用DiscoverStuido所提供的蛋白质三级结构预测之同源模拟
蛋白质三级结构预测(swiss-model同源建模)
利用同源建模预测蛋白质的三级结构首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。
我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行!对于一个未知结构的蛋白质,白质建立结构模型。
那么,我们首先要做的就是找到和我们空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。
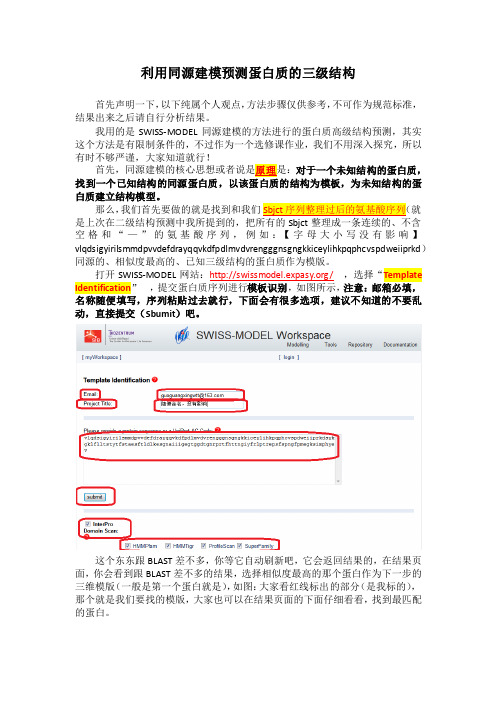
打开SWISS-MODEL网站:/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。
这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。
接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。
找到以后,以FASTA格式显示。
接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择:Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (/sprot )编目号(accession)可以直接通过web界面提交。
discoverystudio 蛋白功能域

discoverystudio 蛋白功能域什么是蛋白功能域,蛋白功能域的发现过程是怎样的,它有哪些重要的应用领域。
一、蛋白功能域是什么蛋白功能域是指蛋白质分子中具有特定生物学功能的结构模块。
蛋白质通常由一个或多个功能域组成,这些功能域负责不同的生物学活动,如结合特定分子、催化化学反应、信号传导等。
蛋白功能域通常具有保守的序列和结构特征,多个蛋白质可能共享相似或相同的功能域。
通过研究蛋白质的功能域可以深入了解蛋白质的结构、功能和进化。
二、蛋白功能域的发现过程1. 蛋白质序列分析首先,研究人员使用生物信息学方法对蛋白质序列进行分析。
他们可以使用数据库中已知功能域的序列模板进行比对,从而识别出目标蛋白质中的功能域。
2. 结构域预测基于蛋白质序列的结构域预测方法能够预测蛋白质中存在的结构域。
其中一种常用的方法是利用多序列比对的结果,寻找序列中的保守区域,并将其作为存在结构域的证据。
3. 结构域数据库挖掘研究人员还可以通过结构域数据库的挖掘来发现新的功能域。
他们可以将已知的结构域序列比对到这些数据库中,从而鉴定相似结构域。
4. 结构域组合预测鉴定出蛋白质中存在的单个结构域后,研究人员可以进一步预测不同结构域的组合方式,以探究蛋白质的功能。
三、蛋白功能域的重要应用1. 药物研发蛋白功能域在药物研发中具有重要作用。
研究人员通过研究蛋白质的功能域,可以了解药物与目标蛋白质之间的相互作用机制,进而设计和开发针对特定功能域的药物。
2. 生物技术领域蛋白功能域的研究对于生物技术领域的开发具有重要意义。
例如,利用功能域的特异性结合能力,可以开发出高效的抗体、酶、激素等蛋白质工具。
此外,将功能域与其他生物分子结合,还可以构建具有特定功能的蛋白质纳米结构。
3. 基因组学和蛋白质组学研究人员利用蛋白功能域的信息对基因组和蛋白质组进行分析和注释,以了解不同蛋白质的功能和相互作用网络。
这有助于揭示生物系统中蛋白质的组成和功能,进一步理解生物学过程。
5 蛋白质三级结构预测

• 令代表核心折叠C中的环到序列S中空位的 映射,显然是通过线索化而确定的。
令f(t)是进行比对的得分函数,其定义如下:
f(t) = g1 (v,t) + g2 (u,v,t) + g3 (,t)
• g1 (v,t) 评价氨基酸残基v所处的位置 • g2 (u,v,t) 评价残基u和v的相对位置,如果u和v 键合,则得 分高; • g3肖 飞
蛋白质三级结构预测的方法
1
2 3
方法比较
同源建模(比较建模)
基础 - 相似的序列结构相近 - PDB结构数据库的快速增长 - 结构基因组学的启动 - 发散进化 特点 - 相对精确可靠
