ESyPred3D蛋白质三级结构预测
蛋白质三级结构预测(swiss-model同源建模)
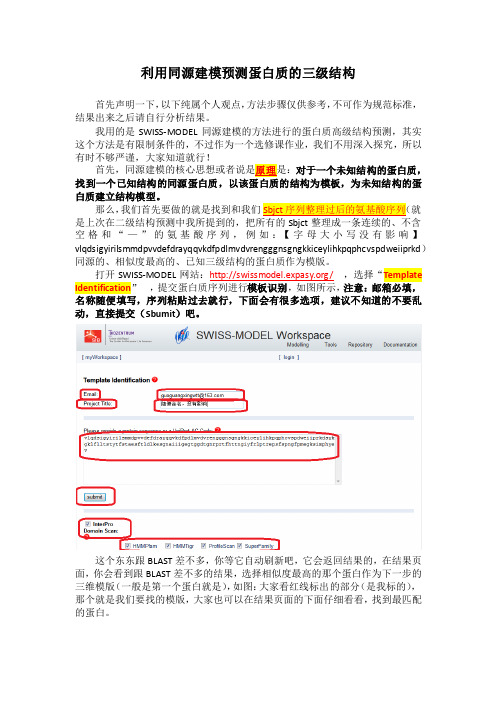
利用同源建模预测蛋白质的三级结构首先声明一下,以下纯属个人观点,方法步骤仅供参考,不可作为规范标准,结果出来之后请自行分析结果。
我用的是SWISS-MODEL同源建模的方法进行的蛋白质高级结构预测,其实这个方法是有限制条件的,不过作为一个选修课作业,我们不用深入探究,所以有时不够严谨,大家知道就行!对于一个未知结构的蛋白质,白质建立结构模型。
那么,我们首先要做的就是找到和我们空格和“—”的氨基酸序列,例如:【字母大小写没有影响】vlqdsigyirilsmmdpvvdefdrayqqvkdfpdlmvdvrengggnsgngkkiceylihkpqphcvspdweiiprkd)同源的、相似度最高的、已知三级结构的蛋白质作为模版。
打开SWISS-MODEL网站:/,选择“Template Identification,提交蛋白质序列进行模板识别,如图所示,注意:邮箱必填,名称随便填写,序列粘贴过去就行,下面会有很多选项,建议不知道的不要乱动,直接提交(Sbumit)吧。
这个东东跟BLAST差不多,你等它自动刷新吧,它会返回结果的,在结果页面,你会看到跟BLAST差不多的结果,选择相似度最高的那个蛋白作为下一步的三维模版(一般是第一个蛋白就是),如图:大家看红线标出的部分(是我标的),那个就是我们要找的模版,大家也可以在结果页面的下面仔细看看,找到最匹配的蛋白。
这里还有一点要作说明,就是上图标出的代码是PDB编号,前四个表示PDB- Code,最后一位表示Chain-ID,具体什么意思,大家有兴趣就去了解一些吧。
接下来,去NCBI串串门吧,在NCBI中搜索上面查到的蛋白的PDB号,一般输入前四位就行啦,注意:搜索蛋白库(Protein)。
找到以后,以FASTA格式显示。
接下来,我们再回到SWISS-MODEL,接下来就是重点和难点啦,在线提交序列进行同源建模分析,这个在线提交不是大家想象的那么容易,这个耗费了我大部分的时间,说到这里我就想画个圈圈诅咒它,大家注意啦~~~~~~~~~~~SWISS-MODEL 是一个自动化的蛋白质比较建模服务器,该服务器提供用户三种模式可选择:Automatic mode(简捷模式): 用于建模的氨基酸序列或是Swiss-Prot/TrEMBL (/sprot )编目号(accession)可以直接通过web界面提交。
蛋白质三级结构的预测和分析方法

蛋白质三级结构的预测和分析方法蛋白质是由氨基酸组成的多肽链,是生命体中重要的组成部分。
蛋白质的功能由其三级结构决定,因此蛋白质三级结构的预测和分析是生物学研究的重要课题之一。
本文将介绍蛋白质三级结构的预测和分析方法。
一、蛋白质序列的预测蛋白质的三级结构是由其氨基酸序列决定的,因此蛋白质序列的预测是蛋白质三级结构预测的第一步。
蛋白质序列的预测分为两种方法:直接预测和间接预测。
直接预测方法是通过实验手段对蛋白质进行测定,并得到其序列。
其中,蛋白质测序是最常用的方法之一,目前已经非常成熟,在实验过程中准确率很高。
但是该方法耗时长、成本高,适用性窄。
间接预测方法则基于蛋白质序列的相似性进行预测,即通过基因组学、区域同源性、数据库、机器学习算法等,对已知的蛋白质序列进行比对和分析,得出未知蛋白质的序列。
其中,BLAST、PSI-BLAST等比对方法,能够在较短时间内对蛋白质序列进行预测,并在很大程度上提高了预测准确率。
此外,还有一些基于机器学习算法的预测方法,如SVM和神经网络方法等。
二、蛋白质结构预测蛋白质结构预测是指通过已知的蛋白质序列,预测出其原子级别的三维结构。
蛋白质结构预测目前主要分为三种方法:实验法、遗传算法和分子动力学模拟法。
实验法主要是通过分析蛋白质结晶体、核磁共振法和质谱分析等实验手段来预测蛋白质的空间结构。
这种方法具有实验数据来源充足、准确性高等特点,但是往往耗时长且成本高昂。
遗传算法是利用生物进化过程的基本原理,在计算机模拟中模拟蛋白质分子构象变化的过程,最终找到能够形成最稳定结构的构象。
