单晶结构分析 用SHELXTL程序进行晶体结构分析的方法
SHELXTL程序进行晶体结构分析的方法

y
z
占有率 温度因子
C1 1 0.155832 0.182795 0.318988 11.00000 0.00001
温度因子: 原子是否正确 过大: 偏重 过小: 偏轻
B 进一步精修:
主体结构的确定
各向异性: ANIS 针对温度因子
ANIS 温度因子用六个参数表示,原子的热振动为 取向的三轴椭球
进行这步操作,可以大幅降低R1值和goof值
e 点refine菜单,系统会提示有Q 峰没命名,并 选择所有Q 峰,顺序与原来顺序相同
•删除原子或(Q)峰的方法: a 光标指在欲删除位置(原子或键) ,点K 键
b 光标指在欲删除位置,右手键点Bonds and Angles,再在对话框中点“Delete”
c 光标指在欲删除位置,右手键点“Delete” d 先选择,再点Select菜单中的Kill selected e 在ins文件删除
Most Disagreeable Reflections (* if suppressed or used for Rfree)
m 是一或两位数,指定氢的类型: =1 叔-H, =2 仲-H, =3(或13) 伯-H, =4 芳-H, =8(或14) X-O-H, =9 X=CH2或X-NH2, =15 笼状B-H
n 是一位数,指定固定的类型: =1 坐标、占有率、位移因子固定, =2 占有率、位移因子固定, =3(或7) 坐标固定, =4 同3,但允许修正X-H的键长(方向固定)
4.INS文件的建立和更新
结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的:
xprep、xshell—refine、xl、xp、edit、copy
单晶结构解析总结.

INS文件的建立和更新
结构解析和精修的过程,是ins文件建立和 不断更新的过程,这主要是下列过程实 现的: xprep、xshell—refine、xl、xp、edit、 copy
参数
R1 残差因子
衡量结构模型与真实结构的差异
wR或wR2 加权重的残差因子(计算方法的差异)
数据好的结构,一般可以可以精修到wR2 <0.15,而
4、XL (各向同性修正)(或差值F峰合成);
(1) 计算更新后的.ins文件或前边XL精修的结果,产生新 的.res(结果文件)和.lst文件(记录精修过程)
(2) 精修的参数 a 原子坐标(general positions
ห้องสมุดไป่ตู้
b 原子的位移参数(atomic displacement parameters)
单晶结构分析电子教案
第五章 用SHELXTL程序 进行结构分析的方法
H H HO HO HO OH O
H H H
OH
一 、 晶体学基本常识介绍
1. 单晶 2. 单晶的培养 3. 晶胞参数
4. 七大晶系、14种点阵、32个点群、
230个空间群
1. 晶体的选择与安置
2. 测定晶胞数据与基本对称性
二
3. 测定衍射强度数据
c 一个总标度因子 一个将实验中获得的衍射强度数 据校正为理论计算得到的F(000)一致的比例参数 d 其它可能参加的精修参数 无序结构中的占有率、消光效应参数、Flack参数等
H原子一般不参与精修,在结构精修中,往往被挷在与
它键合的原子(母原子)上,赋于是母原子1.2 ~1.5倍的 各向同性原子位移参数
几个参数:
•
• •
SHELXTL程序进行晶体结构分析的方法

数据后处理
对计算结果进行进一步的分析和处理,以满足特定需求或提高结果的可用性。
06
未来发展与展望
新技术与算法的引入
01
人工智能与机器学习
利用机器学习算法对晶体结构数据进行训练,提 高预测精度和效率。
02
云计算与分布式计算
借助云计算资源,实现大规模晶体结构分析的计 算和存储。
程序性能的优化与提升
05
常见问题与解决方案
计算速度慢的解决方法
01 优化算法
采用更高效的算法来加速计算过程。
02 使用更强大的计算机
利用具有更高计算能力的计算机进行计算。
