利用 MEGA4 构建分子系统进化树
用MEGA构建进化树

如何用MEGA构建进化树MEGA3.1是一个关于序列分析以及比较统计的工具包,其中包括有距离建树法和MP 建树法;可自动或手动进行序列比对,推断进化树,估算分子进化率,进行进化假设测验,还能联机的Web数据库检索。
下载后可直接使用,主要包括几个方面的功能软件:i)DNA 和蛋白质序列数据的分析软件。
ii)序列数据转变成距离数据后,对距离数据分析的软件。
iii)对基因频率和连续的元素分析的软件。
iv)把序列的每个碱基/氨基酸独立看待(碱基/氨基酸只有0和1的状态)时,对序列进行分析的软件。
v)绘制和修改进化树的软件,进行网上blast搜索。
用MEGA构建进化树有以下步骤:1. 16S rDNA测序和参考序列选取从环境中分离到单克隆,去重复后扩增16S rDNA序列并测序,然后与数据库/blast/Blast.cgi比对,找到相似度最高的几个序列,确定一下你分离的细菌大约属于哪个科哪个属,如果相似度达到百分之百那基本可以确定你分离得到的就是Blast到的那个,然后找一到两个同科的,再找一到两个同目的,再找一到两个同纲的细菌,把序列全部下下来,以FSATA形式整合在TXT文档中,如>TS1GCAGTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAA CACGTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAATACCG GA TAGGACCTCGGGA TGCA TGTTCCGGGGTGGAAAGGTTTTCCGGTGCAGGATGGGCC>gi|117572706|gb|EF028124.1| Rhodococcus sp. Atl25 16S ribosomal RNA gene, partial sequence CGATTAGAGTTTGA TCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAA GTCGAACGATGAAGCCCAGCTTGCTGGGTGGA TTAGTGGCGAACGGGTGAGTAACAC GTGGGTGATCTGCCCTGCACTTCGGGATAAGCCTGGGAAACTGGGTCTAA TACCGGA T>TS2TGCAAGTCGAGCGAATGGA TTAAGAGCTTGCTCTTA TGAAGTTAGCGGCGGACGGGTG AGTAACACGTGGGTAACCTGCCCA TAAGACTGGGATAACTCCGGGAAACCGGGGCTAA TACCGGATAACA TTTTGAACTGCATGGTTCGAAA TTGAAAGGCGGCTTCGGCTGTCACT>gi|56383044|emb|AJ809498.1| Bacillus cereus partial 16S rRNA gene, strain TMW 2.383GA TGAACGCTGGCGGCGTGCCTAA TACATGCAAGTCGAGCGAA TGGATTAAGAGCTTG CTCTTA TGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCCATAAGAC TGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAACYGCATGGTTC ………………………….………………………….参考序列选择有几个原则:a,不选非培养(unclutured)微生物为参比;b,所选参考序列要正确,里面无错误碱基;c,在保证同属的前提下,优先选择16S rDNA全长测序或全基因组测序的种;d,每个种属选择一个参考序列,如果自己的序列中同一属的较多,可适当选择两个参考序列。
mega4使用说明

MEGA的使用产生背景及简介随着不同物种基因组测序的快速发展,产生了大量的DNA序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。
MEGA就是基于这种需求开发的。
MEGA 软件的目的就是提供一个以进化的角度从DNA和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。
现在我们使用的是MEGA4的版本。
它主要集中于进化分析获得的综合的序列信息。
使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。
此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。
此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。
在计算矩阵方面有一些自己的特点:1.推测序列或者物种间的进化距离2.根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树3.考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。
4.随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容可以被保存,复制。
具体使用我们以分析20个物种的血红蛋白为例来具体说明此软件的具体使用情况。
一.启动程序1.运行环境:在Windows 95/98, NT, ME, 2000, XP, vista等操作系统下均可使用。
2.下载安装:可以直接登陆进行下载安装,另外还可以从/tools/phylogeny.php中的链接进去。
3.双击桌面快捷方式图标,进入主界面;或者从开始菜单,单击图标启动。
二.序列分析。
1.启动单击后,会出现如下界面:这里有三个选项,分别对应三种不同的情况:以下分别予以介绍:Create a new alignment :是在你没有任何比对的时候使用,比如你只有一个fasta格式的序列就可以选择这个选项。
Open a saved alignment session:使用它可以打开一个我们已经比对好的序列文件;Retieve a sequence from a file :这种情况同第一种情况相似,只是不用选择是DNA 还是蛋白质序列比对,选择的也是fasta格式的文件,打开后的界面都是一样的。
Mega的使用以及进化树的绘制

