生物信息学-课堂练习生物信息学蛋白质序列分析-课堂练习
生物信息学-课堂练习作业生物信息学蛋白质序列分析-课堂练习
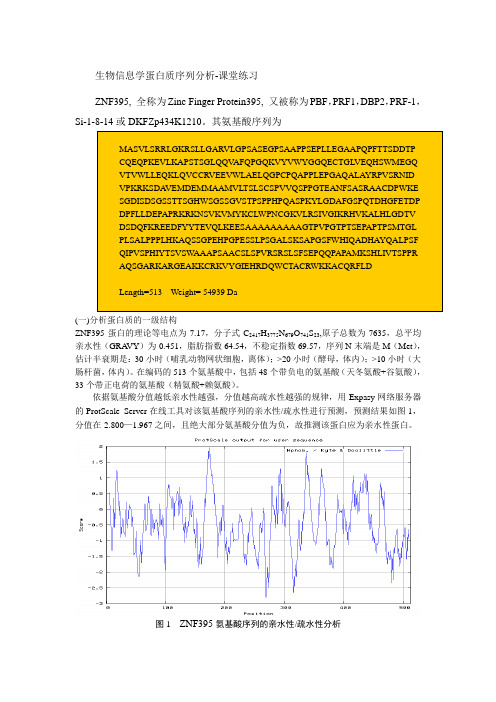
生物信息学蛋白质序列分析-课堂练习ZNF395, 全称为Zinc Finger Protein395, 又被称为PBF ,PRF1,DBP2,PRF-1,Si-1-8-14或DKFZp434K1210。
其氨基酸序列为(一)分析蛋白质的一级结构ZNF395蛋白的理论等电点为7.17,分子式C 2417H 3775N 679O 741S 23,原子总数为7635,总平均亲水性(GRA VY )为-0.451,脂肪指数64.54,不稳定指数69.57,序列N 末端是M (Met ),估计半衰期是:30小时(哺乳动物网状细胞,离体);>20小时(酵母,体内);>10小时(大肠杆菌,体内)。
在编码的513个氨基酸中,包括48个带负电的氨基酸(天冬氨酸+谷氨酸),33个带正电荷的氨基酸(精氨酸+赖氨酸)。
依据氨基酸分值越低亲水性越强,分值越高疏水性越强的规律,用Expasy 网络服务器的ProtScale Server 在线工具对该氨基酸序列的亲水性/疏水性进行预测,预测结果如图1,分值在-2.800—1.967之间,且绝大部分氨基酸分值为负,故推测该蛋白应为亲水性蛋白。
图1 ZNF395氨基酸序列的亲水性/疏水性分析(二)分析蛋白质的二级结构利用SOPMA在线工具对二级结构进行预测,如图2,α螺旋99个占19.30%,延伸链66个占12.87%,β-转角18个占3.51%,无规卷曲330个占64.33%,其二级结构主要由无规卷曲组成。
图2 ZNF395蛋白二级结构预测注:蓝色表示α螺旋;红色表示延伸链;紫色表示无规则卷曲(三)分析膜蛋白质利用在线分析工具TMHMM Server 2.0,对ZNF395氨基酸跨膜结构域进行在线预测和分析,结果表明,该序列编码的蛋白非跨膜蛋白(见图3)。
利用Signal P 3.0 Server在线预测工具对ZNF395蛋白质进行信号肽预测,无信号肽存在(图4)。
生物信息学课后习题

绪论1、生物信息学的概念及其组成部分生物信息学(Bioinformatics):是一门交叉学科,包含了生物信息的获取、处理、储存、分析、解释和应用在内的所有方面,它综合运用了生物学、计算机科学和数学等多方面的知识和方法,来阐述和理解大量生物学数据所包含的生物学意义,并应用于解决生命科学研究和生物技术相关产业中的各种问题。
生物信息学的三个组成部分:①建立可以存放和管理大量生物信息学数据的数据库②研究开发可用于有效分析与挖掘生物学数据的方法、算法和软件工具③使用这些工具去分析和解释不同类型的生物学数据2、生物信息学的主要研究领域①生物数据的建立与搜索②序列比较与相似性搜索③基因组结构注释④蛋白质结构与功能的预测⑤基因组数据分析⑥比较基因组合系统发生遗传学分析⑦功能基因组和蛋白质组学数据分析⑧信号传导、代谢和基因调节途径的构建与描述3、初级数据库二级数据库的概念说出几个数据并说明包含什么数据一级数据库(primary database):数据直接来源于实验获得原始数据,只经过简单的归类、整理和注释。
