分子克隆实验设计
分子克隆实验标准步骤

分⼦克隆实验标准步骤分⼦克隆实验标准步骤⼀、常规分⼦克隆实验流程:⼆、分⼦克隆实验标准步骤(含实验编号):1. PCR 扩增⽬的基因(编号Clone SOP-1)以本实验室常⽤酶KOD-Plus-Neo (TOYOBO )为例体系(50ul ):10×KOD buf 5uldNTP(2mM) 5ulMg 2+ 3ulPrimer1 1ulPrimer2 1ulTemplate50-200ngKOD0.5ulddH 2O up to 50ul程序:95℃2min98℃10s58℃30s 35cycle68℃2kb/min68℃7min12℃∞2.PCR产物的琼脂糖凝胶电泳琼脂糖凝胶的制备(编号Clone SOP-2)琼脂糖溶液的制备:称取琼脂糖,置于三⾓瓶中,按1%-1.5%的浓度加⼊相应体积的TBE或TAE缓液,将该三⾓瓶置于微波炉加热⾄琼脂糖溶解。
胶板的制备:①取有机玻璃内槽,洗净、晾⼲;②将有机玻璃内槽置于⼀⽔平位置模具上,安好挡板,放好梳⼦。
在距离底板上放置梳⼦,以便加⼊琼脂糖后可以形成完好的加样孔。
③将温热琼脂糖溶液倒⼊胶膜中,使胶液缓慢地展开,直到在整个有机玻璃板表⾯形成均匀的胶层。
④室温下静置30min左右,待凝固完全后,轻轻拔出梳⼦,在胶板上即形成相互隔开的上样孔。
制好胶后将铺胶的有机玻璃内槽放在含有0.5~1×TAE(Tris-⼄酸)或TBE(Tris-硼酸)⼯作液的电泳槽中使⽤,没过胶⾯1mm以上。
3.试剂盒回收DNA⽚段(编号Clone SOP-3)以本实验室常⽤DNA凝胶回收试剂盒(天根)为例使⽤前请先在漂洗液PW中加⼊⽆⽔⼄醇,加⼊体积请参照瓶上的标签。
①柱平衡步骤:向吸附柱CA2中(吸附柱放⼊收集管中)加⼊500µl平衡液BL,12,000rpm(~13,400×g)离⼼1min,倒掉收集管中的废液,将吸附柱重新放回收集管中。
(请使⽤当天处理过的柱⼦)②将单⼀的⽬的DNA条带从琼脂糖凝胶中切下(尽量切除多余部分)放⼊⼲净的离⼼管中,称取重量。
分子克隆实验

分子克隆技术实验报告姓名:班级:学号:指导老师:pUC19外源1.5kbDNA片段亚克隆于pUC1301摘要:分别从大肠杆菌菌液中提取质粒pUC19和pUC1301,两种质粒都使用Bam HI+Kpn I两种限制性内切酶进行切割。
回收pUC1301质粒中1.5kb片段,使用连接酶将它与切开的pUC19连接,转化感受态大肠杆菌,利用标记基因amp r、kan r和α—互补筛选阳性克隆。
实验获得了很好的酶切效果,但最后没有筛选出阳性克隆。
关键词:pUC19、pUC1301、亚克隆、限制性内切酶、感受态1前言初始克隆中的外源DNA片段往往较大,含有许多目的基因以外的片段,在诸如表达、序列标签和突变等操作中不变进行,因此必须将目的基因所对应的小片段DNA找出来,这个过程叫做亚克隆。
本实验是将pU1301中一段来自水稻1.5kb的片段亚克隆到pUC19上。
首先需要提取所需的质粒。
在大肠杆菌中质粒又大又小、拷贝数又多又要少,因此分离的方法也多种多样。
在实践中最常见的是通过将裂解法提取高拷贝的小质粒以及基于此方法的纯化技术,其他方法还有试剂盒分离纯化等,本实验通过试剂盒分离纯化E.coil质粒DNA。
将质粒提取出来后需要检测所获质粒的质量,方法有吸光值检测、凝胶电泳检测等。
