利用STRING 数据库进行Cytoscape蛋白互作网络绘制步骤详解
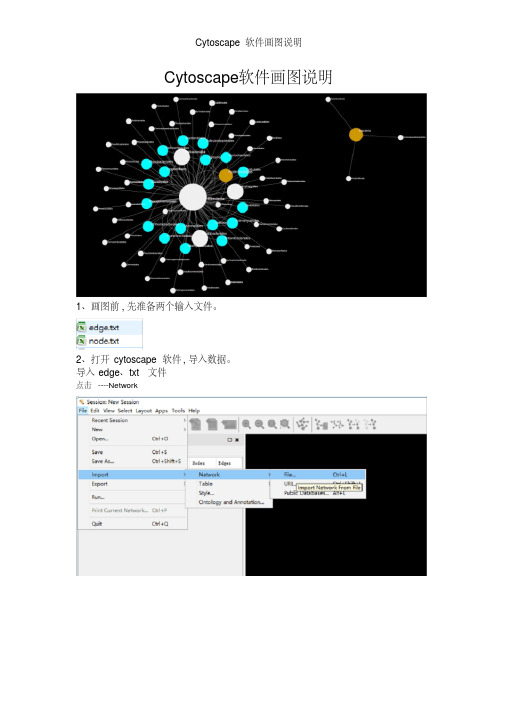
Cytoscape软件画图说明

Cytoscape软件画图说明1、画图前,先准备两个输入文件。
2、打开cytoscape软件,导入数据。
导入edge.txt文件点击File ----Import ----Network点击ok得到原始图形节点1,文件中第一列节点2,文件中第2列连接类型,文件中第3列点击layout ----Apply Preferred Out 改变图形排列方式此处可以用鼠标在画布中拖动图形到合适的位置。
改变画布背景。
点击左侧Contro Panel ----Style---network ---Backgroud paint设置节点之间连线的宽度和颜色Contro Panel ----Style---Edge颜色宽度导入node.txt个文件点击File ----Import ----Table设置节点图形属性Contro Panel ----Style---Node1、node大小通过设置Height和Width来控制大小2、Node性状通过设置Shape来控制3、Node填充色通过设置Fill Color来控制4、Node标签的设置点击Properties -- Label Position点击Label Position来设置标签位置。
这里是一个示范操作,要细致调整标签位置还是要设置Column和Mapping Type两个参数。
设置完成之后图形调节节点之间的距离点击layout ----Scale鼠标拖动最后,画图导出成pdf文件点击File ----Export----Network View as Graphics精品文档。
11欢迎下载欢迎您的下载,资料仅供参考!致力为企业和个人提供合同协议,策划案计划书,学习资料等等打造全网一站式需求。
基于互作网络图——string数据库

数据来源
• 点击关心的互作关系,会给出文献的名称及摘要, 点击pubmed可以跳转到对应的文章。
7
目 录 CONTENTS
1. string数据库 2. 利用string数据库进行分析
8
2.利用string数据库进行分析
• 进入网站 https:///
下载数据库
分析功能
• PS:输入的蛋白有可能在系统中没有信息,因此 数量可能会比输入的少一些。
25
确认蛋白
• 在分析过程中string会自动判断最合适的蛋白,确 认无误后点击continue继续分析
26
结果查看
• 结果显示6个蛋白中有5个蛋白存在互作关系
27
结果优化
• 图中的关系信息太少,点击more,使图片信息更 加丰满。
分析功能
9
操作类型
• 支持多种模式:输入单个蛋白的名称(或氨基酸 序列)查找其互作网络,也可输入多个蛋白的名 称或序列
按蛋白名称
多个蛋白同时 分析 在蛋白后添加 属性和值
按蛋白序列
输入区域
搜索物种、蛋 白家族、例子 或随机互作关 系
选择物种
例子
10
举个例子
• 选择例子1,trpA基因,点击search,开始分析。
• 目前已更新V11.0到(2019.1.19更新),比V10.5 (2017.5.14)的物种、蛋白数量、蛋白间关系要更多。
V10.5
V11.0
4
数据来源
• 数据来源于实验、数据库、文本挖掘、相邻基因、 基因融合、共存、共表达。
5
数据来源
• 点击对应的数据来源,会给出具体互作关系,如 实验的来源,会看到上方给出每个互作关系是通 过如
STRING数据库的蛋白质相互作用(PPI)网络分析

STRING数据库的蛋白质相互作用(PPI)网络分析STRING(Search Tool for the Retrieval of InteractingGenes/Proteins)数据库是一个用于存储蛋白质相互作用(PPI)信息的在线资源。