这种方法通过逐代优化,逐渐提高预测蛋白质结构的准确度,但是也存在时间复杂度高、无法解释性和结果可重复性差等问题。
分子动力学模拟法是运用牛顿力学原理和一些计算模型,对蛋白质分子的运动进行数值模拟,从而得到蛋白质的三维结构。
这种方法的优点在于可以对蛋白质分子动力学过程进行模拟,具有可重复性高、得出结果的信息较多等特点,但是计算时间较长,计算机模拟结果的可信度也需要进一步验证。
蛋白质三级结构预测共22页文档

26、机遇对于有准备的头脑有特别的 亲和力 。 27、自Байду номын сангаас是人格的核心。
28、目标的坚定是性格中最必要的力 量泉源 之一, 也是成 功的利 器之一 。没有 它,天 才也会 在矛盾 无定的 迷径中 ,徒劳 无功。- -查士 德斐尔 爵士。 29、困难就是机遇。--温斯顿.丘吉 尔。 30、我奋斗,所以我快乐。--格林斯 潘。
谢谢你的阅读
❖ 知识就是财富 ❖ 丰富你的人生
71、既然我已经踏上这条道路,那么,任何东西都不应妨碍我沿着这条路走下去。——康德 72、家庭成为快乐的种子在外也不致成为障碍物但在旅行之际却是夜间的伴侣。——西塞罗 73、坚持意志伟大的事业需要始终不渝的精神。——伏尔泰 74、路漫漫其修道远,吾将上下而求索。——屈原 75、内外相应,言行相称。——韩非
5 蛋白质三级结构预测

• 令代表核心折叠C中的环到序列S中空位的 映射,显然是通过线索化而确定的。
令f(t)是进行比对的得分函数,其定义如下:
f(t) = g1 (v,t) + g2 (u,v,t) + g3 (,t)
• g1 (v,t) 评价氨基酸残基v所处的位置 • g2 (u,v,t) 评价残基u和v的相对位置,如果u和v 键合,则得 分高; • g3肖 飞
蛋白质三级结构预测的方法
1
2 3
方法比较
同源建模(比较建模)
基础 - 相似的序列结构相近 - PDB结构数据库的快速增长 - 结构基因组学的启动 - 发散进化 特点 - 相对精确可靠
• 假设待预测三维结构的目标蛋白质为U (Unknown),利用同源模型化方法建立结 构模型的过程包括下述6个步骤: (1)搜索结构模型的模板(T) (2)序列比对 U T (3)建立骨架 (4)构建目标蛋白质的侧链 (5)构建目标蛋白质的环区 (6)优化模型
至于最后建立三维结构模型则是非常困难的
• 线索化的主要思想: 利用氨基酸的结构倾向(如形成二级结构 的倾向、疏水性、极性等),评价一个序 列所对应的结构是否能够适配到一个给定 的结构环境中。
• 建立序列到结构的线索的过程称为线索化, 线索技术又称折叠识别技术。 • 线索化或者折叠识别的目标是为目标蛋白质 U寻找合适的蛋白质模板,这些模板蛋白质 与U没有显著的序列相似性,但却是远程同 源的。
新的趋势 混合预测方法 在比较建模法和折叠识别法中使用从头预 测法来预测部分难以找到模板的片断 在从头预测法中使用二级结构预测的结果 和其他已知结构信息辅助建模
• Meta-predictor 使用多个预测方法 对收集的结果进行综合比较和分析 改进收集的结果
蛋白质三维结构预测

蛋白质三维结构预测第四节蛋白质三维结构预测1、同源模型化方法同源模型化方法是蛋白质三维结构预测的主要方法(Blundell 1987)。
对蛋白质数据库PDB分析可以得到这样的结论:任何一对蛋白质,如果两者的序列等同部分超过30%(对于排列长度大于80),则它们具有相似的三维结构,即两个蛋白质的基本折叠相同,只是在非螺旋和非折叠区域的一些细节部分有所不同。
蛋白质的结构比蛋白质的序列更保守,如果两个蛋白质的氨基酸残基序列有50%相同,那么约有90%的碳原子的位置偏差不超过3 ?。
这是同源模型化方法在结构预测方面成功的保证。
同源模型化方法的主要思想是:对于一个未知结构的蛋白质,首先通过同源分析找到一个已知结构的同源蛋白质,然后,以该蛋白质的结构为模板,为未知结构的蛋白质建立结构模型。
这里的前提是必须要有一个已知结构的同源蛋白质。
这个工作可以通过搜索蛋白质结构数据库来完成,如搜索PDB。
同源模型化方法是目前一种比较成功的蛋白质三维结构预测方法。