要获得目的片段然后亚克隆与其他载体上,需使用两种工具酶:限制性内切酶、连接酶。
限制性酶切酶主要有三类,目前使用最多的是Ⅱ型。
限制性内切酶的使用需要在合适的条件下,当条件不合适时往往会出现星星活性。
限制性内切酶产生的黏性末端将导致载体自连,为了防止其自连,可以使用双酶切、去磷酸化、末端补平等方法。
连接酶是将两段核酸连接起来的酶,常用的有T4DNA连接酶、大肠杆菌DNA连接酶、TaqDNA连接酶、T4RNA连接酶。
目的片段和载体在连接酶作用下连接成一个整体,需借助一定方法导入受体细胞,首先需要制备感受态细胞,这类细胞处在易吸收外源DNA 条件下,然后通过电转化、CaCl2转化法将外源DNA导入受体细胞。
分子克隆(亚克隆)实验总体流程详解

一、扩增1、LB培养基5ml;2、抗生素:1000X,即1:1000比例。
种类根据细菌抗性决定;3、菌体:看浑浊度,1%-5%,取500ul于其中;4、37℃摇床220转,过夜,12-16h。
二、纯化质粒DNA1、1.5ml离心管,编号一定要写清楚;2、加满离心管,离心12000xg. 1min,弃上清。
取三次;3、加Buffer S1 200ul,溶解沉淀,5min;4、加S2(用完立刻盖紧瓶盖,以免CO2中和Buffer中的NaOH)200ul,不能剧烈(以免基因组DNA的污染),上下翻转4-6次,直至形成透亮的溶液,时间少于5min。
目的是使蛋白包裹基因组DNA,游离质粒;5、加S3 280ul,温和充分翻转混合6-8次,12000xg,10min(此步呈白色絮状);*备注:S1:S2:S3=5:5:76、取上清加入制备管(置于2ml离心管),12000xg,1min,去滤液;7、加Buffer W1 500ul,12000xg,1min,弃滤液;8、加Buffer W2 700ul,12000xg,1min,弃滤液。
重复一遍;9、空管离心12000xg,1min;10、制备管移入新的1.5ml离心管,管膜中加60-80ul去离子水,静置1min,12000xg,1min。
(将去离子水加热至65度,将提高洗脱效率)四、跑胶回收:sost回收失败1、2%浓度胶,Loading Buffer如是6X,则加10ul到样品,全部加样到胶孔中。
插入:配胶方法大块胶60ml;小块胶25ml;需要配置大块胶、大孔胶;Agarose 0.6g,TAE60ml,微波中火2min;趁热但不烫手时加入gold view 0.5ul/25ml;倒入槽里。
2、跑胶:单位厘米/5-10v。
所以大槽25cm,150v即可。
小槽100v即可。
3、紫外灯下切胶,纸巾吸进液体,计算凝胶重量(1mg=1ul);4、加3个凝胶体积的凝胶结合液DB(0.1ul视为100ul;如凝胶浓度大于2%,则加入6倍体积溶胶液;凝胶块最大不能超过400ul,超过可多个离心柱);5、56℃水浴放置10分钟,至完全溶解;6、每100mg最初的凝胶重量加入150ul的异丙醇,震荡混匀,回收大于4Kb的片段可不加异丙醇,加入反而降低回收效率;7、将上一步所得溶液加入吸附柱AC中(吸附柱放入收集管中),12000rpm,30-60s,弃液体;8、加700ul漂洗液WB,12000rpm,1min,弃废液;9、加500ul漂洗液WB,12000rpm,1min,弃废液;10、空离心2min,弃废液;11、晾干乙醇,以免抑制下游反应;12、将吸附柱放入新的1.5ml离心管,加入50ul(最少30ul)洗脱缓冲液EB或者去离子水(65-70水浴加热效果更好,或将得到的溶液重新加入离心柱可增加洗脱量);五、连接体系:六、转化1、加感受肽:连接产物=10:1,冰浴30min;2、激活:42℃,90s,不能震动;3、加500ul