述.图3为热解反应式(3) ~式(11)的摩尔反应吉布斯自由能变,由于缺乏Tb(OH)2Cl的热力学数据,暂无法得到热解反应式(1)和式(2)的摩尔反应吉布斯自由能变.600 ℃时热解产物为Tb(OH)2Cl,因而Tb(OH)2Cl需通过一步脱水反应热解生成TbOCl.由此可知,TbCl3的热解首先发生水解反应,生成Tb(OH)2Cl,如式(1)所示,Tb(OH)2Cl 热解脱水生成TbOCl,如式(2)所示.图3a中式(3)和式(4)的ΔG大于-10 kJ/mol,并且随着温度的升高而增大,说明在高温条件下,化学反应式(3)和式(4)不能向右自发进行,即TbCl3高温热解时不能通过一步反应直接转变为TbOCl.图3b为热解反应式(5)~式(11)在600~1 200 ℃范围内的ΔG,其中反应式(5)~式(7) 为 TbCl3 热解时生成TbO2时可能的反应.由图3b可知,ΔG(5)随着温度的升高逐渐增大:当反应过程中有H2O参与反应,热解温度高于700 ℃时ΔG(6)<-20 kJ/mol,并且随着温度的升高而减小;ΔG(7)随着温度的升高也逐渐减小,而且ΔG(7)>ΔG(6),由此可见H2O对TbCl3热解转化为TbO2有着重要作用:H2O的存在使得TbCl3在较低的温度热解时,首先发生水解反应,生成中间物相Tb(OH)2Cl,降低TbCl3转变化TbOCl的活化能,使TbCl3在高温热解时更易被氧化为TbO2;另一方面,H2O的存在促进了TbCl3的热解.反应式(8) ~式 (11)为热解温度高于900 ℃时,TbCl3热解生成Tb7O12可能发生的反应.由图3b可知热解反应式(8) ~式 (11)的ΔG均小于零,ΔG(8)与ΔG(10)在600~1 200 ℃范围内随着温度的升高变化不大,但是ΔG(9)与ΔG(11)随着温度的升高快速减小,说明当温度高于900 ℃时,TbCl3热解按照式(9)与式(11)的方式向右进行的趋势显著增强,式(11)说明H2O的存在有利于TbCl3热解生成Tb7O12.图3 热解反应式(3)~式(11)的摩尔反应自由能变Fig.3 Gibbs free energy per molar varies with thermal decomposition reaction of formula (3)~(11)TbCl3+2H2O=Tb(OH)2Cl+2HCl ,(1)Tb(OH)2Cl=TbOCl+H2O ,(2)TbCl3+H2O=TbOCl+2HCl ,(3)2TbCl3+O2=2TbOCl+2Cl2 ,(4)2TbCl3+2O2=2TbO2+3Cl2 ,(5)4TbCl3+O2+6H2O=4TbO2+12HCl ,(6)2TbOCl+O2=2TbO2+Cl2 ,(7)7TbO2=Tb7O12+O2 ,(8)14TbOCl+5O2=2Tb7O12+7Cl2 ,(9)14TbCl3+12O2=2Tb7O12+21Cl2 ,(10)28TbCl3+3O2+42H2O=4Tb7O12+84HCl .(11)综上所述,TbCl3气溶胶热解时首先发生水解反应,生成Tb(OH)2Cl,如式(1)所示,温度升高Tb(OH)2Cl脱水生成TbOCl,如式(2)所示;在600 ~800 ℃时,TbOCl氧化生成TbO2;热解温度高于900 ℃时,由Tb(OH)2Cl脱水形成的TbOCl氧化生成Tb7O12,如式(9)所示.2.3 热解产物的微观形貌表征以气溶胶为前驱体时,TbCl3热解生成具有化学计量比形式的氧化物TbO2,这可能与气溶胶本身及其热解过程的特点相关.由图4可知TbCl3气溶胶热解产物为规则的球形颗粒.图4a内嵌的高倍扫描电镜图片显示球形颗粒大小约为50 nm,气溶胶热解生成的球形颗粒直径约为0.1~1.3 μm,d50约为0.5 μm(图5).图4 TbCl3气溶胶在不同温度热解时产物的FESEM图Fig.4 FESEM imagines of pyrolysis products of TbCl3aerosol at different temperatures(a)—600 ℃;(b)—700 ℃; (c)—800 ℃; (d)—900 ℃; (e)—1 000 ℃; (f)—1 100 ℃.图5 TbCl3气溶胶不同温度热解产物的粒度分析Fig.5 Particle size analysis of pyrolysis products ofTbCl3 aerosol at different temperatures2.4 TbCl3气溶胶热解过程在超声波作用下TbCl3溶液雾化形成TbCl3气溶胶液滴,其随载气进入管式炉.TbCl3气溶胶进入管式炉后,首先在液滴的表面发生水分的快速蒸发导致液滴内溶质浓度快速达到过饱和状态,使溶质以TbCl3·xH2O(1≤x≤6)的形式从液滴析出,完成液相到固相的转变.由于受限于气溶胶液滴形状,TbCl3·xH2O晶粒从气溶胶液滴中析出后形成了球形颗粒,气溶胶液滴相当于一个承载了TbCl3的软模板.由图4可知,以TbCl3气溶胶液滴为前驱体,在高温下水分蒸发后得到了TbCl3·xH2O纳米颗粒组成的微米级球形颗粒,因此可认为TbCl3的热解是在微米~纳米尺度的范围内完成.热解产物中TbO2的生成可能与形成的TbCl3·xH2O微纳米球形颗粒紧密相关,由于粒径的尺寸效应:①颗粒具有较大的比表面积,提高了热解过程气体的传质速率;②热解反应与温度密切相关,在颗粒的径向上温度的分布相对于宏观块体颗粒更均匀,因此在球形颗粒粒径的径向上热解反应同时进行;③TbCl3气溶胶液滴由流速约为10 L/min的载气送入管式炉,气溶胶液滴在管式炉中的停留时间≤1 s;由于热解产物在高温区停留时间短,生成的TbO2来不及转化为其他形式的氧化铽,因此以TbCl3气溶胶为前驱体热解时,在热解产物中存在具有化学计量比形式的氧化铽TbO2.3 结论1) 以氯化铽气溶胶为前驱体,氯化铽在微纳米尺度范围内热解时极易氧化生成氧化铽.2) 氯化铽热解时首先发生水解反应,生成Tb(OH)2Cl,其脱除一分子水后形成TbOCl,相应的化学反应为TbCl3+2H2O=Tb(OH)2Cl+2HCl,Tb(OH)2Cl=TbOCl+H2O.3) 以氯化铽气溶胶为前驱体时,热解反应发生在微米~纳米的尺度范围内.当热解温度低于800 ℃时,热解产物中存在TbO2和Tb7O12,当热解温度高于800 ℃时,产物中的氧化铽为Tb7O12.4) 氯化铽气溶胶高温热解时生成氧化铽存在两种反应途径:一种为先氧化生成TbO2,其在高温下脱氧生成Tb7O12:2TbOCl+O2=2TbO2+Cl2,7TbO2=Tb7O12+O2;另一种反应途径,由于TbO2热稳定性差,因此热解生成的TbOCl在高温下直接氧化为Tb7O12,即14TbOCl+5O2 = 2Tb7O12+7Cl2.参考文献:【相关文献】[1] Wang R G,Sama V,Li D Q,et al.Hydrothermal synthesis of rare-earth oxide nanocatalysts for automotive exhaust clean-up [J].Advanced MaterialsResearch,2012,512(515):1624-1629.[2] Dabboussi S,Elhouichet H,Bouzidi C,et al.Excitation and emission processes of Tb3+ in porous anodic alumina [J].Applied Surface Science,2009,255(7):4255-4258.[3] Devaiah D,Jampaiah D,Saikia P,et al.Structure dependent catalytic activity ofCe0.8Tb0.2O2-δ and TiO2 supported Ce0.8Tb0.2O2-δ solid solutions for CO oxidation [J].Journal of Industrial and Engineering Chemistry,2014,20(2):444-453.[4] Bhosale R,Kumar A,Almomani F.Solar thermochemical hydrogen production via terbium oxide based redox reactions [J].International Journal ofPhotoenergy,2016,2016(1):1-9.[5] Li M,Gu Q,Han W,et al.Electrodeposition of Tb on Mo and Alelectrodes:thermodynamic properties of TbCl3 and TbAl2 in the LiCl-KCl eutectic melts [J].Electrochimica ACTA,2015,167(1):139-146.[6] Gschneidner K A,Eyring L,Lander G H.Handbook on the physics and chemistry of rare earths [M].Amsterdam:Elsevier,2002:100-130.[7] Johan-Sundstrijm O W.Investigation of the dehydration schemes ofNdCl3·6H2O,TbCl3· 6H2O and DyCl3·6H2O using a fluidized bed [J].Journal of Alloys and Compounds, 1997,249(1/2):224-228.[8] Van Hong V,Sundström J.The dehydration schemes of rare-earth chlorides[J].Thermochimica ACTA.1997,307(1):37-43.[9] 苏勉曾,李根.稀土元素氯化物水合物的脱水过程与水解反应的机理[J].化学通报,1979,43(4):34-38.(Su Mian-zeng,Li Gen.Mechanism of dehydration and hydrolysis of rare earth chloride hydrates [J].Chemistry Online,1979,43(4):34-38.)[10]Vestli M,Maide M,Nurk G,et al.Characterization of doped ceria films as SOFC electrolyte prepared by using ultrasonic spray pyrolysis method [J].ECS Transactions,2013,57(1):1159-1165.[11]吴文远,薛首峰, 边雪,等.超细氧化铈制备工艺研究[J].东北大学学报(自然科学版),2015,36(6):800-804.(Wu Wen-yuan,Xue Shou-feng,Bian Xue,et al.Study on the preparation process for CeO2 superfine powder[J].Journal of Northeastern University( Nature Science),2015,36(6):800-804.)[12]Goulart C,Djurado E.Synthesis and sintering of Gd-doped CeO2 nanopowders prepared by ultrasonic spray pyrolysis [J].Journal of the European CeramicSociety,2013,33(4):769-778.[13]薛首峰,吴文远,边雪,等.氯化镨气溶胶的高温热解机制研究[J].稀有金属,2018,42(6):640-649. (Xue Shou-feng,Wu Wen-yuan,Bian Xue,et al.Mechanism of thermal decomposition of PrCl3 aerosol at high temperature in air atmosphere[J].Chinese Journal of Rare Metals,2018,42(6):640-649.)。