从上述方法介绍也可以看出,因为预测新结构是借助于已知结构的模板而进行的,选择不同的同源的蛋白质,则可能得到不同的模板,因此最终得到的预测结果并不唯一。
假设待预测三维结构的目标蛋白质为U(Unknown),利用同源模型化方法建立结构模型的过程包括下述6个步骤:(1)搜索结构模型的模板(T)。
同源模型化方法假设两个同源的蛋白质具有相同的骨架。
为待预测的蛋白质建立模型时,首先按照同源蛋白质的结构建立模板T。
所谓模板是一个已知结构的蛋白质,该蛋白质的与目标蛋白质U的序列非常相似。
如果找不到这样的模板,则无法运用同源模型法。
(2)序列比对。
将目标蛋白质U的序列与模板蛋白质序列进行比对,使U的氨基酸残基与模板蛋白质的残基匹配。
比对中允许插入和删除操作。
(3)建立骨架。
将模板结构的坐标拷贝到目标U,仅拷贝匹配残基的坐标。
在一般情况下,通过这一步建立目标蛋白质U的骨架。
(4)构建目标蛋白质的侧链。
蛋白质三级结构预测方法简述

文章编号 -7223TMYYJ +1223"23T22K[T21
T!"T
蛋白质三级结构预测方法简述
张 漫 常延琦 7" +天津市兽药监察所 天津
K22Y21"
结构 ’ 进行目标蛋白质的序列与模 板序列的比对 ’ 根据模板蛋白质的 三维结构构建目标 蛋 白 质 的 结 构 ’ 对模建结构进行能量优化 * 分子动 力学优化及可信度评估 ( 首先需要通过序列比较方法在 序列库和结构库中搜索与目标蛋白 质相似性足够高的蛋白质作为模 板 ( 比如可使用 8&# ,N,+&0 到结 构库 8ON +8?;@CAF O>@> N>FP" 中 去 搜索 ’ 也可以通过序列结构穿线法
CG/(’N<* 举 行 的 HC?> 大 会 上 对 所
! " 中国农业大学生物学院 北京 !#$$%&
!+&8( 1 同源模建9:;<=>?>@ABC <;DCEAFG 6
同源模建即比较模建 ’ 是目前在 蛋白质结构预测中最为成功的方 法 ( 应用同源模建的方法进行蛋白 质结构预测 ’ 需要目标蛋白质至少 有一个已知三维结构的同源蛋白 质 ’ 即两者之间要有较高的相似性 ( 同源模建的准确性取决于目标序列 和模板之间的相似性大小 ’ 同时也 与两个蛋白质结构和功能的相似性 相关 ( 如果目标蛋白质与模板序列 的相似性大于 32H ’ 则可以得到 高 精度的同源模建结果 ’ 即主链的均 方根 +*.&" 只有 2I7 F< 左右 ’ 可 以 与中等分辨率的核磁共振或低分辨 率 J, 射 线 衍 射 得 到 的 结 果 相 媲 美 ( 如果相似性界于 K2HL32H ’ 则 可以得到中等精度的结果 ’ 此时约
蛋白质三维结构预测方法

蛋白质三维结构预测方法
嘿,你知道吗?蛋白质的三维结构预测可不是件容易的事儿呀!就好像要解开一个超级复杂的谜题!比如说,想象一下你要拼一个超难的拼图,每一块都得放对位置,这就是蛋白质三维结构预测要做的事情。
现在呢,有好几种方法来对付这个难题。
有一种方法叫同源建模,就像是找一个跟目标蛋白质很像的“模板”,然后根据这个模板来推测结构,就好比你有个好朋友已经完成了拼图,你可以参考他的来拼。
比如说,如果我们要研究一个新的蛋白质,发现它和之前已经研究清楚的某个蛋白质很相似,那我们就能用同源建模啦!
还有一种方法叫从头预测,这可厉害了!完全从零开始,就像你在一片空白的地方自己创造出那个拼图的样子。
哎呀,这得多难啊,但科学家们可牛了呢!比如说对于一些全新的、没有类似模板的蛋白质,就得靠这种办法啦!
另外呢,也有结合实验数据的方法,这就像是有了额外的提示来帮助你完成拼图。
比如说通过 X 射线衍射等实验手段得到一些结构信息,再结合
计算方法来完善预测。
总之啊,蛋白质三维结构预测就像是一场刺激的冒险!有这么多有趣又厉害的方法,难道你不想更深入地了解一下吗?我觉得这些方法都太神奇了,它们为我们揭示蛋白质的奥秘打开了大门呀!。
蛋白质三级结构预测ppt课件

15
完整版课件
Байду номын сангаас
16
完整版课件
17
完整版课件
18
完整版课件
19
作业:对给定的seq1 进行三级结构预测,并用Pdbview可 视化结果
完整版课件
20
感谢亲观看此幻灯片,此课件部分内容来源于网络, 如有侵权请及时联系我们删除,谢谢配合!