PPI网络分析是研究蛋白质之间相互作用的一种方法。
通过分析PPI网络,研究者可以了解蛋白质的功能和作用机制,揭示生物系统的复杂性。
在本篇文章中,我们将探讨PPI网络分析的意义、方法和应用。
PPI网络分析的意义在于帮助我们理解蛋白质的功能和相互作用。
蛋白质是细胞中最重要的功能分子之一,它们通过相互作用形成复杂的网络结构,从而参与调控细胞的生理和病理过程。
通过构建PPI网络并进行分析,我们可以了解蛋白质在细胞中的相互关系,进而找到调控的关键因子。
PPI网络的构建通常基于实验数据或计算机预测。
实验方法包括酵母双杂交、共免疫沉淀和质谱分析等。
这些实验技术可以检测到蛋白质之间的物理相互作用。
计算机预测方法则基于已知蛋白质结构和序列信息,通过算法判断蛋白质之间是否可能相互作用。
STRING数据库整合了多种实验和计算方法生成的PPI数据,提供了更全面的PPI网络信息。
PPI网络分析通常包括网络图的构建和网络特性的分析。
网络图由节点和边组成,其中节点代表蛋白质,边表示蛋白质之间的相互作用。
网络构建可以基于已知的实验数据或计算机预测结果。
网络特性分析包括节点度数、网络连通性、模块化等指标的计算。
这些指标可以帮助我们了解网络的结构和特点。
PPI网络分析的应用非常广泛。
首先,它可以帮助我们预测蛋白质的功能。
蛋白质的功能通常与其相互作用的伙伴密切相关。
通过分析PPI网络,我们可以推断一个未知蛋白质的功能,并为后续实验提供指导。
其次,PPI网络分析还可以帮助我们识别关键的调控通路和靶点。
在许多疾病中,蛋白质相互作用的异常可能是病理过程的关键因素。
通过分析PPI网络,我们可以找到与疾病相关的节点和模块,并设计针对性的治疗策略。
cytoscape使用说明

在数据面板(Data panel) 中选择要显示的节点属性
查看节点属性
选择边属性浏览
选中要显示的边属性
查看边的属性
Agilent Literature Search文献检索插件 /cyto_web/plugins/index.php
检索关键词 是否同时检 索别名 生物种属
从插件菜单(Plugins)中打开cytoprophet插件
首先导入cytoprophet.sif文件
1. 选择整个网络作 为预测对象; 2. 选择MSSC算法; 3. 选择同时预测 DDI Network和G结构域相互作用关系的窗口
Gene Ontology在“功能类”的层面上概括了基因 参与的生命过程。在基因表达谱分析中,GO常用于 提供基因功能分类标签和基因功能研究的背景知识。 Gene Ontology可以用来发掘与基因差异表达现象 关联的“单个特征基因功能类”或“多个特征功能 类”的组合。
/
Cytoscape目前最新版本: 2.7.0
Cytoscape
Cytoscape 2.6.3可由
/download.php?file=cyto2_6_3
下载得到,下载前需要进行简单的注册,输入姓名、 单位、email信息即可。 Cytoscape同时支持Windows、Mac和Linux/Unix。 Cytoscape基于java平台,需首先安装java运行环 境,该软件可由
Cytoscape插件下载
/plugins.php
http://www.psb.ugent.be/cbd/papers/BiNGO/
下载得到的BiNGO.jar存放到 程序安装目录下的plugins
插件安装前:
cytoscape软件画图说明
Cytoscape软件画图说明
1、画图前,先准备两个输入文件。
2、打开cytoscape软件,导入数据。
导入edge、txt文件
点击----Network
节点1,文件中第一列
节点2,文件中
第2列
连接类型,文件
中第3列
点击ok得到原始图形