磁性纳米颗粒材料

表面效应导致小颗粒磁性的减小,例如与块体体积相关的氧化纳米颗粒。这种减少与很多机制有关,例如颗粒表面存在的磁性无感层(magnetically dead layer),倾斜自旋(canted spins)的存在或者表面自旋的类自旋玻璃行为(spin-glass-like behavior)[17]的存在。另一方面,对于金属纳米颗粒,例如钴,也有报道显示尺寸的减小会使磁矩增大。
阻隔时间和有效各向异性常数,颗粒尺寸,应用磁场区域和实验测量时间。.例如,如果使用更短的窗口时间,比方有( )的铁磁共振,可以得到比通过交变磁场测量得到的值更大的阻隔温度。而且,颗粒直径中二个中的一个因素可以改变转换时间从100年到100纳秒。在第一个例子中,颗粒的磁性是稳定的,而在后来的例子中,颗粒集合没有剩磁,是一种超顺磁体。
式子中V=颗粒体积,θ=磁化方向和简单轴(easy axis)的角度。
能量势垒 把两个能量相同的易磁化方向区分开。当减少颗粒的尺寸,热能 超过了能量势垒 ,磁化方向更容易改变。对于这样一个系统( ),其行为像一个顺磁体,而不是原子磁矩,有巨磁矩在每个颗粒里(如图1的d)。这种体系被称为超顺磁体。这种体系没有磁滞,这从不同温度下的数据可以看出。
拟推荐201年度教育部高等学校科学研究优秀成果奖科学技
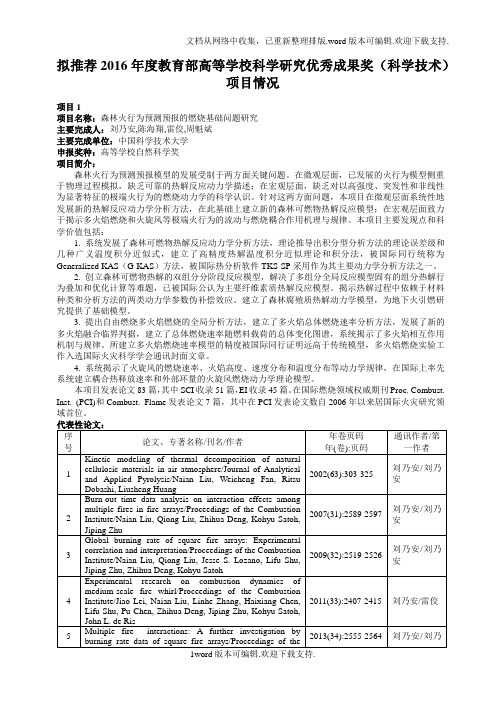
拟推荐2016年度教育部高等学校科学研究优秀成果奖(科学技术)项目情况项目1项目名称:森林火行为预测预报的燃烧基础问题研究主要完成人:刘乃安,陈海翔,雷佼,周魁斌主要完成单位:中国科学技术大学申报奖种:高等学校自然科学奖项目简介:森林火行为预测预报模型的发展受制于两方面关键问题。
在微观层面,已发展的火行为模型侧重于物理过程模拟,缺乏可靠的热解反应动力学描述;在宏观层面,缺乏对以高强度、突发性和非线性为显著特征的极端火行为的燃烧动力学的科学认识。
针对这两方面问题,本项目在微观层面系统性地发展新的热解反应动力学分析方法,在此基础上建立新的森林可燃物热解反应模型;在宏观层面致力于揭示多火焰燃烧和火旋风等极端火行为的流动与燃烧耦合作用机理与规律。
本项目主要发现点和科学价值包括:1. 系统发展了森林可燃物热解反应动力学分析方法,理论推导出积分型分析方法的理论误差级和几种广义温度积分近似式,建立了高精度热解温度积分近似理论和积分法,被国际同行统称为Generalized KAS(G-KAS)方法,被国际热分析软件TKS-SP采用作为其主要动力学分析方法之一。
2. 创立森林可燃物热解的双组分分阶段反应模型,解决了多组分全局反应模型固有的组分热解行为叠加和优化计算等难题,已被国际公认为主要纤维素质热解反应模型。
揭示热解过程中依赖于材料种类和分析方法的两类动力学参数伪补偿效应。
建立了森林腐殖质热解动力学模型,为地下火引燃研究提供了基础模型。
3. 提出自由燃烧多火焰燃烧的全局分析方法,建立了多火焰总体燃烧速率分析方法,发展了新的多火焰融合临界判据,建立了总体燃烧速率随燃料载荷的总体变化图谱,系统揭示了多火焰相互作用机制与规律。
所建立多火焰燃烧速率模型的精度被国际同行证明远高于传统模型,多火焰燃烧实验工作入选国际火灾科学学会通讯封面文章。
4. 系统揭示了火旋风的燃烧速率、火焰高度、速度分布和温度分布等动力学规律,在国际上率先系统建立耦合热释放速率和外部环量的火旋风燃烧动力学理论模型。
煤化学chap8 煤的工艺性质

第一节 煤的发热量
(一)对N、S特殊热效应的校正––恒容高位发热量
从弹筒发热量中扣除稀硫酸和稀硝酸生成热,称为
恒容高位发热量,简称高位发热量,用符号Qgr,
v,
表示:
ad
Q gr, v, ad= Q b, ad-( 94.1S b, ad + Q b, ad )
星蓝海学习网
第一节 煤的发热量
● ~120℃:煤炭脱水、干燥 ● 120~200℃:解吸,脱除吸附的CH4、CO、CO2等气 体。 ● 300℃:低变质程度的煤开始热解,生成CO2、CO等, 生成放出热解水和微量的焦油。
星蓝海学习网
第二节 煤的热解和粘结成焦性质
三、粘结性烟煤热解过程 (2)胶质体的生成和固化阶段(300~550℃)
恒湿无灰基是指煤样含有最高内在水分但不含灰分 的一种假想状态,这时煤样中只含有可燃质和最高内在水 分。煤的恒湿无灰基高位发热量不能直接测定,需用空气 干燥基的高位发热量进行换算,公式如下:
Qgr, maf
Qgr, v, ad
100 (100
100 (100 MHC) M ad ) Aad (100
++ NH2
C4H6
H2
+ 星蓝海学C习4H网6
+ H 2O + CH4
++ N2HH32
+ 2 H2
第二节 煤的热解和粘结成焦性质
●煤热解中的缩聚反应
♣ 胶质体metaplast固化过程的缩聚反应,主要是在热解 生成的自由基之间的缩聚,其结果生成半焦 semicoke/char。 ♣ 半焦分解,残留物之间缩聚,生成焦炭coke。缩聚反 应是芳香结构脱氢。
08-酰胺和胺的人名反应

3
Beckmann重排反应
二、Beckmann重排反应的特点 1 、 反 应 在 强 制 性 的 条 件 下 进 行 ( 高 温 >130 oC , 大 量 的 强 Bronsted酸),不是催化反应。
2、常用的Bronsted酸有:H2SO4, HCl/Ac2O/AcOH等。因此,对 酸敏感的底物不适合这个工艺。
Gabriel伯胺合成法
二、Gabriel伯胺合成法的特点(续) 3、-卤代酮、酯、腈以及-卤代酮酸酯(比如,溴代丙二酸二乙 酯);
4、O-烷基异脲;
5、烷氧基和烷硫基膦盐;
6 、在 Mitsunobu 反应条件下 (DEAD/Ph3P/phthalimide) 的伯醇和仲 醇;
10
Gabriel伯胺合成法
二、Gabriel伯胺合成法的特点(续) 7、带有好几个拉电子基的芳基卤代物(通过SNAr反应制备伯芳香 胺); 8、在Cu(I)作催化剂下的芳基卤代物; 9、环氧化合物(epoxide)和丫啶化合物(aziridines)可以用来制备氨 基醇和二胺; 10 、 ,- 不 饱 和化合 物 可以通 过 邻苯二 甲 酰亚胺 负 离子进 行 Michael加成反应。
26
Hofmann重排反应
二、Hofmann重排反应的特点 1 、次卤酸试剂要新鲜制备,即把氯气或溴素加到 NaOH 或 KOH的水溶液中;
2、在传统的碱性反应条件中,酰胺不能带有碱敏感的官能团, 但是酸敏感的官能团(比如缩醛)不受影响;
3、不能分离得到异氰酸酯中间体,因为在反应条件下马上水 解(溶剂解)得到相应的少一个C的胺,中间经过不稳定的氨基 甲酸;
三、Mannich反应的改进 最初三组分的Mannich反应有好几种改进。 使用预先制备的亚胺盐是最重要的改进,因为这样使反应更快、 区域选择性更高,甚至能在温和条件下进行立体选择性的反 应。
柠檬酸钙分解温度 解释说明以及概述

柠檬酸钙分解温度解释说明以及概述1. 引言1.1 概述柠檬酸钙是一种常见的无机化合物,具有广泛的应用领域。
柠檬酸钙在高温条件下会发生分解,释放出二氧化碳和水。
研究柠檬酸钙的分解温度对于了解其性质以及在工业生产中的应用具有重要意义。
1.2 文章结构本文主要分为四个部分:引言、正文、结果与讨论以及结论。
在引言部分,我们将介绍柠檬酸钙分解温度的定义和意义,并说明本文的目的。
然后,在正文部分,我们将详细探讨影响柠檬酸钙分解温度的因素以及其化学过程。
随后,在结果与讨论部分,我们将呈现实验观察结果,并对其进行深入分析和解释。
最后,在结论部分,我们将总结研究结果并回答研究目的,并提出对柠檬酸钙分解温度进一步研究方向或建议。
1.3 目的本文旨在全面探讨柠檬酸钙分解温度的特性、影响因素以及化学过程。
通过对柠檬酸钙分解温度的研究,我们可以深入了解该化合物的性质,并为其在工业应用中提供参考和指导。
希望本文能够为相关领域的研究者和使用者提供有益信息,并为未来的研究工作提供启示。
2. 正文:2.1 柠檬酸钙分解温度的定义柠檬酸钙分解温度是指柠檬酸钙(CaC6H5O7)在一定条件下分解成其它化合物或元素所需要的温度。
柠檬酸钙是一种白色结晶性粉末,可溶于水和酸,常用作食品添加剂和药物。
2.2 影响柠檬酸钙分解温度的因素多种因素会影响柠檬酸钙的分解温度。
首先,反应物浓度是决定分解温度的重要因素之一。
通常情况下,反应物浓度越高,分解温度越低。
其次,催化剂对柠檬酸钙的分解也具有显著影响。
一些催化剂能够降低分解温度并加速反应速率。
此外,气氛环境对反应也有一定影响。
在不同气氛(如氧化性或还原性气氛)下,柠檬酸钙的分解温度可能不同。
最后,晶体形态和热处理历史也可能对分解温度产生影响。
2.3 柠檬酸钙分解温度的化学过程柠檬酸钙在加热时会发生分解反应,首先是水合物的失去结晶水,然后发生一系列复杂的热分解反应,生成二氧化碳和剩余的钙化合物。