03 并行计算
将计算任务分解为多个子任务,并使用多核处理 器或分布式计算系统同时处理这些子任务。
结果不准确的解决方法
提高输入数据的质量
01
Байду номын сангаас
确保输入数据的准确性,特别是原子坐标和晶胞参数。
晶体结构可以通过X射线晶体学、中子衍射、电子显微 镜等技术进行测定。
晶体结构分析的重要性
晶体结构分析是理解物质性质的基础,有助于揭 示物质的组成、结构和性质之间的关系。
通过晶体结构分析,可以深入了解物质的物理、 化学和生物学性质,为材料科学、药物研发、环 境科学等领域提供重要依据。
晶体结构分析的基本步骤
Shelxtl程序进行晶 体结构分析的方法
目录
• Shelxtl程序简介 • 晶体结构分析基本原理 • Shelxtl程序进行晶体结构分析的方
法 • 实例分析与应用 • 常见问题与解决方案 • 未来发展与展望
01
Shelxtl程序简介
程序起源与历史
起源
Shelxtl程序最初由英国谢菲尔德大学的科学家开发,旨 在提供一种高效、准确的晶体结构分析工具。
单晶结构解析讲义完美版

单晶结构解析讲义完美版晶体结构解析1.Shelxtl 使⽤流程※解析原始⽂件有hkl⽂件(或raw⽂件),包含衍射数据;p4p⽂件,包含晶胞参数※为⼀个晶体的数据建⽴project,该项⽬下所有⽂件具有相同的⽂件名;⼀旦在XPREP 中发⽣hkl⽂件的矩阵转换,则需要输出新⽂件名的hkl等⽂件,因此要建⽴新的project。
※⾸先运⾏XPREP,寻找晶体的空间群※然后运⾏XS,根据XPREP设定的空间群,寻找结构初解※在Xshell中观察初解是否合理,如不合理,需重回XPREP中设定其他的空间群2.Xshell 使⽤流程※找出重原⼦或者确定性⼤的原⼦※找出其余⾮氢原⼦※精修原⼦坐标※精修各项异性参数※找到氢原⼦(理论加氢或差值傅⾥叶图加氢)※反复精修,直到wR2等指标收敛。
最后的R1<0.06(0.08) wR2<0.16(0.18)※通过HTAB指令寻找氢键,判定氢的位置是否合理,并且将相关氢键信息通过HTAB和EQIV指令写进ins⽂件中※将原⼦排序(sort)3.cif ⽂件⽣成和检测错误流程※在步骤1、2完成后,在ins⽂件中加⼊以下三条命令bond $Hconfacta※此时⽣成了cif和fcf⽂件,将cif⽂件拷贝到planton所在⽂件夹中检测错误,也可以通过如下在线检测⽹址:/doc/aaaed6d749649b6648d74737.html /services/cif/checkcif.html※根据错误提⽰信息,修改或重新精修,将A、B类错误务必全部消灭,C类错误尽量消灭。
4.Acta E 投稿准备流程投稿前,请务必切实做好如下⼯作:※按步骤1、2、3解析晶体并⽣成相应cif和fcf⽂件。
※准备结构式图(Chemical structural diagram)、分⼦椭球图(Molecular ellipsoid diagram)和晶胞堆积图(Packing diagram),最好是pdf格式。
单晶结构解析

上一页
下一页
材料科学与化学工程学院
2) 用XS程序定出结构雏形(初始套) 一般先试直接法,再试Pattson法,用什么方法 是通过改变Edit .ins文件中的指令来实现的 直接法:
TITL 020908b in C2/c CELL 0.710730 30.1927 8.5175 13.9108 90.0000 95.1300 90.0000 ZERR 8.00 0.0146 0.0042 0.0071 0.0000 0.0100 0.0000 LATT 7 SYMM -X, Y, 0.5-Z SFAC C H N O Cr UNIT 144 112 24 56 8 TEMP 25 TREF HKLF 4 END
2012-8-26
上一页
下一页
材料科学与化学工程学院 SPACE GROUP DETERMINATION …… Mean |E*E-1| = 0.713 [expected .968 centrosym and .736 non-centrosym] Chiral flag NOT set