• 假设待预测三维结构的目标蛋白质为U (Unknown),利用同源模型化方法建立结 构模型的过程包括下述6个步骤: (1)搜索结构模型的模板(T) (2)序列比对 U T (3)建立骨架 (4)构建目标蛋白质的侧链 (5)构建目标蛋白质的环区 (6)优化模型
至于最后建立三维结构模型则是非常困难的
• 线索化的主要思想: 利用氨基酸的结构倾向(如形成二级结构 的倾向、疏水性、极性等),评价一个序 列所对应的结构是否能够适配到一个给定 的结构环境中。
• 建立序列到结构的线索的过程称为线索化, 线索技术又称折叠识别技术。 • 线索化或者折叠识别的目标是为目标蛋白质 U寻找合适的蛋白质模板,这些模板蛋白质 与U没有显著的序列相似性,但却是远程同 源的。
新的趋势 混合预测方法 在比较建模法和折叠识别法中使用从头预 测法来预测部分难以找到模板的片断 在从头预测法中使用二级结构预测的结果 和其他已知结构信息辅助建模
• Meta-predictor 使用多个预测方法 对收集的结果进行综合比较和分析 改进收集的结果
蛋白质结构预测和模拟方法

蛋白质结构预测和模拟方法蛋白质是生物体内的重要组成部分,对生命活动具有关键作用。
在了解蛋白质功能和相互作用等方面的研究中,蛋白质结构的预测和模拟方法发挥着重要的作用。
本文将介绍蛋白质结构预测的主要方法和蛋白质结构模拟的常见方法。
1. 蛋白质结构预测方法1.1 基于序列的预测方法基于序列的预测方法是根据蛋白质的氨基酸序列推测其结构。
这一方法通过将目标蛋白质的序列与已知结构的蛋白质序列进行比对,从而预测目标蛋白质的结构。
具体方法包括序列比对、蛋白质家族数据库搜索以及机器学习等等。
1.2 基于结构模板的预测方法基于结构模板的预测方法是根据已知结构的蛋白质来预测目标蛋白质的结构。
这一方法通过找到与目标蛋白质具有相似结构的蛋白质,从而预测目标蛋白质的结构。
具体方法包括结构比对、结构模板库搜索以及融合多个结构模板等等。
1.3 基于物理力学的预测方法基于物理力学的预测方法是利用物理力学原理来预测蛋白质的结构。
这一方法通过模拟蛋白质分子内的原子间相互作用,从而预测蛋白质的结构。
具体方法包括分子力学、蒙特卡洛模拟以及分子动力学模拟等等。
2. 蛋白质结构模拟方法2.1 分子力学模拟分子力学模拟是通过计算蛋白质分子内原子之间的相互作用力,来模拟蛋白质的结构和动力学性质。
这一方法可以对蛋白质进行模拟,从而获得与实验结果相一致的结构信息。
2.2 蒙特卡洛模拟蒙特卡洛模拟是通过引入随机性的方法来模拟蛋白质分子的运动和结构。
这一方法通常基于能量最小化原则,通过随机调整蛋白质的构象从而获得可能的结构。
2.3 分子动力学模拟分子动力学模拟是通过数值计算方法,模拟蛋白质分子静态和动态特性的一种方法。
这一方法可以模拟蛋白质的结构和动力学性质,并研究蛋白质在时间和空间尺度上的变化。
3. 蛋白质结构预测和模拟的应用蛋白质结构预测和模拟的方法在生物科学研究中发挥着重要的作用。
首先,它们可以帮助科学家深入了解蛋白质的结构与功能之间的关系。
其次,蛋白质结构预测和模拟方法还可以用于研究蛋白质的折叠机制、稳定性以及相互作用等。
Discovery Studio 2.5操作教程

蛋白质结构预测技术简介简介蛋白质结构的解析对其功能的理解至关重要。
然而,由于技术手段的限制,利用实验方法(主要为X-ray,NMR)解析蛋白质结构投入大、周期长、风险大。
对于某些膜蛋白,只利用现有技术条件,其结构甚至无法解析。
另一方面,随着分子生物学技术的成熟及高通量测序技术的发展,越来越多的基因序列可以轻松被找到。
这造成了现代蛋白质科学中一个奇怪的现象:蛋白质序列数据的累积量及积累速度远远超过蛋白质结构。
这种序列与结构间不平衡的现象极大地限制了我们对蛋白质功能及其相关作用机理的理解。
所以我们需要一种能够简单、快速且相对准确的技术来确定蛋白质的空间结构。
蛋白质建模技术可以很好的解决上面的问题。
该方法利用信息技术的手段,可以直接从蛋白的一级结构(氨基酸序列)预测蛋白质的高级结构(主要为三级结构)。
根据最新一届国际建模大赛(CASP)的分类,目前主要的蛋白质建模方法包括两种:基于模板的建模(Template-based Modeling)和自由建模(Free Modeling)。