具体的化学过程可以描述为:CaC6H5O7·nH2O →CaC6H5O7 + nH2OCaC6H5O7 →3CO2 + 3H2O + CaCO3这些反应可以通过热重分析等实验手段进行观测和分析。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Thermochimica Acta 424(2004)131–142Thermal decomposition (pyrolysis)of urea in an open reaction vesselPeter M.Schaber a ,∗,James Colson b ,Steven Higgins b ,Daniel Thielen b ,Bill Anspach b ,Jonathan Brauer baDepartment of Chemistry and Biochemistry,Canisius College,2001Main Street,Buffalo,NY 14208,USAb OxyChem Technology Center,2801Long Road,Grand Island,NY 14072,USAReceived 30April 2003;accepted 4May 2004Available online 6July 2004AbstractA study was done of the thermal decomposition of urea under open reaction vessel conditions by thermogravimetric analysis (TGA),high performance liquid chromatography (HPLC),Fourier transform-infrared (FT-IR),and an ammonium ion-selective electrode (ISE).Both evolved gases and urea residue were analyzed,and profiles of substances present versus temperature are given.Major reaction intermediates are also identified.Plausible reaction schemes based on product distribution in relation to temperature are proposed.Our data indicate that at temperatures in excess of 190◦C,cyanuric acid (CY A),ammelide and ammeline are produced primarily from biuret.Biuret itself is a result of prior reaction of cyanic acid,HNCO,with intact urea.Cyanic acid is primarily a result of urea decomposition at temperatures in excess of 152◦C.CYA and ammelide first appear at approximately 175◦C,but the reaction rate is very slow.At temperatures in excess of 193◦C,alternate reactions involving the decomposition of biuret substantially increases reaction rates.Several parallel processes compete for the production of products.Production of CY A,ammeline and ammelide appears complete at 250◦C,after which sublimation and eventual decomposition of products occurs.©2004Elsevier B.V .All rights reserved.Keywords:Urea;Pyrolysis;Thermal decomposition;Open vessel1.IntroductionThe thermal decomposition (pyrolysis)of urea to pro-duce cyanuric acid (CY A:2,4,6-trihydroxy-1,3,5-triazine)was discovered by Wöhler approximately 175years ago [1].Details of the reaction mechanism have been studied for many years and recently a plausible temperature dependent reaction scheme,based primarily on product distribution,has been proposed [2].It is apparent that this reaction proceeds by a very complex and diverse pathway.For ex-ample,it is known that the primary decomposition products exhibit high reactivity and undergo a series of secondary reactions.Reaction conditions such as temperature,heating time,closed or open reaction vessel,atmosphere make-up and pressure,have all been shown to affect the outcome of this reaction process in terms of total product distribution and intermediates observed [2–15].Pyrolysis of urea is not∗Corresponding author.Tel.:+17168882351;fax:+17168883112.E-mail address:Schaber@ (P.M.Schaber).just a topic of academic concern.The industrial production of CYA from urea is a widely used process and one that has been practiced for several decades.Industrial interest in CY A is based on its widespread use as a precursor for the production of disinfectants,sanitizers,bleaches and herbicides [16].In addition,ammonia generated from urea decomposition is currently being considered by the diesel engine industry in an effort to develop a selective NO x cat-alytic reduction process (to nitrogen and water)for engine exhaust.There is present,continuing,and growing interest in the details of this reaction and its associated mechanism.Stradella and Argentero [12]recently published a study on the thermal decomposition of urea and related com-pounds with TGA (thermogravimetric analysis)and DSC (differential scanning calorimetry)measurements,together with EGA (evolved gas analysis).Chen and Isa [13],and Carp [15]published related studies using simultaneous TGA,DTA,and MS (mass spectroscopy).These studies focus on the decomposition of urea under purge gas (Ar,He or air)conditions.Tracking of residue species and account-ing for the production of by-products such as ammeline0040-6031/$–see front matter ©2004Elsevier B.V .All rights reserved.doi:10.1016/j.tca.2004.05.018132P .M.Schaber et al./Thermochimica Acta 424(2004)131–142and ammelide and accompanying synthetic details,were not included in these reports.In this study,an analysis of urea decomposition under open reaction vessel conditions utilizing TGA,high performance liquid chromatography (HPLC),Fourier transform-infra red (FT-IR),and an am-monium ion-selective electrode (ISE),is reported.Both evolved gases and urea residue were analyzed and profiles of substances present versus temperature are given.Major reaction intermediates are identified.Although others are considered,plausible reaction schemes based on product distribution in relation to temperature,favoring those with observable intermediates,are proposed.2.ExperimentalUrea was obtained from Aldrich Chemical Co.,Milwau-kee,WI (99%pure,ACS reagent grade),and used without further purification.Biuret was obtained from Fisher Scien-tific Co.,Pittsburgh,PA (99.9%pure,ACS reagent grade)and also used without further purification.CY A,ammelide,ammeline,and