LB培养基;4、37℃,150转摇床,45min;5、铺板子七、检测1、细菌长势良好,对照组不长;2、酶切检测:(1)1ml LB体系:1ml LB培养基+1ul抗生素于1.5mlEP管中;(2)15ul Pcr体系:7.5ul Mix(2X)5.5ul water1ul上游引物1ul下游引物(3)用枪头挑培养皿中的菌体,吹打于1ml LB体系中,再吹打于15ul Pcr体系中;(4)1ml LB体系放入37℃摇床,150rpm;显示4°/4°时,结束。
分子克隆实验
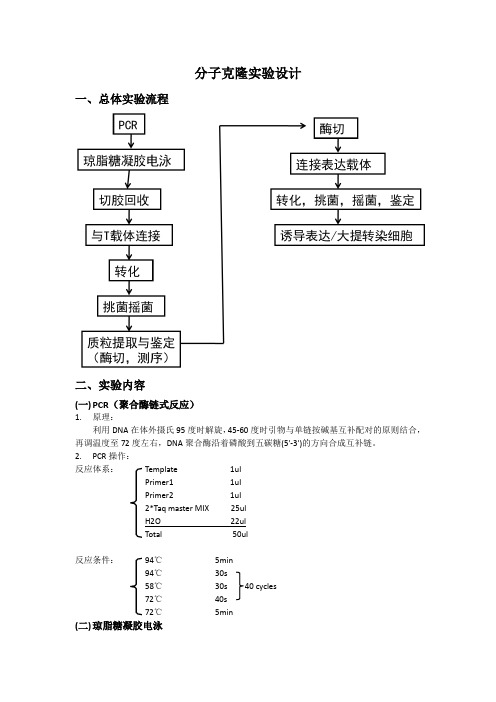
分子克隆实验设计一、总体实验流程二、实验内容(一)PCR(聚合酶链式反应)1.原理:利用DNA在体外摄氏95度时解旋,45-60度时引物与单链按碱基互补配对的原则结合,再调温度至72度左右,DNA聚合酶沿着磷酸到五碳糖(5'-3')的方向合成互补链。
2.PCR操作:反应体系:Template 1ulPrimer1 1ulPrimer2 1ul2*Taq master MIX 25ulH2O 22ulTotal 50ul反应条件:94℃5min94℃30s58℃30s 40 cycles72℃40s72℃5min(二)琼脂糖凝胶电泳1.原理:琼脂糖凝胶具有网络结构,分子通过时会受到阻力,大分子物质在涌动时受到的阻力大,因此在凝胶电泳中,带电颗粒的分离不仅取决于净电荷的性质和数量,而且还取决于分子大小。
2.凝胶电泳步骤:1)称取0.5g琼脂糖至50ml 电泳缓冲液(TAE)中,摇匀;同时装置制胶架。
2)加热至琼脂糖完全融化,期间停止加热摇动3次,即无可见漂浮物,澄清为止。
3)加入1ulEB染液,充分摇匀。
4)缓慢倒入制胶架内,冷却30-60min,至凝胶充分凝固,放入电泳槽中即可使用。
5)使用时,将样品点入相应的加样孔内,即可开始电泳,待指示剂位于整块凝胶2/3位置时,停止电泳,在紫外灯下观察样品条带。
(三)切胶回收(天根生化公司凝胶回收试剂盒)1. 将空小离心管称重。
将回收的带DNA 片段的凝胶放入其中、再次称重,确定凝胶净重。
2. 将离心管中的凝胶捣碎以助于凝胶溶解。
按胶的重量加入三倍体积的Extraction buffer (1mg凝胶加3μl Extraction buffer )。
3. 55℃恒温加热直到凝胶完全融化(5-15 分钟),每隔2-3分钟需将管内混悬液混匀一次。
4. 按照1:1比例加入异丙醇(1mg凝胶加1μl异丙醇),混匀。
5. 将混合液全部移入Spin column、6000rpm离心1分钟,弃去接液管中的液体。
分子克隆技术实验讲义(最终版)

分子克隆技术实验讲义黑龙江大学生命科学学院2016年3月甜菜M14品系BvM14-glyI基因的克隆与鉴定一、实验目的1、熟悉和了解目的基因克隆与鉴定的过程和方法。
2、学习和掌握质粒、T载体的特点。
3、学习和掌握TA克隆的连接体系及操作要点。
4、学习和掌握XcmⅠ酶切制备T载体的过程及方法。