例如GenBank、EMBL、DDBJ、SWISSPORT、PDB二级数据库(secondary database):在一级数据库、实验数据和理解分析的基础上针对特定的目标衍生而来,是对生物学知识和信息的进一步整理。
例如human genome databases GDB转录因子数据库等4、简述核酸序列的测序①DNA测序一般原理DNA测序一般采用全自动的荧光标记链终止反应完成,该法利用了DNA聚合酶能从脱氧核糖核苷酸(dNTP)延伸但不能从双脱氧核糖核苷酸(ddNTP)延伸的特性,通过加入限量的荧光标记过的双脱氧核苷酸来产生有特定终止碱基的嵌套DNA片段,然后通过聚丙烯酰胺凝胶电泳(PAGE)分离并通过扫描仪读取序列(300-800bp)②基因组测序策略—分而治之---shortgun因为测序反应每次只能测300-800bp故先将基因组分割成一定大小的片段,然后对这些片段分别测序,测完后再将这些片段拼接起来—鸟枪法(shortgun)③一次性测序例如:表达序列标签(EST)是其中的代表,它对随机挑选的cDNA克隆进行两端一次测序得到300-500bp的片段,代表cDNA的一部分。
生物信息学习题

一:名词解释1.生物信息学2.NCBI3.PubMed4.生物芯片5.BLAST6.UniProt7.电子克隆8.EMBL二:填空题1.基因芯片可以分为2. 人类基因组全序列分析分两大步骤即制图和测序,并最终绘制出四张图谱:3. 分子系统发生分析主要分为三个步骤即4. 国际上最主要的三大核酸序列数据库分别是5. 蛋白质得分矩阵有7. 文献是掌握科研进展的最直接方式,目前由NCBI维护的大型文献资源是。
3. 用于核酸序列比对中常见的三种得分矩阵,分别为4. 根据生物芯片探针分子类型的不同,可以将生物芯片哪三种,5. 核酸序列分析所获得的信息主要有(举例说明四个)6. 限制性酶切分析是分子生物学实验中的日常工作之一,这方面最好的限制酶数据库是三:选择题1、如果试图确定一个新蛋白质序列属于哪一个蛋白质家族,或该序列可能包含何种结构域或功能位点,应使用:()A: PROSITE数据库 B: DDBJ数据库C: PIR数据库 D: PDB数据库2、构建序列进化树的一般步骤不包括:()A:建立DNA文库 B:建立数据模型 C:建立取代模型 D:建立进化树3、BLAST教案所程序中,哪个方法是不存在的?()A:BLASTP B:BLASTN C:BLASTX D:BLASTQ4. 以下常见的几个物种,哪一个目前还没有完成全基因组测序:()A: 茶树 B: 玉米 C: 水稻 D: 小鼠5、向核酸序列数据库(GenBank/EMBL/DDBJ)提交数据,应该使用下面哪个软件:()。
A: Blast B:Sequin C:SRS D:Swiss-Model6、在蛋白质序列数据库中比较查询手头未知的蛋白质序列,应使用Blast中哪个具体的算法:()。