melamine were obtained from OxyChem’s industrial process and purified in-house.Urea residue in an open reaction vessel was obtained by heating 3.0g samples of urea in a 10mL Pyrex TM beaker on a sand bath until the desired temperature was reached.The sample was then quickly cooled to room temperature in a water bath and the residue collected.Chromatographic analyses of urea residues were con-ducted with HPLC methods previously described in the lit-erature [2].Samples were analyzed for the presence of urea,biuret,CY A,ammelide,ammeline and melamine.An HPLC Mass Table (Table 1)and an HPLC Mass Plot (Fig.1)were constructed from the data obtained.Thermogravimetric analysis (TGA)and measurements of mass losses (and the 1st derivative)versus temperature were determined using a Hi-Res TGA 2950Thermogravimetric Analyzer under N 2(g)purge.The “high resolution”op-tion was routinely used.Typically,30–50mg of sample was placed on a Pt pan and heated at 10◦C min −1.Urea “critical temperatures”,defined as those temperatures corresponding to plateau regions,points of rapidly changing mass,or where phase changes are known to occur (melting points,etc.),were identified from the TGA data plot (Fig.2).Table 1HPLC Mass Table (urea pyrolysis)open reaction vessel a Temperature (◦C)Mass (g)Urea (g)Biuret (g)CY A (g)Ammelide (g)Ammeline (g)Melamine (g)Total %recovery 133100.098.7 1.0––––99.719080.060.520.00.60.5––102.022533.0 6.0 4.615.6 6.10.9–100.525029.00.30.219.97.7 1.30.058101.427528.00.50.318.97.5 1.30.056102.332020.0––12.1 4.9 1.10.04490.73505.0––3.11.00.50.10094.0aThese data were calculated based on the results obtained from HPLC analysis assuming 100.0g of urea initially present.Table 2Urea pyrolysis residue analysis;ammonium ion (NH 4+)analysis with an ion-selective electrode a Temperature (◦C)Concentration of NH 4+ion (ppm)13370190380225260025026027515032512035090aResults in ppm are based upon the mass of the original sample.Table 3Urea pyrolysis off-gas;ammonium ion (NH 4+)analysis with an ion-selective electrode a Temperature range (◦C)Concentration of NH 4+ion (ppm)Room temperature to 133210133–21011900210–2258500225–2551210255–3505200350–400450aResults in ppm are based upon the mass of the original sample.Ammonium ion [NH 4+]analysis of both urea residue and pyrolysis off-gases were accomplished using an ammonium ISE (Orion,Beverly,MA).Typically for urea residues,a 1%aqueous solution was made and analyzed.Resultant con-centration versus temperature data are collected in Table 2.Urea pyrolysis off-gases were generated by placing a 3.0g sample of urea in a three-neck round bottom flask fitted with a thermometer,a thermal watch device,a N 2(g)purge,and an NH 3(g)scrubber (gas trap)consisting of 50.0mL of 12.0M HCl.As the urea sample was heated,the gases that evolved between the desired “critical temperature”points were allowed to pass through the scrubber and collected.Ammonium ion analysis was conducted on diluted scrubber solutions.Results are collected in Table 3.FT-IR spectra of the urea melt were collected using the Applied Systems Inc.(ASI)REACT-1000system (ASI SensIR Technologies,Danbury,CT),fitted with a sili-con (Si)probe.A spectrum was acquired every 2min asP .M.Schaber et al./Thermochimica Acta 424(2004)131–142133Fig.1.HPLC Mass Plot:urea pyrolysis reaction (assume 100.0g of urea initially).“critical temperature”points,between 133and 225◦C,were ramped to and held with the aid of an Omega thermocou-ple (Fig.3).Qualitative analysis was performed using the REACT-1000soft-ware package supplied by ASI.Analy-Fig.2.TGA:urea pyrolysis reaction.sis of urea pyrolysis off-gases were conducted by placing a 1.0g sample of urea into a glass vial and inserting into a tube furnace fitted with a N 2(g)purge and gas collec-tion adapter.The urea sample was heated to the desired134P .M.Schaber et al./Thermochimica Acta 424(2004)131–142Fig.3.FT-IR Si-probe spectra:urea pyrolysis reaction.“critical temperature”and held there for several minutes with the aid of a thermal watch device.Evolved gases were subsequently swept into an IR gas cell fitted with NaCl windows.FT-IR spectra of urea off-gases were obtained with a Nicolet 20SXB Spectrometer (Nicolet,Madison,WI).Condensed materials adhering to the surface of the gas collection adapter were collected and subjected to melting point determination with a 510Melting Point ap-paratus (Brinkmann Instruments,Inc.,Westbury,NY),and HPLC analysis using methods previously referenced in this section.3.Results and discussion 3.1.Process overviewThe TGA of urea measured with a heating rate of 10◦C min −1under N 2(g)purge between 50and 600◦C is given in Fig.2.Three major stages of mass loss are ob-served and calculated to be approximately 72,24,and 4%.The pyrolysis reaction of urea in an open reaction vessel can be divided into four major “reaction”regions.