Systematic absence exceptions: b-c-n-21-N 247 240 237 6 N(I>3s)231 224 221 4 <I> 113.3 120.8 139.2 0.8 <I/s> 28.7 27.3 28.2 9.3 Option Space Group No. [A] P222(1) #17 [B] P2(1)2(1)2 #18 Select Option [B] : -c156 144 187.9 29.5 -a155 141 194.4 29.3 CSD 26 359 -n153 127 108.3 23.5 -216 0 0.1 1.3 --a 74 70 131.0 26.1 --b 74 68 139.3 27.4 --n 76 66 102.7 26.0 CFOM 5.73 2.37 --21 11 3 1.1 5.2
SHELXL结构解析入门教程
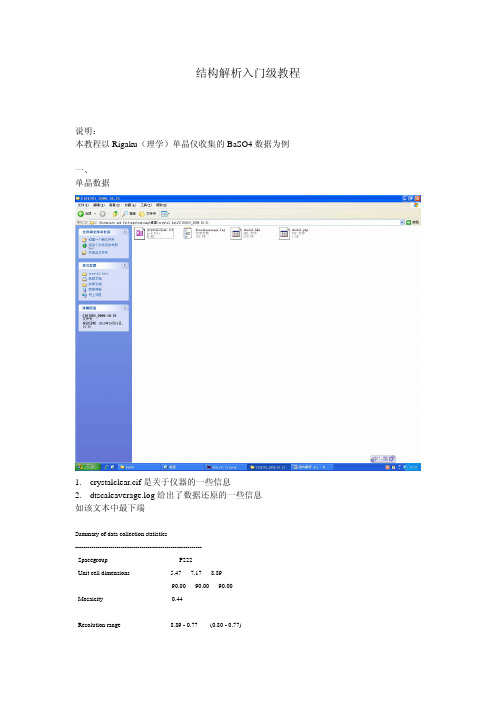
结构解析入门级教程说明:本教程以Rigaku(理学)单晶仪收集的BaSO4数据为例一、单晶数据1.crystalclear.cif是关于仪器的一些信息2.dtscaleaverage.log给出了数据还原的一些信息如该文本中最下端Summary of data collection statistics-------------------------------------------------------------Spacegroup P222Unit cell dimensions 5.47 7.17 8.8990.00 90.00 90.00Mosaicity 0.44Resolution range 8.89 - 0.77 (0.80 - 0.77)Total number of reflections 2726Number of unique reflections 503Average redundancy 5.42 (4.49)% completeness 100.0 (100.0) 完成度要求96%以上Rmerge 0.052 (0.116) R值要求0.1一下Reduced ChiSquared 0.74 (0.58)Output <I/sigI> 20.5 (6.8) 强度10-30较好,过强或过若均不好-------------------------------------------------------------Note: Values in () are for the last resolution shell.3.shelxl.hkl和shelxl.p4p为解结构所需要的数据二、软件介绍所用的的子程序:XPREP:选择空间群,输入元素XS:应用直接法或帕特森法进行初步的解析XL和XP:原子指认和结构精修XCIF:产生结构报表三、结构解析3.1运行XPREPa.新建任务选中shelxl.hkl任意输入,不与之前的重复即可b.运行XPREP强度是否有对称中心输入元素,注意大小写输入新的文件名输入Y 3.2运行XS选中a.hkl,而不再是shelxl.hkl输入新的任务名,不要与之前的重复红线部分稍微关注下。
SHELXTL程序进行晶体结构分析的方法报告

XP中做的图形文件
晶体学信息文件
结构因子文件
记录仪器型号、晶体外观等的文件
SHELXTL结构分析的步骤
1、准备反射点文件 .HKL,格式一般为h, k, l, Io, (I) 。 2、使用 XPREP 程序输入晶胞参数,进行数据统计和检查消光规律以确 定空间群,输入分子式等等,程序结束时输出 .INS。该 .INS文件通常设 定了直接法的现行设置。 3、使用 XS 解析结构,读入 .HKL和 .INS文件,结果输出到 .LST和 .RES文件。 4、使用 XP/XSHELL 读入 .RES文件,建立初始结构模型,结果输出到 .INS文件,该 .INS文件通常设定了最小二乘法修正和 Fourier 合成的设 置。 5、使用 XL 读入 .HKL和 .INS文件,进行结构模型的最小二乘法修正和 Fourier合成(通常为差值Fourier合成),结果输出到 .LST和 .RES文件。 6 、重复 4 , 5过程不断扩展完善结构模型和精修,直到结构修正收敛和 偏离因子最低。 7、生成数据CIF表格。