5、学习和掌握CaCl2法制备大肠杆菌感受态细胞的原理和方法。
6、学习并掌握热激法转化技术的原理和操作步骤。
7、学习并掌握重组子鉴定和筛选的原理及蓝白筛选的原理和方法。
8、学习并掌握碱法小量制备质粒DNA的原理及操作步骤。
二、相关知识(一)T载体的制备pMD18-T Vector是一种高效克隆PCR产物(T-A Cloning)的商业化专用载体,由pΜC 18载体改建而成。
在pΜC 18多克隆位点处的XbaⅠ和SalⅠ识别位点之间插入了EcoRⅤ识别位点,用EcoRⅤ进行酶切反应后,再左两侧的3′端添加“T”而成,可以大大提高PCR产物的连接、克隆效率。
相关知识点:(1)质粒的提取;(2)酶切;(3)PCR等。
(二)DNA的重组与连接(PCR产物的克隆)把DNA片段从某一类型的载体无性繁殖到另一类型载体中,例如从某种质粒克隆到另一种质粒,这个过程称为亚克隆。
所谓重组,就是把外源目的基因“装进”载体的过程,即DNA的重新组合。
为了将目的基因重组于载体分子中,需要将载体DNA和目的基因分别进行适当处理,一般采用内切酶法将载体DNA分子切割成可与外源基因连接的线性分子,使其与相同酶切过的载体分子相互连接,彼此成为配伍末端(compatible end),以产生末端连接。
现在一些生物公司也开发了针对不同插入DNA片段的专用载体,如专门用于克隆PCR产物的载体,大大方便了实验操作。
相关知识点:(1)克隆与亚克隆;(2)DNA重组;(3)内切酶;(4)粘性末端与平末端;(5)连接酶;(6)连接酶的分类及功能等。
(三)大肠杆菌感受态细胞的制备外源基因与载体在体外连接成重组体DNA分子后,需将其导入受体细胞进行扩增和筛选,得到大量、单一的重组体分子,这就是外源基因的无性繁殖,或称为克隆。
分子克隆实验指南

做了快一年的分子克隆,犯了很多错误,经历了很多坎坷,也学到了很多东西。
分子克隆过程中出现问题最多的大概就是连接了,大家抱怨的也最多,我也陷入在个步骤上很久。
经过长时间的摸索我在连接这个问题上有一些体会,在这里跟大家分享一下我的经验,以供和我一样陷入困境及刚开始做克隆的同学参考。
关于连接的提问多集中在连接的体系、DNA的用量、vector和insert的比例、连接酶等。
但是我认为,连接不成功,问题并不一定出在连接这步上。
有很多环节影响连接的成败,如酶切的好不好、回收的质量好坏、连接时的浓度或比例、感受态等。
以下我详细说明。
1,PCR引物的设计。
通俗的不说,需要指出的是设计引物时一定要考虑切点的甲基化问题。
做普通的克隆会涉及到甲基化形式有两种:dam甲基化和dcm甲基化。
常用的大肠杆菌都有这两种甲基化酶。
dam甲基化酶识别GATC位点并甲基化;dcm甲基化酶识别CCWGG 位点(W是A或T)并甲基化。
如果有这两种位点那么多数情况内切酶是切不开了。
容易受甲基化影响的内切酶有:Dpn1(GA/TC)天生就甲基化;Cla1(A TC/GAT)如果前面加个G或后面加个C那么恭喜你,dam甲基化;Xba1(T/CTAGA)如果前面加个GA或后面加个TC也是dam甲基化,等等有好多。
这些容易甲基化的切点设计引物时一定要注意,避免引入甲基化位点。
如果真是避免不了或者后来才发现问题,那么把甲基化的质粒转化到甲基化酶缺陷型大肠杆菌中再提质粒就没有甲基化,可以切了。
甲基化缺陷型菌有:DM1(Invitrogen)、INV110(invitrogen)、JM110等。