“Reaction”regions are dominated by different chemical processes asso-ciated with the mass loss stages observed in the TGA.What follows is a detailed description of the chemical process in each of the urea pyrolysis “reaction”regions.3.2.First “reaction”region (room temperature to 190◦C)Little significant mass loss (0.01%)is observed when heating urea in an open reaction vessel from room tem-perature to its melting point 133◦C.HPLC analysis of the residue at the melting point,gives 98.7%urea and 1.0%biuret (Table 1).The biuret present at this point primarily represents contamination in the original sample.Only a small relative amount of [NH 4+]ion is observed in both the residue and off-gas analysis (Tables 2and 3)and a very small [NCO −]absorption peak at 2156cm −1is noted in the FT-IR spectra (Si-probe)of the melt (Fig.3).These observations are consistent with a very small amount of urea decomposition and vaporization.Recently,Chen and Isa [13]have also observed only a small mass change prior to urea’s melting point,which is consistent with our data.Mass loss begins in earnest at approximately 140◦C as observed from the TGA (Fig.2).The loss observed between 140and 152◦C,is associated primarily with urea vaporiza-tion.(Condensed material adhering to the surface walls of the gas collection adapter (see Section 2),in this temper-ature range was identified as urea from melting point and HPLC determinations.)A more significant mass loss is ob-served between approximately 152and 160◦C and proceeds via two processes;continued urea vaporization and decom-position [9].P.M.Schaber et al./Thermochimica Acta424(2004)131–142135 Urea decomposition:H2N–CO–NH2(m)urea+heat→NH+4NCO−(m) ammonium cynate →NH3(g)ammonia+HNCO(g)cyanic acid(1)At152◦C decomposition begins[4],Eq.(1),accompanied by vigorous gas evolution from the melt.Above152◦C the decomposition rate of urea increases rapidly.At160◦C,the FT-IR Si-probe spectra of the melt,indicates an increase in intensity of the[NCO−]peak from that observed at140◦C (Fig.3).The FT-IR spectrum of gases evolved at160◦C in-dicates the strong presence of NH3(g)(peaks at3333,965 and930cm−1),but little discernible HNCO(g).Stradella and Argentero[12]have,however,observed HNCO(g)in the evolved gases at this temperature.Chen and Isa[13],and Carp[15]have also identified evidence of HNCO(g)pro-duction in their studies.That the ammonium cyanate salt, [NH4+NCO−],is formed as an intermediate in Eq.(1)has recently been confirmed by Carp[15].These data are all con-sistent with urea’s initial decomposition to[NH4+NCO−], which itself decomposes resulting in the evolution of NH3 (g)and HNCO(g).A product of urea decomposition,HNCO,begins to react with intact urea to produce biuret at approximately160◦C, Eq.(2).1This is supported by an increase in the intensity of a peak unique to biuret at1324cm−1,in the FT-IR Si-probe spectra(Fig.3).Biuret production:H2N–CO–NH2(m)urea +HNCO(g)cyanic acid→H2N–CO–NH–CO–NH2(m)biuret(2)Between160and190◦C urea continues to vaporize and decompose and HNCO,continues to react with intact urea to produce biuret.However,complications also begin to enter the system in this temperature range.A small amount of HNCO can now begin to react with biuret[10],Eq.(3),or itself,Eq.(4),to produce CYA,or with urea,Eq.(5),to produce ammelide.From the FT-IR Si-probe data,it appears that the production of both CY A and ammelide commence simultaneously at approximately175◦C.This is supported by an increase in the intensity of respective peaks unique to CYA at1058cm−1,and ammelide,at977cm−1,in the FT-IR Si-probe spectra(Fig.3).CYA production:H2N–CO–NH–CO–NH2(m)biuret+HNCO(g)cyanic acid →CY A(s)cyanuric acid+NH3(g)ammonia(3)via Eq.(4)is a reasonable candidate and one that has been known experimentally for many years.Herzberg and Reid1The actual reaction to form biuret likely involves the interaction of urea with[NCO−]in the melt[11].The form of the equation used here is for convenience,and will be used throughout this document.However, it should be realized that reactions in the melt involving HNCO are likely to proceed via[NCO−]interaction.[17]have observed that if HNCO(g)exceeds a critical vapor pressure,3HNCO(g)cyanic acid→CY A(s)cyanuric acid(4)spontaneous and rapid polymerization to CY A can oc-cur.This critical vapor pressure can be quite low,espe-cially in the presence of a metal surface.2Another po-tential route to CY A involves the cyclicization of triuret (H2N–CO–NH–CO–NH–CO–NH2)with the evolution of NH3[3].Chen and Isa[13]identify the existence of urea trimer under TGA conditions.However,since no triuret is observed in these studies,its existence under open reaction vessel conditions is questionable.Ammelide production:2HNCO(g)cyanic acid+H2N–CO–NH2(m)urea→ammelide(s)+2H2O(g)(5) The intermediate for Eq.