前者又包括两种方法:同源建模法(Homology Modeling)和“穿线法”(Threading)。
后者主要以从头计算法(ab initio)为主。
所有的建模方法中,以同源建模法(Homology Modeling)使用最为广泛,预测结果的准确性最大。
同源建模的理论基础为蛋白质三级结构的保守性远远超过一级序列的保守性。
因此,人们可以通过使用一个或多个已知结构的蛋白(模板蛋白,template)来构建未知结构蛋白(目标蛋白,target)的空间结构。
其主要的步骤包括:1.搜索用于建模的template(s)2.将target与templates进行比较3.将步骤(2)中的比较信息用于建模Discovery Studio为用户提供了一整套利用Homology Modeling方法自动预测蛋白质空间结构的工具。
用户只需要提供蛋白质的氨基酸序列就可以轻松完成模型构建及模型可信度评估的工作。
Discovery Studio官方教程--预测蛋白质聚集、水溶性、粘度、可开发性等性质
预测蛋白聚集位点(Protein Aggregation)教程介绍抗体等具有治疗功能的蛋白,如果处于比较高的浓度下,会有发生聚集的趋势。
这会导致抗体的活性下降,并引起免疫反应。
抗体的聚集趋势计算是一种衡量蛋白表面氨基酸聚集倾向性的指标。
具有比较高的聚集趋势得分的位点表明了该区域的氨基酸倾向于发生聚集。
因此这些位点的预测,使得我们可以通过氨基酸定点突变的方法来改造蛋白,增强其稳定性。
本教程使用Calculate Aggregation Scores对一个全长IgG1抗体分子(PDB号为1h2h)进行蛋白聚集位点的预测计算,并分析预测的结果。
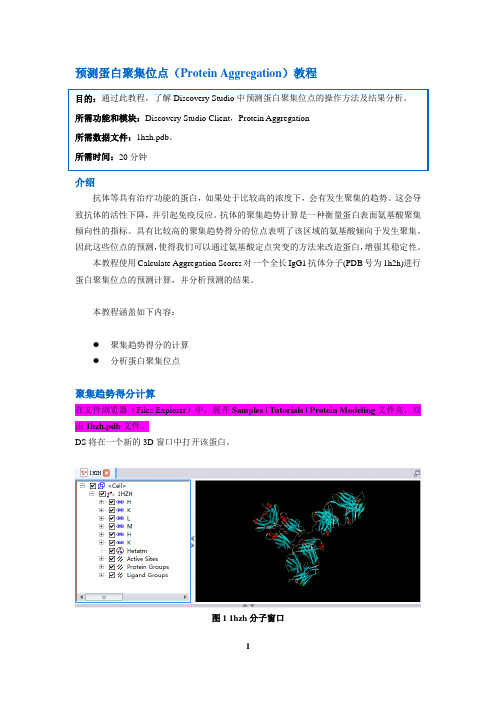
本教程涵盖如下内容:●聚集趋势得分的计算●分析蛋白聚集位点聚集趋势得分计算在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Protein Modeling文件夹,双击1hzh.pdb文件。
DS将在一个新的3D窗口中打开该蛋白。
图1 1hzh分子窗口Ctrl+H打开Hierarchy窗口,然后选中Water,点击Delete删除蛋白结构中的结晶水分子。
在工具浏览器(Tools Explorer)中,展开Simulation | Change Forcefield,将Forcefield设为CHARMm Polar H,然后点击Apply Forcefield,这将为蛋白赋上CHARMm Polar H力场。
图2 Apply Forcefield设置界面在工具浏览器(Tools Explorer)中,展开Macromolecules | Predict Protein Aggregation工具面板,点击Calculate Aggregation Scores。
在弹出的参数设置界面中,将Input Typed Protein设为1hzh:1HZH,将Cutoff Radius设为5,7,10。
点击Run运行该任务。
该任务在奔腾4,2Gb内存和2.8GHz的计算机平台上运行大约需要3分钟。
蛋白质三级结构预测方法简述

文章编号 -7223TMYYJ +1223"23T22K[T21
T!"T
蛋白质三级结构预测方法简述
张 漫 常延琦 7" +天津市兽药监察所 天津
K22Y21"
结构 ’ 进行目标蛋白质的序列与模 板序列的比对 ’ 根据模板蛋白质的 三维结构构建目标 蛋 白 质 的 结 构 ’ 对模建结构进行能量优化 * 分子动 力学优化及可信度评估 ( 首先需要通过序列比较方法在 序列库和结构库中搜索与目标蛋白 质相似性足够高的蛋白质作为模 板 ( 比如可使用 8&# ,N,+&0 到结 构库 8ON +8?;@CAF O>@> N>FP" 中 去 搜索 ’ 也可以通过序列结构穿线法
CG/(’N<* 举 行 的 HC?> 大 会 上 对 所
! " 中国农业大学生物学院 北京 !#$$%&
!+&8( 1 同源模建9:;<=>?>@ABC <;DCEAFG 6