2,PCR产物。
两种方式:一种纯化后直接酶切连接;一种连T载体再往下切连接。
我个人强烈建议第二种,连T载体。
因为PCR产物直接酶切我觉得有两个缺点:①由于两头把手太短,虽说有保护碱基,但我觉得还是不如从质粒上往下切好切,而且容易切坏、切碎;②无法从电泳上看出来切没切开,因为也就切下了几个十几个bp,带形没啥变化。
分子克隆3—在大肠杆菌中表达克隆化基因

在大肠杆菌中表达克隆化基因导言方案 1.用IPTG诱导启动子在大肠杆菌中表达克隆化基因方案 2.用T7噬菌体启动子在大肠杆菌中表达克隆化基因方案 3.用λ噬菌体P L启动子在大肠杆菌中表达克隆化基因方案 4.用碱性磷酸酶启动子(pho A)和信号序列在大肠杆菌中分泌表达外源蛋白------附加方案:phoA融合蛋白的亚细胞定位方案 5.谷胱甘肽琼脂糖亲和层析纯化融合蛋白方案 6.直链淀粉树脂亲和层析纯化麦芽糖结合融合蛋白方案7.用固化Ni2+吸收色谱纯化带有组氨酸标签的蛋白------替代方案:用pH递降的缓冲液从金属亲和柱洗脱多聚组氨酸标签蛋白------附加方案:NTA- Ni2+琼脂糖再生方案8.从包涵体中纯化表达蛋白------附加方案:重折叠从包涵体提取的溶解蛋白方案9.9信息栏克隆基因的表达大肠杆菌表达系统LacZ融合离液剂导言方案 1.用IPTG诱导启动子在大肠杆菌中表达克隆化基因(一)引言带异丙基-β-D硫代半乳糖苷(IPTG)诱导启动子的质粒表达外源蛋白的总量可以达到全菌蛋白的30%以上。
这些质粒非常适于实验室小规模操作,但IPTG价格昂贵,不能用于大规模制备外源蛋白。
(二)实验方案A.材料1.缓冲液和溶液a)考马斯亮蓝染色溶液或银染溶液b)IPTG (1mmol/L)c)1*SDS凝胶加样缓冲液d)10% SDS-PAGE2.载体和细菌菌株a)带有lacI q或lacI q1等位基因的适于转化的大肠杆菌菌株带有lacI q等位基因的IPTG诱导表达质粒可以转化实验室的任何大肠杆菌菌株(如JM1, DH5F’,TG1等)。
b)IPTG诱导的表达载体c)阳性对照质粒(表达已知大小的LacZ融合蛋白的IPTG诱导载体)3.培养基(如何配制?---分子克隆附录及科技出版社。
)a)含氨苄(50μg/ml)的LB琼脂平板b)含氨苄(50μg/ml)的LB培养基c)含氨苄(50μg/ml)的LB培养液4.专用设备a)沸水浴b)振荡培养箱5.细胞和组织6.附加试剂B.方法含重组表达载体的大肠杆菌菌株的构建1.PCR2.含3.重组质粒4.通过诱导靶蛋白表达的优化5.对照菌和重组菌分别挑取1~2个菌落,接入1ml含氨苄青霉素(50μg/ml)的LB培养液,适当温度(20~37℃)培养过夜(~16h)。
分子克隆的实验报告(3篇)

第1篇一、实验目的本实验旨在学习分子克隆技术的基本原理和操作步骤,掌握目的基因的扩增、克隆及表达,为后续相关研究奠定基础。
二、实验原理分子克隆技术是指将目的DNA片段从供体细胞中分离出来,通过体外重组、转化和转导等方法,将其插入到克隆载体中,再将其引入宿主细胞进行复制和扩增。
本实验采用无缝克隆技术,通过T5核酸外切酶、DNA聚合酶和DNA连接酶三种酶的共同作用,实现单片段或多片段与载体连接。