蛋白质三级结构预测(Swissmodel)
完整版课件
1
Swissmodel基于同源建模的方法 是目前三级结构预测当中基于同源建模方法做的最好 的一个之一 优点是:如果能得到结果往往很可靠 缺点是:很多时候得不到结果
完整版课件
2
例:给定的example 进行三级结构预测
完整版课件
3
主页
完整版课件
4
完整版课件
5
选用 Automated mode
完整版课件
6
完整版课件
7
输入序列、Email、姓名、序列名字,然后提交
完整版课件
8
等待结果,结果发到邮箱中
完整版课件
9
完整版课件
10
三级结构可视化(Pdbviewer)
完整版课件
11
完整版课件
12
完整版课件
13
完整版课件
14
完整版课件
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
BIOINFORMATICSVol.18no.92002Pages1250–1256ESyPred3D:Prediction of proteins 3D structuresChristophe Lambert ∗,Nadia L´eonard,Xavier De Bolle and Eric DepiereuxFacult´es Universitaires Notre-Dame de la Paix,Unit´e de Recherche en Biologie Mol´eculaire,Rue de Bruxelles,61,B-5000Namur,BelgiumReceived on February 1,2002;revised on March 7,2002;accepted on March 18,2002ABSTRACTMotivation:Homology or comparative modeling is cur-rently the most accurate method to predict the three-dimensional structure of proteins.It generally consists in four steps:(1)databanks searching to identify the struc-tural homolog,(2)target–template alignment,(3)model building and optimization,and (4)model evaluation.The target–template alignment step is generally accepted as the most critical step in homology modeling.Results:We present here ESyPred3D,a new automated homology modeling program.The method gets benefit of the increased alignment performances of a new alignment strategy.Alignments are obtained by combining,weighting and screening the results of several multiple alignment programs.The final three-dimensional structure is build using the modeling package MODELLER.ESyPred3D was tested on 13targets in the CASP4experiment (C ritical A ssessment of Techniques for Proteins S tructural P rediction).Our alignment strategy obtains better results compared to PSI-BLAST alignments and ESyPred3D alignments are among the most accurate compared to those of participants having used the same template.Availability:ESyPred3D is available through its web site at http://www.fundp.ac.be/urbm/bioinfo/esypred/.Contact:mbert@fundp.ac.be;http://www.fundp.ac.be/∼lambertcINTRODUCTIONThree-dimensional (3D)protein structure is an important source of information to better understand the function of a protein,its interactions with other compounds (ligands,proteins,DNA,...)and to understand phenotypical effects of mutations (Tramontano,1998).The 3D protein struc-ture can be predicted according to three main categories of methods (Rost and O’Donoghue,1997):(1)homology or comparative modeling (described below);(2)fold recogni-tion (predicting the global fold of a protein);(3)ab initio techniques (trying to model the 3D structure of proteins using only the sequence and a force field).∗To whom correspondence should be addressed.Homology modeling is historically the first (Browne et al.,1969)and the most accurate method (Sanchez and Sali,1997).It was shown during the last CASP experiment (Venclovas et al.,1999)(C ritical A ssessment of Techniques for Proteins S tructural P rediction)that the critical steps are:(1)template selection,(2)target–template alignment step,(3)modeling of regions not present or significantly different from those in template and (4)modeling of side chains.Among these critical steps,it is commonly accepted that the target–template alignment step is the most critical (Mosimann et al.,1995;Martin et al.,1997).It is known that above 50%of identity rate between target and template,pairwise alignments provide accurate models.Between 30%and 50%of identity,multiple align-ments between target,template and similar proteins must be used and the pairwise alignments between target and template must be extracted from this multiple alignment.Below 30%of identity rate,only heuristic combinations of multiple alignments,experimental data and know-how of an expert are able to generate an accurate model.A large number of techniques have been developed to predict 3D structures of proteins by homology modeling.For the target–template alignment step,most of them use PSI-BLAST (Altschul et al.,1997),PileUp (Wisconsin Package Version 9.1,Genetics Computer Group (GCG),Madison,Wisc.),ClustalW (Thompson et al.,1994),3D-PSSM (Fischer et al.,1999),SAMT99(Karplus et al.,1997),or also the alignment producing the best model out of a collection computed from various alignment programs (Yang and Honig,1999).Our laboratory developed the MATCHBOX multiple sequence alignment software in the early 1990s (De-piereux and Feytmans,1992)and it has proved to be one of the most accurate in terms of specificity (Depiereux et al.,1997).Much effort has been consented into im-proving alignment accuracy by adding information such as secondary structure predictions,solvent accessibility predictions,specific scoring matrices and combination with ClustalW.In all cases,it was only possible to slightly improve multiple alignment accuracy (unpublished re-1250cOxford University Press 2002ESyPred3D:Prediction of proteins3D structuressults).Meanwhile,no significant improvements in align-ment performance have been published by other groups. Furthermore,no alignment method