同源模建即比较模建 ’ 是目前在 蛋白质结构预测中最为成功的方 法 ( 应用同源模建的方法进行蛋白 质结构预测 ’ 需要目标蛋白质至少 有一个已知三维结构的同源蛋白 质 ’ 即两者之间要有较高的相似性 ( 同源模建的准确性取决于目标序列 和模板之间的相似性大小 ’ 同时也 与两个蛋白质结构和功能的相似性 相关 ( 如果目标蛋白质与模板序列 的相似性大于 32H ’ 则可以得到 高 精度的同源模建结果 ’ 即主链的均 方根 +*.&" 只有 2I7 F< 左右 ’ 可 以 与中等分辨率的核磁共振或低分辨 率 J, 射 线 衍 射 得 到 的 结 果 相 媲 美 ( 如果相似性界于 K2HL32H ’ 则 可以得到中等精度的结果 ’ 此时约
Discovery Studio官方教程--基于MODELER构建蛋白酶模型

基于蛋白序列构建同源蛋白模型(MODELER)教程介绍蛋白质结构的解析对其功能的理解至关重要。
然而,由于技术手段的限制,利用实验方法(主要为X-ray,NMR)解析蛋白质结构投入大、周期长、风险大。
对于某些膜蛋白,只利用现有技术条件,其结构甚至无法解析。
另一方面,随着分子生物学技术的成熟及高通量测序技术的发展,越来越多的基因序列可以被轻松找到。
这造成了现代蛋白质科学中一个奇怪的现象:蛋白质序列数据的累积量及积累速度远远超过蛋白质结构的解析。
这种序列与结构间不平衡的现象极大地限制了我们对蛋白质功能及其相关作用机理的理解。
因此,我们需要一种能够简单、快速且相对准确的技术来预测蛋白质的空间结构。
同源建模技术可以很好的解决上面的问题。
该方法利用信息技术的手段,可以直接从蛋白的一级结构(氨基酸序列)预测蛋白质的高级结构(主要为三级结构)。
根据最新一届国际建模大赛(CASP)的分类,目前主要的蛋白质建模方法包括两种:基于模板的建模(Template-based Modeling)和自由建模(Free Modeling)。
前者又包括两种方法:同源建模法(Homology Modeling)和“穿线法”(Threading)。
后者主要以从头计算法(ab initio)为主。
所有的建模方法中,以同源建模法(Homology Modeling)使用最为广泛,预测结果的准确性最为可靠。
同源建模的理论基础为蛋白质三级结构的保守性远远超过一级序列的保守性。
因此,人们可以通过使用一个或多个已知结构的蛋白(模板蛋白,template)来构建未知结构蛋白(目标蛋白,target)的空间结构。
Discovery Studio为用户提供了一整套利用Homology Modeling方法自动预测蛋白质空间结构的工具。
用户只需要提供蛋白质的氨基酸序列就可以轻松完成同源模型的构建及模型可信度评估的工作。
DS的Homology Modeling主要基于MODELER程序。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(一)摘要:利用Discover Stuido所提供的蛋白質三級結構預測之同源模擬(Homology Modeling)的方法,可以很快速及有效率的獲得離子通道蛋白之三維結構,再深入探討分析其結構所提供的資料,進一步了解離子通道蛋白的功能,可用來指引生物實驗,例如site - directed mutagenesis、rational drug design 和protein - protein interaction 等等。
模擬之後的結構再透過Discover Stuido所提供之LigandFit、LibDock 以及CDOCKER等計算方法,進行藥物與通道蛋白的對接,再經過Discover Stuido生物軟體計算預測藥物可能在通道上可能的結合位置以及可能相互作用的關鍵胺基酸。
(二)研究動機:細胞膜上的離子通道是所有生命體的基本要件,很多疾病,例如一些神經系統疾病和心血管疾病就是由於細胞上的離子通道功能發生紊亂或蛋白結構的缺陷所造成。
例如本實驗室研究項目之ㄧ的離子通道「NMDA受器」,在哺乳類動物中樞神經系統中扮演重要角色,從興奮性突觸的訊息傳遞、突觸的可塑性、到學習與記憶的整合此通道受器都參予其中,許多神經性疾病如急性腦中風或慢性的阿茲海默症、帕金森氏症、精神分裂症、癲癇等等都認為和NMDA受器有關,因此增加對它們的認識是幫助了解許多疾病狀態的重要基礎,目前許多離子通道已成為製藥界開發新藥的目標,離子通道的研究還有非常大的潛力和應用空間。