三、实验材料1. 试剂:限制性内切酶、DNA连接酶、T5核酸外切酶、DNA聚合酶、dNTPs、Taq DNA聚合酶、PCR引物、载体DNA、目的基因DNA、质粒提取试剂盒、琼脂糖凝胶电泳试剂盒等。
2. 仪器:PCR仪、凝胶成像仪、电泳仪、紫外灯、超净工作台、离心机、恒温水浴锅、移液器等。
四、实验步骤1. 目的基因扩增(1)设计引物:根据目的基因的序列设计特异性引物,引物长度一般在18-25bp,5'端添加限制酶切位点。
(2)PCR反应:配制PCR反应体系,加入引物、模板DNA、dNTPs、Taq DNA聚合酶等,进行PCR反应。
2. 载体线性化(1)酶切:使用限制性内切酶对载体DNA进行酶切,获得线性化的载体。
(2)去磷酸化:对单酶切得到的线性化载体进行去磷酸化处理。
3. 目的基因与载体连接(1)同源臂连接:将目的基因PCR产物和线性化载体进行同源臂连接,确保目的基因正确插入载体。
(2)连接反应:配制连接反应体系,加入目的基因PCR产物、线性化载体、DNA连接酶等,进行连接反应。
4. 转化与筛选(1)转化:将连接产物转化至宿主细胞中。
(2)筛选:通过抗生素筛选、酶切鉴定和测序等方法筛选出含有目的基因的克隆。
5. 目的基因表达(1)重组质粒提取:从筛选出的阳性克隆中提取重组质粒。
(2)重组质粒转化:将重组质粒转化至表达宿主细胞中。
(3)表达产物检测:通过Western blot、ELISA等方法检测目的蛋白的表达水平。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
需要合成多种序列的引物,彼此间只有 一个或几个碱基差异。这样的混合引物 称简并引物。
⑧套嵌引物(nested primers) 设计多组引物,结合位点依次位于前一组 引物之间。增加扩增产物的特异性。 1 3
4
引物设计现在多用专业软件
2
如Primer5.0等
4.序列比对。 序列比对包括部分完全相同序列查找和 序列相似性排列两类。与限制酶分析相似, 由于这类软件的算法没有多大变化,内核 大都是相同的,所以其结果输出和易用性 也成了更重要的标准。 推荐软件: BLAST 同类参考软件: GeneDoc 3.2,MagAlign 4.03(Lasergene Suite), Vector NTI Suite 6.0等
三、实验操作和数据分析阶段 本阶段由于各实验室的设备和方法的不同 差异太大,如若用到软件,请参考机器配 备之软件。但也有一些共同的要求和使用 的软件。 1.单向电泳条带定量分析。 推荐软件:BandScan 4.50 参考软件:band leader 3.0 2.据统计分析作图。可能在某些情况下需要 用到这类软件。由于专业的统计软件也很 多,比较出色。我们只推荐一个比较适合 生物的软件SigmaPlot 2000。
• 8.RNA二级结构预测及分析。 似乎人们在二级结构预测方面没有什么明 确的模型。因此,对这类软件只能以参考 参考的心态来使用了,不能报任何希望。 推荐软件:RNAdraw 1.1b2 参考软件:RNAStructure 3.5
• 9.蛋白结构分析。 不要指望软件能给你带来一个有三维结构 的蛋白,所谓的分析只能给你一个蛋白某 些片段有形成什么结构的趋向。结果判定 还是要靠人本身。
• 3.序列拼装。这是多基因方面用得比较多的 程序,在单基因的研究中有时也会用到。 推荐软件:SeqMan 4.03 (Lasergene Suite) 参考软件:Vector NTI Suite 6.0