can be qualified as the absolute most reliable one.Indeed,benchmarks(Briffeuil et al.,1998;Thompson et al.,1999)have shown that comparative performances of alignment programs are deeply dependent on the set of aligned sequences.In this work,we tackle the target–template alignment problem by developing a specific program to align target and template sequences in homology modeling.Matching of homologous segments is improved by incorporation of the results of several multiple alignment programs.Results are scored to optimize the performances and screened to remove incompatible matches.Several algorithmic prob-lems have required specific developments in order to gen-erate and efficiently screen the database of the various and often incompatible alignments proposed by the different algorithms.This new alignment strategy is included into our ESyPred3D program(http://www.fundp.ac.be/urbm/ bioinfo/esypred/)that predicts the3D structure of proteins using the homology modeling approach.SYSTEM AND METHODSOur automatic program(ESyPred3D)implements the four steps of the homology modeling approach(Eisen-haber et al.,1995):(1)databanks searching to identify the structural homolog,(2)target–template alignment, (3)model building and optimization and(4)model eval-uation(not implemented at the time of the CASP4ex-periment).ESyPred3D was run on an SGI Octane Dual processor225MHz workstation under IRIX6.5. Identifying the structural homologTofind homologs to the target sequence,PSI-BLAST2.0.14 (downloaded from NCBI and run locally)is run using the latest possible version of the NR databank(NCBI).The chosen template is the sequence from the latest version of the PDB databank with the lowest expected value after four iterations.The cutoff for the expected value is0.0001 (-hflag).If no template is found with these criteria,the program stops.Aligning sequences and constructing the3D model According to Thompson et al.(1999),the quality of the alignment of sequences highly depends on the context of the alignment.The results obtained for a given pair of sequences may be different depending on the set of sequences submitted to multiple alignment programs.So, after fetching all sequences retrieved by PSI-BLAST,two sets of sequences are generated in order to create two different computational conditions for running multiple alignment programs:the set A contains the50best hits including the target and the template(the number of sequences is limited to50to reduce computing time).The set B is a subset of at least seven sequences,including the target and the template,produced by dropping too redundant sequences with the PURGE program(provided with the Gibbs package(Neuwald et al.,1995)).The BLAST score(using the-bflag)to select or eliminate sequences during the PURGE operation is250.The building of the target–template alignment is per-formed in these steps(see Figure1):(a)Matching.Both sets of sequences(A and B)arealigned byfive alignment programs emerging fromtwo benchmarks(Briffeuil et al.,1998;Thompson etal.,1999).These programs are:ClustalW,Dialign2(Morgenstern,1999),Match-Box(Depiereux et al.,1997),Multalin(Corpet,1988)and PRRP(Gotoh,1996).Ten multiple alignments are generated,eachone including the target and template sequences.Then,the pairwise alignments between the targetand template sequences are extracted,leading to tendifferent pairwise alignments between the target andthe template.(b)Database building.Each position of the alignmentsis stored in a database,all the redundant results,i.e.the same amino acid placed at the same positionby different programs,being scored in a frequencytable.(c)Screening.The position with the highest score istaken as thefirst anchor point to build thefinaltarget–template alignment.Incompatible results(seeFigure2.),aligning regions located up-and down-stream anchor points,are removed from the database.The process is pursued,new anchor positions beingdetermined,and incompatible regions being elimi-nated,until all results are selected or removed.Thefinal target–template alignment is thus composedby the most frequent aligned positions,under thecondition of compatibility.Thisfinal pairwise alignment is used by the MODEL homology modeling routine of MODELLER release4 (Sali and Blundell,1993;Sali et al.,1997)to build a 3D model of the target protein.This routine includes the satisfaction of spatial and geometric restraints and a very fast molecular dynamic annealing:no other refinements were applied.Participation to the CASP4experimentESypred3D server participated to the CASP4experiment (see complete results at / casp4/;group218:LAMBERT-CHRISTOPHE).All the models generated by MODELLER were submitted to the CASP4contest without any geometric or energetic evaluation.The number of homology modeling targets used during the CASP4competition was too small to take1251mbert et al.ESyPred3D target-template alignmentFig.1.Flowchart of the ESyPred3D target –template alignment method.See the text for details.Fig.2.Example of compatible and incompatible results on two hypothetical sequences.Three cases are reported:(a)Alignments I –I and I –L are not compatible because the same amino acid in sequence 1is aligned to two amino acids in sequence 2.