A:BLASTX B:tBLASTN C:BLASTP D:BLASTN7、下列中属于一级蛋白质结构数据库的是:()A:EMBL B:DDBJ C:PDB D:SWISS-PROT8、下面不属于SWISS-PROT蛋白质数据库的注释范畴的是:()A: 与其它蛋白质的相似性 B: 蛋白质的二级结构C: 由于缺乏该蛋白质而引起的疾病 D: 核酸的功能描述9、下列属于蛋白质二级结构预测的软件程序是()A: BLASTX B:SOPMA C:DNAstar D:GO10. 如果做DNA结构分析,应该考虑用下面哪个数据库:()A:GenBank B: PIR C:NDB D:UniProt四:简单题1.简述Entrez的设计概念和使用方法?2. 简述生物大分子PDB存储的生物分子种类和数据结构特点?3.简述生物信息学的研究意义?4 简述蛋白质序列分析的基本内容以及常用的软件?5. 简述Swiss-Prot的数据结构?6、简述序列多重比对的意义?7、简述生物信息学的发展历史?五:论述题1.论述蛋白质相互作用研究的意义,传统的实验方法和计算预测方法的应用?2.论述后基因组时代生物信息学面临的挑战和研究策略?3.论述生物信息学的应用?4. 论述如何利用基因芯片数据做聚类分析。
2-蛋白质序列特征分析-生物信息学

TMPRED在线网页
生命科学学院
用TMPRED分析P51684序列所得到生的命可科能学学院 的7个跨膜螺旋区
生命科学学院
用TMPRED分析P51684序列所得到的7个可 能的跨膜螺旋区的相关性列表
含有卷曲螺旋结构最知名的蛋白质有原癌蛋白 (oncoprotein)c-fos和jun,以及原肌球蛋白 (tropomyosin)。
生命科学学院
利用COILS分析蛋白质的卷曲螺旋
COILS是由Swiss EMBNet维护的预测卷曲螺旋的在 线工具,该软件是基于Lupas算法,将查询序列在一个由 已知包含卷曲螺旋蛋白结构的数据库中进行搜索,同时也 将查询序列与包含球状蛋白序列的PDB次级库进行比较, 并根据两个库搜索得分决定查询序列形成卷曲螺旋的概率。 COILS也可以下载到本地进行运算。
生命科学学院
序列特征分析
Analysis of Sequence Characterristics
一、蛋白质结构 蛋白质的一级结构
生命科学学院
蛋白质的一级结构决定二级结构 蛋白质的二级结构决定三级结构
蛋白质的二级结构
生命科学学院
H表示螺旋 E表示折叠 B表示β桥 G表示3-螺旋 I表示π螺旋 T表示氢键转角 S代表转向
或者全部由碳原子和氢原子组成,因此这类氨基酸不太可 能与水分子形成氢键; 2. 极性氨基酸(polar amino acid),其测链通常由氧原子或 氮原子组成,它们比较容易与水分子形成氢键,因此也称 为亲水氨基酸; 3. 带电氨基酸(charged amino acids),这类氨基酸在生物 pH环境中带有正电或负电。
生命科学学院
生物信息学习题

第六章 分子系统发生分析(问题与练习)
1、构建系统发生树,应使用
A、BLAST
B、FASTA
C、UPGMA
D、Entrez
2、构建系统树的主要方法有
、
、
等。
3、根据生物分子数据进行系统发生分析有哪些优点?
4、在 5 个分类单元所形成的所有可能的有根系统发生树中,随机抽取一棵树是反映真实关
系的树的可能性是多少?从这些分类单元所有可能的无根系统发生树中,随机选择一棵
库
8、TreeBASE 系统主要用于
A、发现新基因 B、系统生物学研究 C、类群间系统发育关系研究 D、序列比对
二、 问答题
1、 为什么说 SWISS-PROT 是最重要的蛋白质一级数据库?
2、 构建蛋白质二级数据库的基本原则是什么?
3、 构建蛋白质二级数据库的主要方法有哪些?