(5)could be either biuret,via Eq.(2),or dicyanic acid(H2N2C2O2).Biuret is observed in the reaction sequence and if it also serves as an intermedi-ate to ammelide,Eq.(5)is then very similar in essence to that represented in Eq.(7).Dicyanic acid would be very un-stable at these temperatures and if behaving as the interme-diate,would not be expected to accumulate to a significant extent.However,since not a hint of dicyanic acid was de-tected in this study,its contribution to ammelide production is questionable under the reaction conditions imposed. The conceptually simplest route to ammelide involves the direct reaction of CY A with available NH3(ammination), Eq.(6).This is a seeming logical path but one that has been determined to occur only under conditions of high pressure [18]or temperatures above300◦C[19].In the industrial preparation of CY A using a kiln,less ammelide is in general, CY A(s)cyanuric acid+NH3(g)ammonia→ammelide(s)+H2O(g)(6)produced when NH3is removed from the system.Although this would seem consistent with Eq.(6),the situation is likely more reflective of the different environmental aspects of lab-oratory versus industrial preparation of CY A,where temper-ature gradients in the kiln may produce conditions favorable to the production of ammelide in this fashion.A more likely laboratory route is one that parallels the formation of CY A, that is,the direct reaction of biuret with HNCO,Eq.(7), but with the formation of ammelide[20,21].Other possible routes to ammelide have been suggested,however,theyH2N–CO–NH–CO–NH2(s)biuret+HNCO(g)cyanic acid→ammelide(s)+H2O(g)(7)2Under TGA conditions,or with the use of a purge gas,this route is expected to be somewhat curtailed.However,Eq.(4)may be significant in the industrial preparation of CY A where metal kilns are often used.136P.M.Schaber et al./Thermochimica Acta424(2004)131–142typically require the presence of a precursor such as ammi-nated bi-or triuret,guanidine[10],or cyanamide[19].These proposed precursor species are stable in this“reaction”re-gion and if present,evidence of their existence would be expected.Our HPLC data give no indication any exist in measurable amounts in the residue(see Table1;note ap-proximately100%recoveries at these temperatures),nor are any detected in the FT-IR data.Under the conditions imposed,the reaction scheme most likely to produce ammelide,Eq.(7),operates in parallel to the one most likely to produce CYA,Eq.(3).Both prod-ucts are produced at about the same overall rate,and begin to appear at approximately the same temperature(175◦C). This suggests the reactions producing them share common reactants,(i.e.biuret and HNCO)and possess similar acti-vation energies.In addition,since relatively small amounts of CYA and ammelide are observed in the residue(Table1), whatever route adapted,reaction rates are rather sluggish for their production in this temperature range.Up to190◦C,the net mass loss for the system is pre-dominately the result of urea decomposition.By this same temperature,urea mass has decreased by38.2%,biuret mass has increased by19.0%and has reached a maximum ac-cording to HPLC analysis(Table1).The FT-IR of evolved gases is dominated by NH3due primarily to the contin-ual and increased decomposition rate of urea.Peaks in the 2100–2300cm−1region indicate the presence of HNCO, or of volatile salts of HNCO3such as ammonium cyanate, [NH4+NCO−],or hydronium cyanate,[H3O+NCO−][22]. Continued build up of[NH4+NCO−]in the melt is indicated by both the FT-IR Si-probe data(Fig.3),and the[NH4+]ion analysis of the residue(Table2).However,although build-ing,the amount of[NH4+]ion in the residue(380ppm)is still not significantly large.These data indicate that much of the NH3formed escapes the system without further reaction and that HNCO has either reacted to produce biuret,CY A, or ammelide,or escaped the system in the vapor form.In addition,the vaporization of volatile HNCO salts,has just begun.3.3.Second“reaction”region(between190and250◦C) As temperature exceeds190◦C,alternate reaction se-quences begin to dominate the process.At approximately 193◦C,the increased evolution of gases from the urea melt reflects the beginning of biuret decomposition(biuret melts with decomposition at193◦C),Eq.(8).A corresponding mass maximum for biuret(Table1),and1st derivative peak in the urea TGA near this temperature are observed(Fig.2). However,the gas evolution rate increase is not only the result of biuret and continuing urea decomposition.3In our studies,unequivocal identification of the HNCO salts cannot be made at this temperature due to poor resolution in this region of the FT-IR spectrum.Biuret decomposition:H2N–CO–NH–CO–NH2(m)biuret→H2N–CO–NH2(m)urea+HNCO(g)cyanic acid(8)(The urea produced by biuret decomposition in Eq.