(b)Alignments P –P and A –A are not compatible.P in sequence 1is at the right of A but P in the second sequence is the left of A.(c)Alignment I –I and P –P are compatible.The prolines are both at the right of the isoleucines.a very robust statistical conclusion about the performances of our method.However,results obtained provide a first estimation of performances.For more statistical results,see the continuous evaluation of servers performed by EV A (Eyrich et al.,2001)(/eva/).For the purpose of the CASP4experience,two models were built for 13comparative modeling targets (Table 1.)for which ESyPred3D was able to predict a 3D structure:(1)The first model was built using the complete strategydescribed above(ESyPred3D)(models T0xxxTS2181).(2)The second model was built using the same strategyas ESyPred3D but by using the rough sequence-structure alignment provided by PSI-BLAST (models T0xxxTS2182).Scoring schemes used to compare target structures to modelsTo compare ESyPred3D models to PSI models and ESyPred3D models to models of other CASP4partici-pants,the AL0and the GDT TS scores were chosen.Both scores where calculated using the LGA (Local –Global Alignment)program (Zemla,2000).AL0AL0is the number of correctly aligned residues in the target –template alignment.This score is very signi ficant in this case because our method was designed to generate op-timal alignment performances.This number is evaluated by,at first,making a structural alignment of the prediction and the target structure with the DALI-server,and then,counting the number of residues in the model for which the closest residue in the target is the correct one (the distancebetween their α-carbons being less than 3.8˚A).GDT TSThe Global Distance Test,G DT d i ,is the number of α-carbons of a prediction not deviating from more than d i ˚Afrom the α-carbons of the targets,after optimal super-imposition.This optimal superimposition is computed in such a way that the number of residues (α-carbons)that can fit under the distance cutoff d i is maximum.If NT is the total number of residues of the target,GDT TS (GDT Total Score)computed according to the formula given below is the mean fraction of residues of the target not deviating from the prediction after fouroptimal superimpositions with 1.0,2.0,4.0,8.0˚A α-carbon distance cutoffs.The GDT TS score represents the overall quality of the model.This score was used to evaluate the complete procedure of ESyPred3D:identify-ing the structural homolog,aligning target to template and building the 3D model.G DT T S =100∗d iG DT d iN T4d i ∈{1.0,2.0,4.0,8.0}1252ESyPred3D:Prediction of proteins3D structures Table1.Homology modeling targets for the CASP4experimentTarget Description PDB codeT0090ADP-ribose pyrophosphatase,E.coli1g0s,1g9q,1ga7 T0092Hypothetical protein HI0319,H.InfluenzaeT0099No descriptionT0103Pepstatin insensitive carboxyl proteinase,Pseudomonas sp.1ga6T0111Enolase,E.coliT0112Ketose Reductase/Sorbitol Dehydrogenase,B.argentifolii1e3jT0113Short chain3-hydroxyacyl-coa dehydrogenase,rat1e3w,1e3s,1e6w T0117Deoxyribonucleoside kinase,D.MelanogasterT0121MalK,T.litoralis1g29T0122Tryptophan Synthase alpha subunit,P.furiosus1geqT0123Beta-lactoglobulin,pig1exsT0125Sp18protein,H.fulgens1gakT0128Manganese superoxide dismutase homolog,P.aerophilumRESULTS AND DISCUSSIONThe performance of our homology modeling server is analyzed in three steps.In thefirst section,ESyPred3D alignments are compared to PSI-BLAST alignments.In the second section,ESyPred3D alignments are compared to those of other participants having used the same template.Since our alignment method is specifically designed for homology modeling,in the third section, ESyPred3D models are compared to those of other CASP4 competitors in order to evaluate the global performance of our homology modeling strategy.Alignment performances of ESyPred3D models compared to those of PSI-BLAST modelsTable2contains AL0scores for all models.Out of the 13models,ESyPred3D obtains nine AL0scores greater than PSI-BLAST and only one AL0score significantly lower(more than two amino acids incorrectly aligned) than PSI-BLAST,for T0112.Two reasons explain the poor alignment for T0112:(1)The number of homologs found by PSI-BLAST wasso large that the non-redundant set could not becomputed with PURGE.