1.MEGA构建系统进化树的步骤2.CLUSTALX进行序列比对1.MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
如图:2. 打开MEGA软件,选择"Alignment" - "Alignment Explorer/CLUSTAL",在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开,如图:。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可,如图:。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes,如图:。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” -“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Minimun-evolution”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“Maximun-parsimony”,如图:在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny” –“UPGMA”,如图:6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。
MEGA 软件——系统发育树构建方法

• 双击图标
,
• 下载下来的序列片段保存文件为FASTA格式,打开方式为TXT格式。 • 将Blast对比后所Download的序列筛选后,构建系统发育进化树。 • 构建系统发育树需要测序所得序列1个,Blast对比得出序列5-10个, 构建出的为单枝系统发育进化树。通过这个对比可以确定出所测 定的序列最相似物种,当相似度为99%甚至100%时,基本可以确 定所测定的基因序列所属物种。
ME待测PCR产物送测序后,一个星期左右会得到生物 公司发的邮件,里面包含测序结果的附件,下载后得到文件压缩 包 ,将文件包解压,可以看到文件夹里有文件
• 测序公司会提供一款解读软件Chromas,免安装类型,能够直接 打开测序结果并通过软件直接进入NCBI数据库进行Blast搜索。
• 可培养真菌可以选定对比物种种属后构建大型系统发育进化树, 不可培养真菌则可直接构建系统发育进化树。
• 双击
后打开MEGA软件
MEGA4的中文使用说明
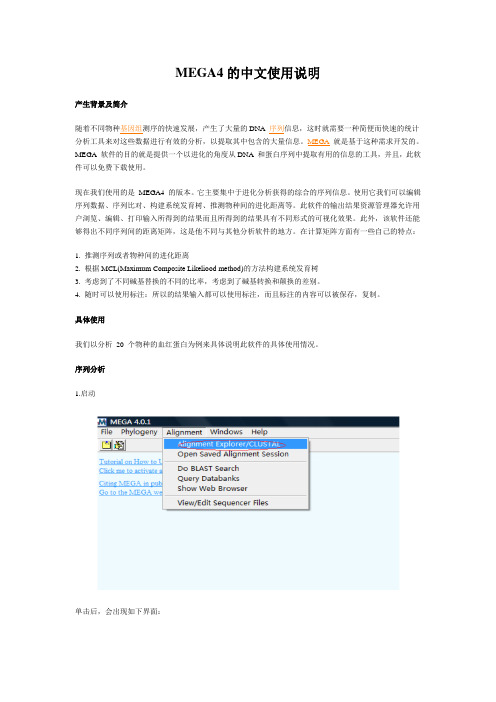
MEGA4的中文使用说明产生背景及简介随着不同物种基因组测序的快速发展,产生了大量的DNA 序列信息,这时就需要一种简便而快速的统计分析工具来对这些数据进行有效的分析,以提取其中包含的大量信息。
MEGA就是基于这种需求开发的。
MEGA 软件的目的就是提供一个以进化的角度从DNA 和蛋白序列中提取有用的信息的工具,并且,此软件可以免费下载使用。
现在我们使用的是MEGA4 的版本。
它主要集中于进化分析获得的综合的序列信息。
使用它我们可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。
此软件的输出结果资源管理器允许用户浏览、编辑、打印输入所得到的结果而且所得到的结果具有不同形式的可视化效果。
此外,该软件还能够得出不同序列间的距离矩阵,这是他不同与其他分析软件的地方。
在计算矩阵方面有一些自己的特点:1. 推测序列或者物种间的进化距离2. 根据MCL(Maximum Composite Likeliood method)的方法构建系统发育树3. 考虑到了不同碱基替换的不同的比率,考虑到了碱基转换和颠换的差别。
4. 随时可以使用标注:所以的结果输入都可以使用标注,而且标注的内容可以被保存,复制。
具体使用我们以分析20 个物种的血红蛋白为例来具体说明此软件的具体使用情况。
序列分析1.启动单击后,会出现如下界面:这里有三个选项,分别对应三种不同的情况:以下分别予以介绍:Create a new alignment :是在你没有任何比对的时候使用,比如你只有一个fasta 格式的序列就可以选择这个选项。
Open a saved alignment session:使用它可以打开一个我们已经比对好的序列文件;Retieve a sequence from a file :这种情况同第一种情况相似,只是不用选择是DNA 还是蛋白质序列比对,选择的也是fasta 格式的文件,打开后的界面都是一样的。
以第一种情况为例说明,点击如出现下界面:这里我们分析的是蛋白序列所以选择No。
MEGA4.0构树步骤