4、 叙述 SCOP 数据库对蛋白质分类的主要依据
第八章 后基因组时代的生物信息学(问题与练习)
1、 比较生物还原论与生物综合论的异同 2、 简述“后基因组生物信息学”的基本研究思路 3、 后基因组生物信息学的主要挑战是什么? 4、 功能基因组系统学的基本特征是什么? 5、 说明后基因组生物信息学对信息流动的最新理解 6、 列举几种预测蛋白质-蛋白质相互作用的理论方法 7、 解释从基因表达水平关联预测蛋白质-蛋白质相互作用的理论方法 8、 解释基因保守近邻法预测蛋白质-蛋白质相互作用的理论方法 9、 解释基因融合法预测蛋白质-蛋白质相互作用的理论方法 10、解释种系轮廓发生法预测蛋白质-蛋白质相互作用的理论方法
1、蛋白质得分矩阵类型有 、
、、
和
等。
2、对位排列主要有局部比对和 三、运算题 1、画出下面两条序列的简单点阵图。将第一条序列放在 x 坐标轴上,将第二条序列放在 y
生物信息学中的蛋白质序列分析与预测研究

生物信息学中的蛋白质序列分析与预测研究蛋白质是生命体中至关重要的分子,它们在细胞功能和结构的调控中发挥着重要的作用。
蛋白质的序列决定了其结构和功能,因此蛋白质序列的分析和预测成为生物信息学研究的重要方向之一。
本文将重点介绍蛋白质序列分析和预测的方法与技术,以及在生物学研究中的应用。
蛋白质序列的分析是指根据蛋白质的氨基酸序列,通过一系列的计算和分析方法,对其结构和功能进行研究的过程。
蛋白质序列分析的方法有很多,其中最常用的包括:比对分析、同源建模、序列特征分析和亚细胞定位预测。
首先,比对分析是蛋白质序列分析的基础方法之一。
通过将待分析的蛋白质序列与已知的蛋白质序列数据库进行比对,可以找到与之相似的序列,进而推测蛋白质的结构和功能。
比对分析常用的工具有BLAST和PSI-BLAST等,它们通过比较序列之间的相似性和一致性,确定序列的保守区域和结构域,从而揭示蛋白质的功能。
其次,同源建模是一种根据已知蛋白质的结构来预测未知蛋白质的结构的方法。
在同源建模中,通过比对已知蛋白质的结构与待预测蛋白质的序列,找到与之相似的蛋白质结构作为模板,并利用模板的结构信息,预测待预测蛋白质的结构。
同源建模的常用工具有SWISS-MODEL和Phyre2等。
同源建模不仅可以预测蛋白质的三维结构,还可以提供结构功能的启示,从而推测其功能。
另外,序列特征分析也是蛋白质序列分析的重要方向之一。
序列特征分析通过对蛋白质序列中的特定模式、保守区域和功能位点进行分析,揭示蛋白质的结构和功能。
常用的序列特征分析方法包括信号肽预测、跨膜区域识别、功能位点预测和蛋白质域识别等。
这些方法通过分析蛋白质序列中的特定特征,揭示蛋白质的功能和结构。
最后,亚细胞定位预测是蛋白质序列分析的一个重要方向。
蛋白质在细胞中的定位决定了其在细胞内发挥的功能,因此准确预测蛋白质的亚细胞定位对于理解其功能至关重要。
亚细胞定位预测通过分析蛋白质序列中的亚细胞定位信号和保守区域,预测蛋白质的亚细胞定位位置。
《生物信息学》练习题剖析

1、在Genbank中查找以下6个植物蛋白序列:protein1:NP_974673.2; protein2: NP_187969.1; protein3: NP_190855.1; protein4: NP_565618.1; protein5: NP_200511.1; protein6: NP_191407.1 (以FASTA格式)。
(1)用EBI上的ClustalW2工具对其进行多序列比对,分析各蛋白序列之间的同源性。
序列比对结果比对结果表明:protein1:NP_974673.2和protein4: NP_565618.1的亲缘关系最近。
(2)利用Phylip软件,选择距离法构建其进化树(要求写出具体的建树步骤)。
1.将蛋白序列保存为FASTA格式,存于txt文档;2.用Clustalx打开txt文本,保存为*.phy文件;3.用seqboot程序打开phy文件,输出结果文件*_seqboot4.用protdist程序打开*_seqboot文件,输出为*_protdist文件5. 用neighbor程序打开*_protdist文件,输出为*_neighbor文件6. 用consense程序打开*_neighbor文件,输出为*_consense文件7.用dratree程序打开*_consense文件得到进化树。
(注:由于seqboot软见无法正常运行,因此进化树无法显示)(3)任意选取其中的一个蛋白进行蛋白质一级序列分析、二级结构预测及三维结构的模拟。