3.其它文件
晶体结构报表文件 4.INS文件的建立和更新 结构解析和精修的过程,是ins文件建立和不 断更新的过程,这主要是下列过程实现的: xprep、xshell—refine、xl、xp、edit、copy
res lst plt cif fcf pcf tex
xs、xl、refine产生的文件
单晶结构分析电子教案
用SHELXTL程序 进行结构分析的方法
H H HO HO HO OH O
H H H
OH
SHELXTL有以下程序: windows 界面
XPREP:为结构解析准备 .INS文件程序。
单晶结构解析

两个必要文件(由XPREP程序产生) name.hkl, name.ins
结构解析和精修的过程,是ins文件建立和不断更新的过程, 这主要是下列过程实现的:XPREP、XS、XL、XP
其它文件
res lst plt cif fcf pcf tex
xs、xl、refine产生的文件 记录xs、xl、refine过程和结果的文件 XP中做的图形文件 晶体学信息文件 结构因子文件 记录仪器型号、晶体外观等的文件
画图
XCIF
打印表格
二、数据处理--XPREP
运行步骤:
1.从name.hkl文件(若存在)或name.raw文件中读入衍射 点;2.从name.p4p或键盘获得单胞参数及误差 3.判断晶格类型 4.寻找最高对称性
5.确定空间群
6.输入分子 7.建立 name.hkl和 name.ins
* XPREP的主要功能和应用
读入、更改、 •单击进入XPREP程序 合并衍射数据 •根据程序的提示输入晶胞参数 •选择可能的晶格 程序则显示以下菜单: 计算显示 Patterson截面 寻找更高 的对称性 确定或输 入已知的 空间群
[D] Read,Modify or Merge DATDSETS [P] Contour PATTERSON Secions [H] Search for HIGHER mertric symmetry [S] Determine or input SPACE GROUP
•XS计算结果的评估
# 直接法,RE越小越好,一般大于0.3,就预示 着不成功,可以尝试用Patterson法来解
N
O
C u (N O )2 3
+
O N
E tO H
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
信 息 区
命 令 区
DFIX 1.43 0.02 o5 c27 WGHT 0.074900 1.631200 FVAR 0.169240 TEMP 25
原子表 命 坐标、占有率、温度因子 令 区
Co1 5 0.639436 0.182296 0.778299 11.000000 0.037950 = 0.043120 0.052330 -0.002120 0.007180 -0.002370 Co2 5 0.132299 0.088237 0.782543 11.000000 0.061270 = 0.080610 0.082660 -0.004140 0.011300 0.005080 N1 3 0.816545 0.181404 0.857527 11.000000 0.048210= 0.044720 0.042580 -0.001460 0.010450 -0.001390 N2 3 0.641818 0.098803 0.864065 11.000000 0.055810 = 0.050870 0.070080 0.000700 0.013560 -0.009990 …………………………………………………………………. HKLF 4 衍射点文件类型 END
记录xs、xl、refine过程和结果的文件