MEGA构建系统进化树的步骤1. 将要用于构建系统进化树的所有序列合并到同一个fasta格式文件,注意:所有序列的方向都要保持一致( 5’-3’)。
2. 打开MEGA软件,选择”Alignment” –“Alignment Explorer/CLUSTAL”,在对话框中选择Retrieve sequences from a file, 然后点OK,找到准备好的序列文件并打开。
3. 在打开的窗口中选择”Alignment”-“Align by ClustalX” 进行对齐,对齐过程需要一段时间,对齐完成后,最好将序列两端切齐,选择两端不齐的部分,单击右键,选择delete即可。
4. 关闭当前窗口,关闭的时候会提示两次否保存,第一次无所谓,保存不保存都可以,第二次一定要保存,保存的文件格式是.meg。
根据提示输入Title,然后会出现一个对话框询问是否是Protein-coding nucleotide sequence data, 根据情况选择Yes或No。
最后出现一个对话框询问是否打开,选择Yes。
5. 回到MEGA主窗口,在菜单栏中选择”Phylogeny”-“Bootstrap Test of Phylogeny”-“Neighbor-joining”,打开一个窗口,里面有很多参数可以设置,如何设置这些参数请参考详细的MEGA说明书,不会设置就暂且使用默认值,不要修改,点击下面的Compute按钮,系统进化树就画出来了。
6. 最后,使用TreeExplorer窗口中提供的一些功能可以对生成的系统进化树进行调整和美化。
另外,还可以用Word进一步编辑MEGA构建的进化树。
一般说来,MEGA适用于对少量的序列进行比对和画Tree,如需处理大量或海量的序列数据,建议使用ARB。
用BioEdit合并序列:1、打开BioEdit,点击“File”->”New Alignment”;2、“File”->”import”->”Sequence Alignment file”,将全部要合并的序列导入;3、”File“->”Save“ or “Save as”,保存为.fas格式文件。
进化树构建方法-MEGA

利用MEGA 来构建进化树(molecular evolutionary genetics analysis 分子进化遗传分析)打开mega5,选择Align----edit/built alignment----create a new alignment—OK选择DNA/protein出现新的对话框Open------选择已经保存好的用clustalx 经过比对保存的以.aln格式的文件打开之后,出现下面的页面双击文件名可以进行修改的。
我的就是从这里开始修改把A,B,C 都去掉,只留号码就好右键菜单点击delete 删除带※的那一行。
得到下面的图示,点击保存,重新起名字。
之后点击此图内的Alignment 选择Align by clustalW即可。
默认设置即可,点击OK就进行比对了,此后会出现一个过渡对话框,显示的是两两比对和多序列比对的过程之后回到初始页面,就是这个页面之后点File---点开,把刚才保留的文件点开然后出现下面的页面多了几个内容,点击TA的那个框框。
之后出现这样的框框图片然后在主程序中选择phylogeny---construct/test neighbor-joining tree,然后出现下面的页面黄色框框处的的参数是可以改变的,该图为我已经改变好的,把Bootstrap 的值改为1000 Methods根据文献上的参考改为了Kimura2-parameter model.之后点击compute,就出现了,而且还带有必需的支持率即自展值,是用来检验你所计算的进化树分支可信度的。
简单地讲就是把序列的位点都重排,重排后的序列再用相同的办法构树,如果原来树的分枝在重排后构的树中也出现了,就给这个分枝打上一分,如果没出现就给0分,这样经过你给定的repetitions 次(至少1000次)重排构树打分后,每个分枝就都得出分值,计算机会给你换算成bootstrap值。
重排的序列有很多组合,值越小说明分枝的可信度越低,最好根据数据的情况选用不同的构树方法和模型。
怎样使用MEGA建立进化树