选择protein3: NP_190855.1一级结构网址:/tools/protparam.htmlNumber of amino acids: 456 氨基酸数目Molecular weight: 51154.5 相对分子质量Theoretical pI: 8.69 理论 pI 值Amino acid composition 氨基酸组成Ala (A) 30 6.6%Arg (R) 28 6.1%Asn (N) 15 3.3%Asp (D) 27 5.9%Cys (C) 5 1.1%Gln (Q) 18 3.9%Glu (E) 28 6.1%Gly (G) 37 8.1%His (H) 16 3.5%Ile (I) 16 3.5%Leu (L) 42 9.2%Lys (K) 32 7.0%Met (M) 5 1.1%Phe (F) 17 3.7%Pro (P) 16 3.5%Ser (S) 46 10.1%Thr (T) 21 4.6%Trp (W) 8 1.8%Tyr (Y) 19 4.2%Val (V) 30 6.6%Pyl (O) 0 0.0%Sec (U) 0 0.0%(B) 0 0.0%(Z) 0 0.0%(X) 0 0.0%正/负电荷残基数Total number of negatively charged residues (Asp + Glu): 55Total number of positively charged residues (Arg + Lys): 60Atomic composition: 原子组成Carbon C 2270Hydrogen H 3531Nitrogen N 645Oxygen O 686Sulfur S 10Formula: C2270H3531N645O686S10 分子式Total number of atoms: 7142 总原子数Extinction coefficients: 消光系数Extinction coefficients are in units of M-1 cm-1, at 280 nm measured in water.Ext. coefficient 72560Abs 0.1% (=1 g/l) 1.418, assuming all pairs of Cys residues form cystines Ext. coefficient 72310Abs 0.1% (=1 g/l) 1.414, assuming all Cys residues are reducedEstimated half-life: 半衰期The N-terminal of the sequence considered is M (Met).The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro).>20 hours (yeast, in vivo).>10 hours (Escherichia coli, in vivo).Instability index: 不稳定系数The instability index (II) is computed to be 48.99This classifies the protein as unstable.Aliphatic index: 75.26 脂肪系数Grand average of hydropathicity (GRAVY): -0.554 总平均亲水性/tools/protscale.html蛋白质亲疏水性分析所用氨基酸标度信息Ala: 1.800 Arg: -4.500 Asn: -3.500 Asp: -3.500 Cys: 2.500 Gln: -3.500 Glu: -3.500 Gly: -0.400 His: -3.200 Ile: 4.500 Leu: 3.800 Lys: -3.900 Met: 1.900 Phe: 2.800 Pro: -1.600 Ser: -0.800 Thr: -0.700 Trp: -0.900 Tyr: -1.300 Val: 4.200 : -3.500 : -3.500 : -0.490分析所用参数信息Weights for window positions 1,..,9, using linear weight variation model:1 2 3 4 5 6 7 8 91.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00edge center edge跨膜结构预测结果(没有跨膜结构)信号肽分析:二级结构预测三级结构预测网站/~phyre2、在拟南芥基因组数据库中(/)查找编号分别为At4G33050, At3G13600,At3G52870或At2G26190基因,针对所查找的基因进行初步的生物信息学分析(每人任选其中一个基因)。
《生物信息学》练习题

1、在Genbank中查找以下6个植物蛋白序列:protein1:NP_974673.2; protein2: NP_187969.1; protein3: NP_190855.1; protein4: NP_565618.1; protein5: NP_200511.1; protein6: NP_191407.1 (以FASTA格式)。
(1)用EBI上的ClustalW2工具对其进行多序列比对,分析各蛋白序列之间的同源性。
序列比对结果比对结果表明:protein1:NP_974673.2和protein4: NP_565618.1的亲缘关系最近。
(2)利用Phylip软件,选择距离法构建其进化树(要求写出具体的建树步骤)。