XP中做的图形文件
晶体学信息文件
结构因子文件
记录仪器型号、晶体外观等的文件
SHELXTL的主要子程序和文件
XS XPREP XP
*.plt *.sav *.ps
*.hkl
*.ins
*.HELL
*.lst
*.cif *.fcf
用这种方法时,首先利用重原子的特征峰,即 Harker峰,求出重原子坐标,再通过Fourier合成 获得其它原子的坐标 Harker峰,或Harker截面,就是同一套等效的 原子组成的Patterson峰,由于平方效应,重原子的 Harker峰会显得十分突出,寻找起来一般比较容易。 因此,可以轻松地从分析重原子的Harker峰,得到 重原子的坐标
一、SHELXTL文件 1. 文件名 一般,同一结构,所有文件都用相同的名 (不能超过8个字符),只是扩展名不同 2. 两个必要文件(由XPREP程序产生) *.hkl文件: 所有的衍射点,每一点一行。 格式为:h k l F2 σ (F2)
-4 4 -4 -4 2 -2 -2 -2 -2 2 -2 -2 21.189 19.539 17.129 20.459 3.050 2.120 2.710 3.120
设臵好ins文件 ,点击XS,就可自动进行计算
•XS计算结果的评估
# 直接法,RE越小越好,一般大于0.3,就预示 着不成功
# 打开Xshell,检查计算出的原子或(Q) 峰位 臵是否构成合理的化学结构
# 精修(refint)一次后,R1值是否大小于0.5; 或继续精修R1值是否很快降低
a 将|Fo|转化为归一化结构因子|Eo|
b 建立可以利用正切公式的三相角及四相角关系
c 赋于起始相角 d 利用正切公式精修相角
e 计算诊断指标,判断各套相角的质量 f 采用诊断指标最佳的相角数据计算解析电子密度图,即E图
* Patterson法是其本人1934年提出,通常只用来 解析含有重原子的结构
指令 END EQIV ESEL EXTI EXYZ FLAT FMAP FREE FVAR HFIX HKLF HTAB
含
义
指令输入结束 提供分子内或分子间键合原子的对称操作码 限制E值的下、上限 对晶体消光效应参数进行精修 让两个或多个原子具有相同的坐标 限制指定原子在相同的平面上 所计算Fourier图的类型
c 用Select菜单(或背景右手键点“Select‖)可 选择部分和所有的原子或Q 峰,顺序与原顺序相同 d 按住“Shift‖键,左手键拖出一个选择框并同 时轻开,可选择一个或多个原子
e 点refine菜单,系统会提示有Q 峰没命名,并 选择所有Q 峰,顺序与原来顺序相同
•删除原子或(Q)峰的方法: a 光标指在欲删除位臵(原子或键) ,点K 键 b 光标指在欲删除位臵,右手键点Bonds and Angles,再在对话框中点“Delete‖ c 光标指在欲删除位臵,右手键点“Delete‖
指令
含
义
SYMM 所属空间群的对称操作 TEMP 衍射数据收集的温度 TITL 样品的编号(或名称)和空间群
UNIT
晶胞中每种原子的总个数
WGHT 指定所用权重
ZERR 晶胞中分子个数和晶胞参数的标准偏差
三、 SHELXTL结构分析的步骤
1.项目的设立 打开SHELXTL程序:点project 输入文件名,查找到hkl 文件并打开 new,
不计算指定原子对的键长、键角 全比例系数 限制H原子在理想位置上 衍射数据的格式 计算氢键
指令 ISOR L.S. LATT
含 义 限制指定原子的位移参数类似于各向同性 指定XL中用最小二乘法进行精修的轮数 晶格的类型.依次为:P I R F A B C,无心为负值
MOVE 移动或转换坐标 MPLA 计算平面 OMIT PART PLAN SFAC SIMU SIZE 忽略指定的衍射点或限定theta角范围 划分成键原子的范围(用于无序结构) 计算和列出Q峰的数目 晶体中存在的原子的种类 限制指定范围内的原子有相同的位移参数 晶体的大小
c 如果独立单元只是分子的一部分,原子不好取 舍,可用右手键菜单中的GROW指令把分子长全处理