点击layout ----Apply Preferred Out 改变图形排列方式
此处可以用鼠标在画布中拖动图形到合适的位置。
改变画布背景。
点击左侧Contro Panel ----Style---network ---Backgroud paint
设置节点之间连线的宽度与颜色Contro Panel ----Style---Edge
导入node、txt个文件点击----Table 宽度颜色
设置节点图形属性
Contro Panel ----Style---Node
1、node大小
通过设置Height与Width来控制大小
2、Node性状
通过设置Shape来控制
3、Node填充色
通过设置Fill Color来控制
4、Node标签的设置
点击Properties -- Label Position
点击Label Position来设置标签位置。
这里就是一个示范操作,要细致调整标签位置还就是要设置Column与Mapping Type两个参数。
设置完成之后图形
调节节点之间的距离
点击layout ----Scale
鼠标拖动
最后,画图导出成pdf文件点击View as Graphics。
STRING网站Cytoscape软件制作精美蛋白互作网络图(PPI)

STRING网站Cytoscape软件制作精美蛋白互作网络图(PPI)之前小编为大家推送了利用DAVID网站进行差异基因的GO和KEGG分析,链接:DAVID&Metascape:专注于基因功能注释和富集通路分析的网站。
而基因功能注释后就可以寻找蛋白表达之间的关系了,在生信分析中,常常会使用STRING网站+Cytoscape软件来制作蛋白互作网络图(PPI)。
今天小编奉上一部PPI制作教程,让我们一起细细咀嚼吧!首先我们进入STRING网站的官网(网址为/),界面很简单明了,左侧一栏是网站的输入方式,右侧一栏是我们的蛋白输入框。
通常我们在做PPI时选择输入方式是“Multiple proteins”,即多个蛋白的输入。
选择好蛋白的输入方式后,输入一列差异基因,STRING网站对基因的限制条件是不超过2000个基因,格式也是每行一个基因名(或者说是蛋白名字吧,总之就是差异基因所表达的蛋白)。
简而言之,这个页面上的操作的步骤是先选择“Multiple protenins”,然后输入基因名,最后选择“Homo sapiens”。
然后点击“SEARCH”选项,千万别停下来,继续点击“CONTINUE”选项。
这样就做好了一张PPI图,但是看上去还是比较杂乱的,因此我们需要通过Cytoscape软件对PPI图进一步做美化处理。
下拉当前页面,可以看到一行菜单,Legend菜单里面是关于网络图中Nodes和Edges的注释,了解一下。
Settings菜单功能比较重要,比如我们对Edges的选择,“confidence”是通过线条的粗细来反映蛋白之间相互作用的强弱。
如果我们制作的网络图比较分散,我们可以通过设置“minimum required interaction score”将conbined_score调高来调整PPI,使图形看上去更紧密。
如果我们觉得网络图上的蛋白数量较少,我们可以通过设置蛋白数量的上限,比如我们将其设置为“no more than 50 interactors”,看下效果。
STRING蛋白网络分析操作流程

STRING蛋白网络分析操作流程蛋白网络分析是一种系统生物学方法,用于研究蛋白质相互作用网络。
它可以帮助科学家解析蛋白质相互作用、信号传导和代谢途径,从而更好地理解细胞的功能和调控机制。
以下是一个关于蛋白网络分析的操作流程,包括数据准备、网络构建和分析、结果解释和验证等几个主要步骤。
1.数据准备:首先,需要收集和整理相关的蛋白质相互作用数据。