(四)、文章写作和结果发表 本阶段主要是对所获得的结果整理,并发 表在合适的地方。 1.文献引用。 推荐软件:EndNote 5.0 参考软件:Reference Manager 9.0
即2.751011 bp的基因组中有一次完全 与19个核苷酸的序列相同的机会(或 机会是约2×10-11)。
一般引物设计为长15—30bp。
③引物的碱基序列 3‘端必须与模板正确配对,5’端可以不配对。
primer
3’GCTAGCTACTTAAG5’ 5’ATGCAGAAACTGATCGATCGATCGAT3’
目前应用的蛋白质结构预测的算法
1. 2. 3. 同源预测(一级结构决定高级结构) 结构与结构相对比(DALI算法) 当前最先进的结构预测方法: 结构类识别(fold recognition) 先建立一个已知的结构类数据库(fold library),将待测序列“穿过”该数据库构成的座 标,并根据事先确定的物理限制,逐个位置移动 (threading, sequence-structure alignment) ,并 一个函数(sequence-structure fitness alignment) 判断序列与结构类的符合程度,找出未知序列在 目标结构上的能量最优和构象最稳固的比对位置。 对计算机要求很高。
Taq
模板
ACGTACGTA 5’
(5)1个循环的结果
(4)新一轮循环开始 变性—复性—延伸。
理论上25-30次循环就可以合成225-230条DNA。
引物(primer) ①位置 与待扩增的模板DNA区段的两3’端序列 互补( 5‘端相同)的短DNA。
3’
3’
②引物的长度
理论计算:
19=2.751011。 4
手工计算:
Tm=(G+C)4 + (A+T)2 一般估计: 当引物中的(G+C)含量低于50%时, 复性温度低于55 oC。
实际复性温度选择低于Tm值5 oC。
适当提高复性温度可以提高引物与模板结 合的特异性,减少非特异产物的出现。
⑥引物的浓度
一般使用终浓度各0.5mol/L。
⑦简并引物
如果引物的序列是从基因产物蛋白质的 氨基酸序列反推出来的,就必须考虑密 码的简并性。
• 2.生成出版质量的图片。发表时,对用到的 分子结构的图片质量要求较高,直接从 RasMol等软件中截取的图片质量不够精美。 推荐软件:Pov-Ray for Windows 3.1 参考软件:molmol 2.5.1,mole 1.1.8 3.序列递交。结果中有新的序列需要递交到 各大数据库中必须符合各数据库的要求。 除了下面推荐的软件外,也可以通过写好 一种格式后,用格式转换软件转换为其他 种类格式,以向不同的数据库递交序列。 推荐软件:Sequin 3.32
PCR技术的原理
模板DNA变性
引物DNA复性
引物DNA延伸
(1的氢键断裂, DNA两条链分开成为单链。 5’ ATCTTGAAC 3’ TAGAACTTG 95oC
TGCATGCAT 3’ ACGTACGTA 5’
5’ ATCTTGAAC 3’ TAGAACTTG
template 5’端根据需要可设计成某个内切酶的切 点顺序、RNA聚合酶识别序列、突变位 点或生物素标记等,方便与以后操作。
④引物的碱基组成 1) 尽可能提高G+C含量,以提高引物与模 板的结合力 2) 避免连续相同碱基排列或内部回文序列.