* 如果仍得不到较为合理的初始结构,则应试
验用三种方法中的另一法,或试验用不同的ESEL值
* 如果试验用各种方法,仍不行,那就要考虑
是不是出现了如下的问题:
结构的解析中的若干问题
A 晶胞的错误确定 用CCD探测器,仪器可以自动确定晶胞(分别 在matrix、sadabs和xprep中),但有时会不正确, 因而晶系和空间群会因之错误 解决方法:在xprep中重新确定晶胞;实在不 行,需用收集数据重新做晶胞 B 空间群的错误指认
继续尝试直接法:
TITL 020908b in C2/c CELL 0.710730 30.1927 8.5175 13.9108 90.0000 95.1300 90.0000 ZERR 8.00 0.0146 0.0042 0.0071 0.0000 0.0100 0.0000 LATT 7 SYMM -X, Y, 0.5-Z 归一化结构因子E SFAC C H N O Cr 的 下、上限值, UNIT 144 112 24 56 8 可试探性设臵 TEMP 25 ESEL 1.5 3.5 TREF HKLF 4 END
单晶结构分析电子教案
第五章 用SHELXTL程序 进行结构分析的方法
H H HO HO HO OH O
H H H
OH
new open copy archive delete exit
Xl XH
edit edit .res edit .ins edit .cif edit .prp edit .lst copy .res→.ins
…………………………………..
标题 晶胞参数 X光波长 空间群 TITL 041203e in P2(1)/c *.ins 文件:组成上可分成五部分 CELL 0.71073 11.5870 19.3080 17.2144 90.000 108.471 90.000 晶格类型 ZERR 4.00 0.0069 0.0112 0.0102 0.000 0.009 0.000 对称操作码 LATT 1 单胞中 SYMM -X, 0.5+Y, 0.5-Z 单胞中 分子数目 SFAC C H N O Co S 原子数目 晶胞参数的标 准偏差 UNIT 108 104 40 20 8 16 原子类型
2) 用XS和XSHELL程序定出结构雏形(初始套) 一般先试直接法,再试Pattson法,用什么方法 是通过改变Edit .ins文件中的指令来实现的
直接法:
TITL 020908b in C2/c CELL 0.710730 30.1927 8.5175 13.9108 90.0000 95.1300 90.0000 ZERR 8.00 0.0146 0.0042 0.0071 0.0000 0.0100 0.0000 LATT 7 SYMM -X, Y, 0.5-Z SFAC C H N O Cr UNIT 144 112 24 56 8 TEMP 25 TREF HKLF 4 END
d 先选择,再点Select菜单中的Kill selected
e 在ins文件删除
• 恢复删错的原子或(Q)峰的方法:
点―Edit>Restore Killed Atom‖,先选择要恢 复的原子(或Q峰),再点“Restore‖ • 命名(标记)原子或(Q)峰的方法:
a 先选择,再用Lablels菜单中的Group label
*.tex
二、常用的XS和XL指令 指令 含 义 ACTA 产生cif文件 AFIX 将原子坐标强制性地固定在指定位臵上, 或在指定位臵上产生原子 ANIS 将各向同性换成各向异性精修 BOND 计算键长、键角(加$H包括H的键长、键角) BIND 计算指定原子对的键长、键角 CONF 计算扭转角 DELU 限制指定原子具有相似的位移参数 DFIX 限定指定原子对间的距离 EADP 给两个或多个原子指定相同的位移参数
b 光标指在要命名的位臵,用右手键的Edit c 光标指在欲命名位臵,右手键点Bonds and Angles,再在对话框中点“Rename‖
•常用 的小技巧:
a 解析和精修结构 的初期,可把C、O、N都定为C