(8),is unstable in this“reaction”region and will itself decompose further to HNCO(g)and NH3(g),Eq.(1)).It also results from gas producing,auto-condensation reactions associated with biuret decomposition,Eqs.(9)and(10),to produce CY A and ammelide.CY A production:2H2N–CO–NH–CO–NH2(m)biuret→CY A(s)cyanuric acid+HNCO(g)cyanic acid+2NH3(g)ammonia(9)Ammelide production:2H2N–CO–NH–CO–NH2(m)biuret→ammelide(s)+HNCO(g)cyanic acid+NH3(g)ammonia+H2O(g)(10) Although Eqs.(3),(4)and(9)all contribute to the produc-tion of CY A,Eq.(3)is expected to predominate at lower temperatures[9](first“reaction”region),Eqs.(4)and(9) become more competitive at higher temperatures(second “reaction”region)where biuret begins to decompose.It is also expected that Eq.(4)will begin to play an increased role in CY A production if,or when,critical vapor pressure is achieved[17].A similar situation exists with respect to the production of ammelide.Eq.(7)is expected to predomi-nate at lower temperatures(first“reaction”region),Eq.(10) above193◦C(second“reaction”region).However,only small amounts of CY A and ammelide are observed prior to the decomposition temperature of biuret.At this point the production rate for both CY A and ammelide increase rapidly.The close relationship between the onset of biuret decomposition and increased production rates suggests that both CY A and ammelide are produced largely from biuret and in parallel fashion within the second“reaction”region [8].In addition,Eqs.(9)and(10)involve stoichiometric larger amounts of gaseous products than reaction candidates previously considered.The large increase in gas evolution physically observed in the urea melt above193◦C and the large amount of[NH4+]detected(11,900ppm)in the py-rolysis off-gas analysis between133and210◦C(Table3) are consistent with both continued urea and biuret decom-position and the production of CY A and ammelide by ki-netically faster mechanisms.TGA and DTA data collected on biuret itself indicates ready transformation to CY A upon heating[8,23].Stradella and Argentero[12]estimate that about50%of biuret is converted to CY A.At temperatures exceeding the melting point of biuret,conversion to CY A most probably involves Eqs.(4)and(9).At lower tem-peratures an alternative reaction path,be it minor,is one reminiscent of the production of CY A from urea and in-volves the initial decomposition of biuret to urea and HNCO.P.M.Schaber et al./Thermochimica Acta424(2004)131–142137Fig.4.TGA:biuret pyrolysis reaction.(Although the melting point of biuret is193◦C,our TGA analysis indicates mass loss prior to this temperature(Fig.4). These data are consistent with other investigators including Nelson et al.[8,23]).Biuret decomposition is followed by reaction of intact biuret with HNCO to produce CY A as in the urea process.The beginning of ammeline production is also observed in the second“reaction”region as noted by its detection in the HPLC analysis of the residue at250◦C(Table1).Al-though CY A and ammelide are most likely produced via parallel processes,ammeline could be produced either by ammination of ammelide,Eq.(11),or as suggested by Ha-effele et al.[20],by the direct reaction between HNCO and remaining urea,Eq.(12),or some modification thereof.No other species is detected in our analysis that could serve as a precursor to ammeline.However,at all temperatures up to350◦C,the mass amounts of ammelide exceed ammeline in the residue(Table1).Although this is not proof of a lin-ear process,it has been used as supporting evidence[24]. As previously mentioned,however,ammination reactions are only likely to occur at elevated pressures[18]or tem-peratures[19].The intermediate for Eq.(12)is most likely biuret.Ammeline production:ammelide(s)+NH3(g)→ammeline(s)+H2O(g)(11)or2HNCO(g)cyanic acid+H2N–CO–NH2(m)urea→ammeline(s)+2H2O(g)(12) If this occurs,Eq.(12)can be written in the following com-bined form assuming the previous occurrence of Eq.(2). This is a very likely scenario:4H2N–CO–NH–CO–NH2(m)biuret+HNCO(g)cyanic acid→ammeline(s)+2H2O(g)(12a) The evolution of gases begins to visibly slow as a white precipitate forms at temperatures exceeding210◦C.Con-version of the melt into a“sticky”solid matrix is complete at225◦C,and CY A becomes the major component of the residue.Analysis of trapped off-gases for[NH4+]ion col-lected between210and225◦C,gives8500ppm(Table3). This is an indication that decomposition of urea and biuret4Eq.(12a)is very similar to Eqs.(3)and(7).The latter two reactions are relatively slow and most likely continue to be so even at elevated temperatures.Eq.