(2)Four regions of T0112shared only a very low sim-ilarity with homologues.So the different alignmentprograms produced contradictory results in these re-gions,and only a poor alignment could be estab-lished by our method.From thisfirst evaluation,we can conclude that the quality of the target–template alignment is generally better by using ESyPred3D alignment methodology than using the target–template alignment provided by PSI-BLAST. Comparison of ESyPred3D with those of the participants having used the same templateThe number of groups that have used the same templates as ESyPred3D is strongly variable(from two to65groups, Figure3).In Figure3,using AL0scores,ESyPred3D models obtained one time thefirst place,five times the second place,three times the third place and one times the fourth place.ESyPred3D models are then ten times in the top four places out of the13targets.Taking into account that a group that performs better than ESyPred3D model for one target is rarely the same that performs better for another target,one can conclude that our methodology is among the most efficient.Comparison of ESyPred3D models with those of all CASP4participantsIn this section,the complete strategy of ESyPred3D is evaluated and the performances are compared to those of other CASP4participants using the GDT TS score. The number of models submitted for each target was always above200.So,to enable a rapid interpretation of the distributions of scores,we have computed the third quartile of these distributions.The third quartile(Q3)of a distribution is the value such that75%of values in the list are less or equal to it.All information provided in Figure4has been normalized by the Q3value,for each target.For each target,Figure4contains:(1)the GDT TS score of ESyPred3D models;(2)the GDT TS score of the1253mbert et al.Table2.Scores for ESyPred3D and PSI-BLAST models.The last column shows templates that lead to the best models presented at CASP4 Model RMSD(allαcarbons)GDT TS AL0Template Templates leading to the best modelsT0090TS2181 6.5230.15411tum1mutT0090TS2182 6.4423.37301tum1mutT0092TS218114.7035.69731d2g1xva,1d2hT0092TS2182 5.2234.69661d2g1xva,1d2hT0099TS2181 5.5452.23261qly1a0n,1ad5,2hck,1qcf,2srcT0099TS2182 5.5650.00211qly1a0n,1ad5,2hck,1qcf,2srcT0103TS218111.9538.591281sbh1mee,1supT0103TS218212.4133.761131sbh1mee,1supT0111TS2181 2.2983.553831one1pdz,1pdy,1ykf,4-7enlT0111TS2182 2.2682.393811one1pdz,1pdy,1ykf,4-7enlT0112TS2181 5.3554.311741hdy1teh,1ykfT0112TS2182 4.1359.191971hdy1teh,1ykfT0113TS2181 3.3281.862141hdc1hdc,2hsdT0113TS2182 3.6880.492071hdc1hdc,2hsdT0117TS21818.2456.851141qhi1e2k,1kim,1ki2-7T0117TS2182 3.8755.711091qhi1e2k,1kim,1ki2-7T0121TS2181 3.3541.941431b0u1b0uT0121TS2182 3.3840.931411b0u1b0uT0122TS2181 2.4379.152031cw21a5a,1a5b,1beu,1cw2T0122TS2182 2.4174.581901cw21a5a,1a5b,1beu,1cw2T0123TS2181 4.1563.911022a2u2a2g,1bebT0123TS2182 3.7565.471022a2u2a2g,1bebT0125TS2181 4.1561.13743lyn2lis,3lynT0125TS2182 4.0760.40753lyn2lis,3lynT0128TS2181 1.7486.731851abm1b06,1sssT0128TS2182 1.6587.321871abm1b06,1sssbest model received by CASP4organizers and(3)the third quartile is equal to1.0because of the normalization. Figure4shows that ESyPred3D built three models with scores close to the best model,indeed the second place was obtained for targets T0103,T0121and T0122 (see full tables at /casp4/). ESyPred3D predicted eight models above Q3values,i.e. in the top25%of participants.Further analysis of the data at the CASP4web site shows that there are few groups that have reached such a number of scores values above the Q3. It is also important to note that the group that obtained the best model for one target is rarely the same that submitted the best model for another target.The analysis of GDT TS scores(Figure4)showed that seven targets(T0090,T0092,T0099,T0112,T00117,T0123and T0128)obtained values significantly lower than those of the best models.For targets T0090,T0092, T0099,T0117,T0123and T0128the low values of GDT TS are due to the selection of a template that was not fully adequate.Indeed,for these targets,the alignment performances remain good when comparing only to groups that used the same template,as shown by the AL0score in Figure3.Although in our methodology the template selection process has to be improved,it is important to note that a completely inadequate template was never chosen.The result of T0112is due to the quality of the alignment as you can see in Figure3.The fact that eight models from13are above the Q3 shows that our alignment method combined with the PSI-BLAST template selection and the use of MODELLER1254ESyPred3D:Prediction of proteins 3Dstructures102030405060708090100T 0090T 0092T 0099T 0103T 0111T 0112T 0113T 0117T 0121T 0122T 0123T 0125T 0128TargetsA L 0 (i n % o f t h e l e n g t h )Fig.3.AL0scores for targets studied in this work.Two series are reported for each target:the score of ESyPred3D models (black bullets)and the scores of models of other CASP4participants having used the same template (blank bullets).AL0scores are expressed as a fraction of the length of thetarget.T 0090T 0092T 0099T 0103T 0111T 0112T 0113T 0117T 0121T 0122T 0123T 0125T 0128TargetsG D T _T S (i n % o f Q 3)Fig.4.GDT TS scores for targets studied in this work.Three points are reported for each target:the score of the model that obtain the best score (bold line),the ESyPred3D model (box)and the third quartile value (dotted line).All values are expressed as a fraction of the third quartile value.The group IDs of best predictors with their selected templates are also reported.to obtain the 3D model is a good strategy.Even if the template selection or the alignment quality is not optimal,the global quality of the ESyPred3D modeling strategy remains good.CONCLUSIONA new alignment methodology for homology modeling of proteins has been developed.The program has been tested on 13targets of the CASP4for its alignment performances and for the general quality of the provided models.Our alignment strategy produced better results com-pared to PSI-BLAST alignments and ESyPred3D align-ments are among the most accurate comparing to partic-ipants having used the same template.Furthermore,our ESyPred3D program provides models that are among the best of the CASP4experiment.Nevertheless,our alignment methodology could be im-proved.Thompson et al.