怎样使用MEGAt 立进化树如何使用MEGA4.0#立进化树 1、首先是双击软件打开如下图所示|M| ijaKMr3 valj 141 Mrhr ArgrwricQt iVvta“qplii :护 忏冲 i 二客H - I 号筍需.廿星"LIF M ■ H 、-| II ■ DKi -Mjrsrze: H r« r-r r ^c>az^ LCS2、现在是处于DNA序列,而我们要做蛋白质的进化树的话,就如下操作M4. Aligmr>&nl Explof頁H L lQnmt*Ftji Editm e祁3、接下来我们要进行序列的输入,点击左边那个红箭头,贝U出现下面的窗口刚M4: Alfgnment Explorer匚;日屯EJrt S«ar di Aflgmnenl Wfrb $e<)□ d| D ◎日「蹇輻酋1 41象Protein S^quer匚弊1|主曲色"匕色丄4、然后右击sequenee 1,修改名字,如改成DPVFrotejn Sequence?5、然后从Word里复制蛋白质序列,然后在下面的位置粘贴G 辱CopfPTCtfiT X CU,書 f sterna6则可出现如下图的序列了□ QCW1C3 iRWfl Wq^ri[ V^i>n irequ^Ki 幷册枷・1話皿讥曲佰i"—喇・ct Mgeirc 惟■ sy7、然后点击窗口上的保存图标保存 8、重复从3开始,直到你的序列输入完9、序列输入元后进行最后的保存,方法如下垂邑trit 5|讨之斗和"1 of op«r * dow亠 P TOUMT 1 <io-jrr<n接下来打开册b M 罗哥 H*lpi t X t tt b要输入ul7两次保存名字一然后关闭这个窗口出现下面这个窗口■■■MM Jfc接下来就可以建立各种样式的进化树〜乜 MdngHie-r jein^ IMJL* &? Wrigym 佔抽杓也山-« UW3ML ■> ■小h,鼻 陆01申*貝Trfl 和 Hi^Tgrn^ ,HnNkk T ivn HnM "d i-Oi^4*cflArs R 协 FriWrt '^l^diCNHE軒I 匚 fkrti tiiitanr-ri : hy A 护产就 匸沁”-嗯,只是把过程写出来,方便大家建立进化树,不足的地方,大家补充好〜。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
利用MEGA 4构建分子系统进化树-图示
1、利用Clustal X软件对序列进行多重比对,保存的文件为aln格式。
2、利用MEGA 4软件将aln格式文件转换为meg文件,操作如下:
File按钮下的Convert To MEGA Format命令
点击后出现对话框,如下:
点击OK按钮,出现以下界面:
点击“保存”按钮,则aln文件成功转换为meg文件并保存在同一目录下。
3、关闭转换文件窗口,回到MEGA 4程序的主窗口,如下图:
点击“Click me to activate a data file”按钮,选择之前转换好的meg文件并打开,如下图:
选择所输入的数据类型(核酸or蛋白),之后点击OK即可。
此时,在MEGA4主程序窗口的底部出现了我们所输入的文件名(如下图),之后就可以构建分子系统进化树了。
4、通常选择邻接法(neighbor-joining,NJ)构建分子系统进化树。