由於生物資訊近幾年來發展迅速,尤其基因體計劃的進行更增加了資料庫中的數量,包括核酸、蛋白質及結構等資料庫。
一般說來,蛋白質三維結構主要以實驗方法來決定,例如X-射線繞射法或NMR光譜法。
事實上,技術上的困難使得蛋白質三維結構決定的速度相當地緩慢,因此發展出利用電腦並依據蛋白質的序列來預測其三級結構的方法。
這些方法包含以物理化學原理的ab-initio methods 及以資料庫提供的序列和結構知識衍生的蛋白質摺疊認識fold- recognition methods(亦稱threading 穿針引線法),和同源模擬法(homology modeling)。
我們想透過Discover Stuido中的homology modeling方法來預測離子通道蛋白經胺基酸點突變之後可能的構型變化,以及預測相關藥物與通道蛋白之間可能的作用位置,並且可以有效率的篩選出可能作用的關鍵胺基酸,再透過電生理的方式來探測藥物對離子通道的行為模式,有助於進一步且有效率的了解離子通道可能的分子機制。
(三)研究方法與步驟:同源模擬法五個主要的步驟:(1)資料庫搜尋及選擇模版(templates)(2)多重序列排列(multiple sequence alignment)(3)骨架(framework)的建構,環狀結構(loop)模擬及側鏈(side-chain)模擬(4)能量最小化(energy minimization)(5)結構合理性評估Homology Modeling的第一個步驟是搜尋蛋白質及結構資料庫,例如Swiss-Prot或PDB,期望能在與此未知結構蛋白質(target)有序列相關的蛋白質中,獲得一個或一個以上具有三維結構的蛋白質作為模版(template)。
模版的首要條件是其蛋白質序列與未知結構蛋白質序列之間的一致性必須大於30%,相似度越高,模擬出來的準確性也越高。
假如可作為模版的結構很多時,則從這些結構中選擇出更適當的結構作為模版,來提高新結構的準確性。
接著透過Discover Stuido生物軟體的運算即可模擬出可能的蛋白質結構,最初的結構必須進行模型修飾(model refinement)的過程,藉由分子力學(molecular mechanics)的能量最小化來修正不利的非共價碰觸及達到理想的鍵結幾何和能量最低的狀態(configuration)。
能量最小化是一連串的計算,包括鍵長、鍵角、二面角、靜電作用力(electrostatics)和凡得瓦力作用力等位能參數(E total = E stretching+ E bending+ E dihedral+ E out-of-plane+ E cross terms+ E VdW+ E coulombic)。
能量最小化常用的力場CHARMM,AMBER,CVFF,CFF91或GROMOS 等,Discover Stuido主要是利用CHARMM來計算最初的結構的能量,再調整結構中上述的各項參數直到最低總能量。
當能量最佳化之後,要進行結構合理性評估,包括stereochemistry、energy profile、residue environment、structure similarity 等等。
a. stereochemistry:運用PROCHECK,AQUA或SQUID等方法評估結構是否符合的一般的常規,例如鍵長,鍵角,主鏈二面角,側鏈χ1 二面角,和非共價碰觸等,以及包裝、溶劑可接觸性、厭水性及親水性胺基酸分佈、主鏈氫鍵等結構上的特性。
b. energy profile:ProsaII 或VERIFY3D 是根據三維輪廓(3D profile)和平均力的統計位能來計算結構的能量分佈。
c. residue environment:Profiles-3D 程式是以演算法將三維結構還原為一維的表示方式來計算胺基酸序列和三維結構之間的相容性,可用來評估新模型與模版中每一個胺基酸的環境狀態是否相同。
d. structure similarity:首先判斷新結構是否具正確的摺疊,通常新結構在幾何上(geometrical)會比較接近模版結構。