这些数据可以来自公开数据库,如STRING(Search Tool for the Retrieval of Interacting Genes/Proteins)、BioGRID、IntAct等。
可以根据研究对象的物种和特定的研究领域选择相应的数据库。
确保所选择的数据可靠性和准确性。
2.数据预处理:在进行网络构建之前,需要对原始数据进行预处理,以去除噪音和无效的信息。
这通常包括去除重复的相互作用、修复格式错误和标准化数据格式等。
此外,还可以根据研究的目的进行数据筛选和过滤,如选择特定阈值的相互作用等。
3.网络构建和分析:接下来,使用预处理后的数据构建蛋白网络。
最常用的方法是基于相互作用的网络,其中蛋白质与其他蛋白质之间的相互作用表示为网络中的节点和边。
可以使用网络分析软件,如Cytoscape等,进行网络可视化和分析。
可以计算网络的节点度、聚集系数、连通性等基本拓扑特征,并进行模块和子网络的发现,以揭示蛋白质相互作用网络的组织结构和功能模块。
4.结果解释:对于网络中的重要节点和模块,可以使用注释数据库(如Gene Ontology)来解释其生物学功能和关联的代谢途径。
这可以帮助科学家理解蛋白质网络的功能和调控机制,从而提出相关的假设和研究问题。
5.结果验证:为了验证通过蛋白网络分析得到的结果,可以采用多种实验技术,如免疫共沉淀、基因敲除、RNA干扰等。
这些实验可以用来验证网络预测的相互作用和功能,进一步验证和揭示蛋白质网络的生物学意义和调控机制。
需要注意的是,蛋白网络分析是一个复杂的过程,需要综合运用多种工具和技术,并结合实验验证来解析蛋白质相互作用网络。
利用生物大数据技术开展蛋白质互作网络分析的方法与技巧

利用生物大数据技术开展蛋白质互作网络分析的方法与技巧引言:在生物学研究中,蛋白质互作网络是理解细胞内分子之间相互作用的关键工具。
生物大数据技术的出现为我们提供了大规模蛋白质互作网络数据和分析工具。
本文将介绍利用生物大数据技术开展蛋白质互作网络分析的方法与技巧。
一、数据获取蛋白质互作网络分析的第一步是获取蛋白质互作网络的数据。
目前,公共数据库如STRING、BioGRID、HPRD、MINT等提供了大量的蛋白质互作数据。
研究人员可以通过访问这些数据库,根据自己的研究兴趣和需要获取相关数据。
此外,也可以利用生物大数据技术从大规模基因组学研究中获得蛋白质互作网络数据。
二、数据预处理蛋白质互作网络数据的预处理对于后续的分析至关重要。
预处理的目的是去除噪声和无关信息,提高分析的准确性和可靠性。
预处理的步骤包括数据清洗、数据归一化和数据筛选等。
数据清洗主要是去除无效数据和错误数据,如重复数据、无效交互等。
数据归一化用于消除数据集之间的差异,将数据调整到统一的尺度上。
数据筛选是根据研究目的和假设筛选出与研究有关的蛋白质互作数据。
三、网络分析方法基于生物大数据的蛋白质互作网络分析有多种方法可选,本文将介绍常用的两种方法:网络可视化和网络分析工具。
1. 网络可视化网络可视化是将蛋白质互作网络数据以图形方式呈现,帮助研究人员直观地理解蛋白质之间的相互关系。
常用的网络可视化工具有Cytoscape、Gephi等。
这些工具可以根据互作关系绘制节点和边,节点代表蛋白质,边代表蛋白质之间的互作关系。
网络可视化不仅可以展示整体的蛋白质互作网络,还可以根据需求进行扩展和收缩,分析特定的互作子网络。
2. 网络分析工具网络分析工具是用于发现蛋白质互作网络的特征和模式的工具。
常用的网络分析工具有CentiScaPe、MAGI和STRING等。
这些工具可以对蛋白质互作网络进行拓扑结构分析、模块检测、节点中心性分析等。
通过网络分析工具,可以发现互作网络中的关键节点、功能模块和互作模式,为深入理解蛋白质相互作用提供重要线索。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用STRING 数据库进行蛋白互作预测步骤详解一、简介