1.将蛋白序列保存为FASTA格式,存于txt文档;2.用Clustalx打开txt文本,保存为*.phy文件;3.用seqboot程序打开phy文件,输出结果文件*_seqboot4.用protdist程序打开*_seqboot文件,输出为*_protdist文件5. 用neighbor程序打开*_protdist文件,输出为*_neighbor文件6. 用consense程序打开*_neighbor文件,输出为*_consense文件7.用dratree程序打开*_consense文件得到进化树。
(注:由于seqboot软见无法正常运行,因此进化树无法显示)(3)任意选取其中的一个蛋白进行蛋白质一级序列分析、二级结构预测及三维结构的模拟。
选择protein3: NP_190855.1一级结构网址:/tools/protparam.htmlNumber of amino acids: 456 氨基酸数目Molecular weight: 51154.5 相对分子质量Theoretical pI: 8.69 理论 pI 值Amino acid composition 氨基酸组成Ala (A) 30 6.6%Arg (R) 28 6.1%Asn (N) 15 3.3%Asp (D) 27 5.9%Cys (C) 5 1.1%Gln (Q) 18 3.9%Glu (E) 28 6.1%Gly (G) 37 8.1%His (H) 16 3.5%Ile (I) 16 3.5%Leu (L) 42 9.2%Lys (K) 32 7.0%Met (M) 5 1.1%Phe (F) 17 3.7%Pro (P) 16 3.5%Ser (S) 46 10.1%Thr (T) 21 4.6%Trp (W) 8 1.8%Tyr (Y) 19 4.2%Val (V) 30 6.6%Pyl (O) 0 0.0%Sec (U) 0 0.0%(B) 0 0.0%(Z) 0 0.0%(X) 0 0.0%正/负电荷残基数Total number of negatively charged residues (Asp + Glu): 55Total number of positively charged residues (Arg + Lys): 60Atomic composition: 原子组成Carbon C 2270Hydrogen H 3531Nitrogen N 645Oxygen O 686Sulfur S 10Formula: C2270H3531N645O686S10 分子式Total number of atoms: 7142 总原子数Extinction coefficients: 消光系数Extinction coefficients are in units of M-1 cm-1, at 280 nm measured in water.Ext. coefficient 72560Abs 0.1% (=1 g/l) 1.418, assuming all pairs of Cys residues form cystines Ext. coefficient 72310Abs 0.1% (=1 g/l) 1.414, assuming all Cys residues are reducedEstimated half-life: 半衰期The N-terminal of the sequence considered is M (Met).The estimated half-life is: 30 hours (mammalian reticulocytes, in vitro).>20 hours (yeast, in vivo).>10 hours (Escherichia coli, in vivo).Instability index: 不稳定系数The instability index (II) is computed to be 48.99This classifies the protein as unstable.Aliphatic index: 75.26 脂肪系数Grand average of hydropathicity (GRAVY): -0.554 总平均亲水性/tools/protscale.html蛋白质亲疏水性分析所用氨基酸标度信息Ala: 1.800 Arg: -4.500 Asn: -3.500 Asp: -3.500 Cys: 2.500 Gln: -3.500 Glu: -3.500 Gly: -0.400 His: -3.200 Ile: 4.500 Leu: 3.800 Lys: -3.900 Met: 1.900 Phe: 2.800 Pro: -1.600 Ser: -0.800 Thr: -0.700 Trp: -0.900 Tyr: -1.300 Val: 4.200 : -3.500 : -3.500 : -0.490分析所用参数信息Weights for window positions 1,..,9, using linear weight variation model:1 2 3 4 5 6 7 8 91.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00 1.00edge center edge跨膜结构预测结果(没有跨膜结构)信号肽分析:二级结构预测三级结构预测网站/~phyre2、在拟南芥基因组数据库中(/)查找编号分别为At4G33050, At3G13600,At3G52870或At2G26190基因,针对所查找的基因进行初步的生物信息学分析(每人任选其中一个基因)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