計算r.m.s.d.的數值常用於結構相似性的比較,亦即將新結構與模版結構重疊後,計算Cαatoms 的距離求出r.m.s.d.的數值,r.m.s.d.越低則結構越相似。
最後,分析新模型與模版的結構和參考文獻所得的資訊,進一步地修正新結構,來增加其合理性及可信度。
當離子通道蛋白完成結構最佳化之後,先採用Discover Stuido所提供之 Ligand Fit 計算方式,掃描表面的結構觀察蛋白質可能的凹陷區塊,再將可能的凹陷區塊與藥物分子進行對接,再透過LibDock 及 CDOCKER 做精準的計算,以求得更精確的作用模式。
(四)預期結果:透過同源模擬法以及能量最小化的方式,我們將會模擬出研究中未知構型的離子通道蛋白,再加入小分子藥物進行對接,計算出各種自由能的評估數據,篩選出最合理的交互作用,透過電生理的功能性分析相互佐證。
圖一:利用Homology Modeling模擬的結果圖二:利用圖一模擬之後的結果,加入藥物進行對接,並於圖中顯示出與藥物作用的關鍵胺基酸(五)參考文獻:1.Fiser A, Giian D R and Šali A. Modeling of loops in protein structures.Protein Sci., 2000, 9, 1753-1773.2.Al-Lazikani B, Jung J, Xiang Z and Honig B. Protein structureprediction. Curr.Opin. in Chem. Biol., 2001, 5, 51-56.3.Guex N, Diemand A and Peitsch M C. Protein modelling for all. TIBS,1999, 24, 364-367.4.Sippl M J. Recognition of errors in three-dimensional structures ofproteins. Proteins, 1993, 17, 355-362.5.Luthy R, Bowie J U and Eisenberg D. Assessment of protein modelswith three-dimensional profiles. Nature, 1992, 356, 83-85.6.Allen FH. 3D search and research using the Cambridge structuraldatabase. Chem. Des. Autom. News, 1993, 8, 31–37.7.Barton, GJ. ALSCRIPT: a tool to format multiple sequence alignments.Protein Eng, 1993, 6, 37–40.8.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H,Shindyalov IN and Bourne PE. The protein data bank. Nucleic Acids Res (Online), 2000, 28, 235–242.9.Johnson MS and Lehtonen JV. Comparison of protein three-dimensional structures. Bioinformatics, Sequence, Structure andDatabanks, 2000, 15–50.10.Johnson, MS, May ACW, Rodionov, M.A. and Overington JP.Discrimination of common protein folds: application of proteinstructure to sequence/structure comparisons. Methods Enzymol,1996, 266, 575-598.11.孫慶姝. Homology Modeling(Knowledge - Based StructureModeling).中央研究院生物醫學科學研究所鄒文雄與黃明經蛋白質結構之電腦預測Chemistry (The Chinese Chem. Soc. Taipei), 1997, 55, 101-109.。