STRING (Search Tool for the Retrieval of Interacting Genes/Proteins )数据库(/)是一个搜寻已知蛋白质之间和预测蛋白质之间相互作用的系统,这种相互作用既包括蛋白质之间直接的物理的相互作用,也包括蛋白质之间间接功能的相关性。
STRING 数据库除了包含有实验数据、从PubMed 摘要中文本挖掘的结果和综合其他数据库数据外,还有利用生物信息学的方法预测的结果。
所应用的生物信息学的方法有:染色体临近、基因融合、系统进化谱和基于芯片数据的基因共表达。
系统中利用一个打分机制对这些不同方法得来的结果给予一定的权重,最终给出一个综合的得分。
二、使用方法
1. 打开STRING 网站/
图 2.1
2. 支持多种类型文件来搜索,如名称、蛋白序列、多个名称、多条蛋白序列。
这里,我们
以多个猪转录本Ensembl的ID称为例。
图 2.2
3.输入转录本名称,按“GO!”,进入下个界面;
图 2.3
4.点击“GO”之后会出现如下界面:
图 2.4
5.匹配到的蛋白会自动勾选出来,如图显示
图 2.5
6. 按“Continue”,获得蛋白网络图;如图2.6 所示,可以选择不同的表达类型。
图 2.6
7.不同颜色的圆点,代表不同的蛋白;图2.7 所示为蛋白的注释信息。
图 2.7
8.如图2.8 所示,为蛋白互作关联分析结果,线条的粗细表示关联程度的强弱。
图 2.8
9.保存数据分析的结果;
10.利用Cytoscape去画基因蛋白互作关系;
首先我们要确定与基因互作的蛋白节点关系,这个关系可以从之前的结果页面下载,选择Other fomats:
11.点击Other fomats之后会出现一系列的下载选项,选择下载Text Summary:
12.summary结果展示,前两列即为节点文件,导入Cytoscape即可作图:
14.用cytoscape作图操作流程:
首先我们要安装cytoscape,cytoscape程序官方网站:/
点击进去之后用户可以根据自己的实际情况选择不同的版本,安装没有特别需要注意的地方,一直点击NEXT就可以了,这里我们以cytoscape2.8.3版本进行演示。
1)安装好了点击运行,会出现如下界面:
2)点击左上角的File选项中的Import选项,再选择文本格式输入:
3)选择文本格式输入之后,会出现如下界面,点击Select File(s):
4)选择已经准备好的节点文件,总共两列,每一行代表一种对应关系:
5)选择好文件之后会在界面中展示文件中的内容,如下所示:
6)然后选择互作中的第一列元素,也就是Select Source node column:
7) 再选择第二列元素,也就是Target Interaction的输入数据:
8)其中的Interaction Type选择默认,选择好了之后点击Import:
9)cytoscape会根据我们输入的节点文件自动绘制出互作网络图:
10)根据需要我们可以将网络图认为调整得更加美观(直接点击图中的红色圆圈可以随意拖动),下图为调整后的图形:
11)保存图片:点击File-Export-Current Network View As Graphics(或者直接快捷键:Ctrl+shift+P)
12)选择保存类型,有好多种类型可以自行选择,选择之后点击OK:
13.更多的数据展示,请登录/ 查阅。
三、参考文献
Franceschini et al., STRING v9.1: protein-protein interaction networks, with increased
coverage and integration. Nucleic Acids Res. 2013 Jan。