生物信息学蛋白质序列分析-课堂练习
ZNF395, 全称为Zinc Finger Protein395, 又被称为PBF,PRF1,DBP2,PRF-1,Si-1-8-14或DKFZp434K1210。
其氨基酸序列为
结构域分析:http://www.expasy.ch/prosite/
(一)分析蛋白质的一级结构
分析蛋白质的pI、Mw、氨基酸组成:Tools and software packages------Identification and characterization-----ProtParam
http://www.expasy.ch/tools/protparam.html
分析蛋白质的疏水性:Primary structure analysis-----ProtScale
http://www.expasy.ch/tools/protscale.html
分析蛋白质的重复序列:Primary structure analysis-----REP
http://www.embl-heidelberg.de/~andrade/papers/rep/search.html
(二)分析蛋白质的二级结构
预测蛋白质的?-螺旋和?-折叠结构:Secondary structure prediction-----nnPredict
/~nomi/nnpredict.html
蛋白质的其它二级结构:Secondary structure prediction-----SOPMA
(三)分析蛋白质的三级结构
molecular modeling:“tertiary structure prediction ”栏目选择选择一个分析工具,email服务
(四)分析膜蛋白质
预测膜整合蛋白的跨膜区: Topology prediction------SOSUI
http://bp.nuap.nagoya-u.ac.jp/sosui/
分析膜锚定蛋白的GPI位点:Post-translational modification------big-PI Predictor
http://mendel.imp.ac.at/sat/gpi/gpi_server.html
(五)分析蛋白质的翻译后修饰
分析信号肽及其剪切位点: Post-translational modification prediction----SignalIP
http://www.cbs.dtu.dk/services/SignalP/
分析糖链连接点:分析O-连接糖蛋白,Post-translational modification prediction----NetOGlyc
http://www.cbs.dtu.dk/services/NetOGlyc/
分析N-连接糖蛋白,Post-translational modification prediction----NetNGlyc
(六)分析蛋白质的亚细胞定位
Topology prediction----PSORT-----WoLF PSORT
/
(七)分析化学因子作用蛋白质的位点
“Identification and characterization ”------“Other prediction or characterization tools”栏目选择“PeptideCutter” 软件
http://www.expasy.ch/tools/peptidecutter/
1.蛋白基本理化性质分析
利用Expasy 软件包中的ProtParam工具(http://www.expasy.ch/tools/protparam.htmL) 进行蛋白的氨基
酸组成、分子质量、等电点及疏水性等理化性质的分析。
2. 利用PROF法(/)网站工具和(http://npsa-pbil.ibcp.fr)网站HNN软件预测蛋白的二级结构。
利用Scan Prosite(/)网站工具对蛋白进行结构域分析。
利用Swiss-Model数据库软件预测该蛋白的三级结构,结果用蛋白质三维图象软件Rasmol2.7查看。
3.蛋白亚细胞定位与功能分析
利用CBS(http://www.cbs.dtu.dk/services/TMHMM-2.0/)网站工具对蛋白进行跨膜分析。
采用PredictNLS (/predictNLS/) 对蛋白进行核定位信号分析,预测利用(http://www.cbs.dtu.dk/services/SignalP/)网站工具预测蛋白的信号肽。
利用PSORT(http: //psort.nibb.ac.jp/form2.htmL)网站工具进行蛋白亚细胞定位预测。
利用CBS (http://www.cbs.dtu.dk/services/ProtFun/)网站工具预测蛋白的功能。