(12a)is probably even slower than either Eq.(3) or Eq.(7)at all temperatures,and is likely the reason ammeline is not observed until this point.(Biuret+HNCO reaction rates:Eq.(3) producing CY A>Eq.(7)producing ammelide>Eq.(12a)producing ammeline.)138P.M.Schaber et al./Thermochimica Acta424(2004)131–142Table4HPLC Mass Table(biuret pyrolysis)aTemperature(◦C)Mass(g)Biuret(g)CY A(g)Ammelide(g)Ammeline(g)Melamine(g)Total(%)recovery 22565.028.634.5 2.860.26–101.826051.0 6.6340.8 3.830.36–101.227539.00.0835.5 3.470.35–101.0a These data were calculated based on the results obtained from HPLC and TGA analysis assuming100.0g of urea initially present.continue in this temperature range.The continued produc-tion of CY A and ammelide,Eqs.(9)and(10),are also ex-pected to contribute to the NH3(g)production as well.Analysis for[NH4+]ion gives a maximum in the residueat225◦C.This represents a substantial increase from thevalue observed at190◦C(Table2).Accumulation of[NH4+]is a direct consequence of system precipitation.As the ureamelt is transformed into a solid matrix,the rate at whichNH3(g)and HNCO(g)can diffuse from the system is di-minished.These species are either trapped in the solid ma-trix,or interact with each other,Eq.(13),and exist in theresidue in ionic form.The fact that ionic formation occurs issupported by the[NH4+]analysis of the residue from thesestudies,and by the MS data collected by Carp[15].Just asdecomposition species are“tied up”in ionic formAmmonium cyanate production:NH3(g)ammonia +HNCO(g)cyanic acid→NH4+NCO−(s)ammonium cyanate(13)in the matrix,5it is also likely that the remaining urea and biuret are likewise converted to ionic cyanurates via inter-action with CYA,Eqs.(14)and(15).Urea cyanurate production:H2N–CO–NH2(m)urea+CY A(s)→H2N–CO–NH3+CY A−(s)urea cyanurate(14)Biuret cyanurate production:H2N–CO–NH–CO–NH2(m)biuret+CYA(s)→H2N–CO–NH–CO–NH3+CY A−(s)biuret cyanurate(15)The narrow plateau region observed near225◦C in the urea TGA corresponds very well to a more demonstrable one exhibited by biuret at approximately the same temper-ature(Figs.2and4,respectively).In the biuret system, the residue is primarily composed of biuret and CY A,with lesser amounts of ammelide and ammeline(Table4).Since all the product species are thermally stable at225◦C,a plateau region is observed in both the urea and biuret TGA at this temperature.FT-IR data collected on the off-gases resulting from the urea residue at this temperature illustrates that gaseous emissions have greatly decreased.Only a small5Eq.(13)is in essence the reverse of the latter portion of Eq.(1).amount of NH3(g),HNCO(g)and HNCO salt speciesare emitted from the residue matrix.Emission thereforecontinues,but at a much reduced rate[12].Melamine is positively identified for thefirst time at250◦C6(Table1).This represents a departure from thetemperature and level at which this product isfirst observedunder TGA conditions(350◦C,and in substantially smalleramounts)[2].The direct ammination of ammeline,Eq.(16),is one possible route to melamine.Once again,althoughthis may be the simplest conceptual route,it is one likelyonly to be observed atMelamine production:ammeline(s)+NH3(g)→melamine(s)+H2O(g)(16) elevated pressures[18]or temperatures[19].However,openreaction vessel conditions as applied here may produce lo-calized temperature conditions favoring this process to alimited extent.Another possibility is cyanamide(H2NC≡N) trimerization[10],Eq.(17).Although cyanamide has notbeen isolated from urea decomposition under normal3H2NC≡N(g)cyanamide→melamine(s)(17)pressure[11],it could be produced via an alternate decom-position route associated with urea dehydration,Eq.(18), or by ammination of cyanic acid[10],possibly within the solid matrix,Eqs.(19)and(19a),and behave as a very reac-tive intermediate at these temperatures.The small amounts of melamine observed may be reflective of small Cyanamide production:H2N–CO–NH2(m)urea→H2O(g)+H2NC≡N(g)cyanamide(18)6This is somewhat of an anomaly since melamine is reported to melt with accompanying sublimation below250◦C.It is therefore necessary to assume that if melamine forms,it sublimes simultaneously as it is being produced.The sublimation of melamine may therefore also contribute to the mass loss observed between225and250◦C in the TGA(Fig.2). Accumulation of measurable amounts of melamine,result from production that is kinetically faster that sublimation.It should also be noted that melamine is observed at250,275,and350◦C,but not at320◦C(Table1). This would suggest different mechanisms for its production in different temperature ranges.At lower temperatures(below300◦C),Eq.(17),or some other unidentified reaction,results in production of melamine.At higher temperatures,Eq.(16)is expected to predominate.。