5’GGCAGTCTGCCAGTCTAC3’
发卡结构 T CTGCCAGTCTAC3’ GACGG5’
6.结构域(motif)查找。 与限制酶的分析相似,这也是一个文本查 找的内容,关键在于以何种方式显示查询 结果。 推荐软件:Primer Premier 5.0 7.序列查找下载工具。 推荐软件:Vector NTI Suite 6.0; PubMed-Entrez Searching 参考软件:Network Entrez
• 10.克隆策略图谱。 几乎所有人都用手工或用绘图软件来画出 整个克隆过程,各种位点只能大概估计, 与核酸的序列之间的关系当然无从谈起。 现在终于有可以解决问题的软件登陆了。 推荐软件:Gene Construction Kit 2.0或 NoeClone(必须掌握其中的一个)
Gene Construction Kit 2.0 模拟电泳
2.制酶分析。 可能是最简单的功能了,用普通的文本 搜索也能找出相应的序列位点。正因如此, 我们希望软件在结果输出上有更完美的表 现。另外几乎所有的软件都没有考虑在酶 切位点前应该有保护碱基,所以该分析通 过,并不能在实验中总能通过。 推荐软件: Vector NTI Suite 6.0 同类参考 软件:DNAssist 1.0 ,Primer Premier 5.0,webcutter 2.0(在线
图1.4 重组质粒pTRE-p53的限制性酶切分析与预测的模式比较。 Fig.1.4 Predicted Gel Electrophoresis Patterns can be Used to Analyze Real Gels. Banding pattern in lane 3-8 math lane 2,3 and not lane 4,5 indicating antisense clones of pTER-p53. Lane 8-10 correspond to lane 4,5; Lane 1 to lane 1.
3)避免形成引物二聚体(dimer) 两个引物之间不能有两个以上的连续 碱基序列互补。 primer1 5’GGTCTGCCAGTCTAC3’
3’CAGGACTTAGTCACT5’ primer2 Sense primer Upstream primer Forward primer Primer1 vs vs vs vs Antisense primer Downstream primer Reverse primer Primer2
模板
TGCATGCAT 3’ ACGTACGTA 5’
模板(template)
(2)DNA模板与引物复性
引物(primer):
40-65oC
人工合成的单链DNA小片断,碱基顺序分别 与所要扩增的模板DNA双链的3‘端互补。 模板
5’ ATCTTGAAC 引物
5’ ATCTTGAAC 3’ 3’ TAGAACTTG
• 12.翻译/ORF Finding 推荐软件:Gene Construction Kit 2.0 参考软件:Primer Premier 5.0,Vector NTI Suite 6.0
• 13.格式转换。 各种软件为了自己软件的需要有不同的格 式,对我们使用数据带来了困难。格式转 换软件可以将不同格式数据转换以方便使 用。 推荐软件:Seqverter 1.3 参考软件:Visual Sequence Editor 1.1, Genestudio
• 5.质粒作图。 这也是最常用的序列显示功能。我们要求 软件能对已知序列自动作图,对未知序列 但关键部位位置清楚的质粒也能作图。主 要是标示各种酶切位点、基因序列、特定 结构域序列。 推荐软件:Gene Construction Kit 2.0 参考软件:Winplas 2.6 ,pDRAW32 1.0.33等
The pTRE-p53 Cloning Project
Plasmid
Gain p53 cDNA fragment from pC53SN3( BamH I digestion ) and clone in each orientation into the BamH I site in pTRE. This gives the two constructs below.
二、序列分析、实验设计模拟阶段
1.综合性软件。 这类软件要求对本阶段上述的大部分 功能均具备,而且这类软件在一些专项分 析上仍有绝对优势。 推荐软件:DNASTAR 4.03 同类参考软件:Vector NTI Suite 6.0,Omiga 2.0、DNASIS 2.5,DNATools 5.1