(1999)and Briffeuil et al.(1998)benchmarks showed that all alignment programs have different level of performance.We plan to use this in-formation to improve the computing of the alignment,by weighting each multiple alignment method with the numeric representation of the mean performance of the method.Additional information such as secondary struc-ture predictions can also be used in the box selection in order to improve the alignment quality.The template problem remains troublesome in homol-ogy modeling,especially when the target and template sequences are sharing a low identity rate.To improve the template selection,the use of better parameters or better scoring matrices for PSI-BLAST (like the one de-scribed in Kann et al.(2000))need to be investigated.In the same way,PSI-BLAST can also be replaced by SAM-T99(Karplus et al.,1998)or other programs.The intrinsic quality of the possible template structures (NMR,resolution,...)and the selection of multiple templates will also be taken into account to improve our modeling strategy.The model evaluation step of our homology modeling methodology has not yet been developed.Geometric and energetic evaluation of the model can be done using ANOLEA (Melo and Feytmans,1997),PROCHECK (Laskowski et al.,1993)or Verify3D (Luthy et al.,1992).The results of these evaluations will be used to change our target –template alignment or to select a more appropriate template.The process will be iterated in order to find the template that provides the best evaluated model.A similar iteration procedure has been used by the Blundell group in the CASP4.ACKNOWLEDGMENTSWe thank the organizers and assessors of the CASP4experiment for their valuable contributions to the structure prediction field.Christophe Lambert holds a specializedgrant from the ‘Fonds pour la Formation `ala Recherche dans l ’Industrie et dans l ’Agriculture ’(F.R.I.A.).We particularly want to thank Guy Baudoux,Katalin de Fays and Johan Wouters for helpful and fruitful discussions.REFERENCESAltschul,S.F.,Madden,T.L.,Sch ¨affer,A.A.,Zhang,J.,Zhang,Z.,Miller,W.and Lipman,D.J.(1997)Gapped BLAST and PSI-BLAST:a new generation of protein database search programs.Nucleic Acids Res.,25,3389–3402.Briffeuil,P.,Baudoux,G.,Reginster,I.,De Bolle,X.,Vinals,C.,Feytmans,E.and Depiereux,E.(1998)Comparative analysis of1255mbert et al.seven multiple protein sequence alignment servers:clues to enhance predictions reliability.Bioinformatics,14,357–366. Browne,W.J.,North,A.C.and Phillips,D.C.(1969)A possible three-dimensional structure of bovine alpha-lactalbumin based on that of hen’s egg-white lysozyme.J.Mol.Biol.,42,65–86. Corpet,F.(1988)Multiple sequence alignment with hierarchi-cal clustering.Nucleic Acids Res.,16,10881–10890. Depiereux,E.,Baudoux,G.,Briffeuil,P.,Reginster,I.,De Bolle,X., Vinals,C.and Feytmans,E.(1997)Match-Box server:a multiple sequence alignment tool placing emphasis on put.Appl.Biosci.,13,249–256.Depiereux,E.and Feytmans,E.(1992)Match-Box:a fundamentally new algorithm for simultaneous alignment of several protein put.Appl.Biosci.,8,501–509. Eisenhaber,F.,Persson,B.and Argos,P.(1995)Protein structure prediction:recognition of primary,secondary,and tertiary structural features from amino acid sequence.Crit.Rev.Biochem.Mol.Biol.,30,1–94.Eyrich,V.A.,Marti-Renom,M.A.,Przybylski,D.,Madhusudhan,M.S., Fiser,A.,Pazos,F.,Valencia,A.,Sali,A.and Rost,B.(2001)EV A: continuous automatic evaluation of protein structure prediction servers.Bioinformatics,17,1242–1243.Fischer,D.,Barret,C.,Bryson,K.,Elofsson,A.,Godzik,A.,Jones,D., Karplus,K.J.,Kelley,L.A.,MacCallum,R.M.,Pawowski,K., Rost,B.,Rychlewski,L.and Sternberg,M.(1999)CAFASP-1: critical assessment of fully automated structure prediction methods.Proteins,(Suppl3),209–217.Gotoh,O.(1996)Significant Improvement in Accuracy of Multiple Protein Sequence Alignments by Iterative Refinement as Assessed by Reference to Structural Alignments.J.Mol.Biol., 264,823–838.Kann,M.,Qian,B.and Goldstein,R.A.(2000)Optimization of a new score function for the detection of remote homologs.Proteins, 41,498–503.Karplus,K.,Barrett,C.and Hughey,R.(1998)Hidden Markov models for detecting remote protein homologies.Bioinformatics, 14,846–856.Karplus,K.,Sjolander,K.,Barrett,C.,Cline,M.,Haussler,D., Hughey,R.,Holm,L.and Sander,C.(1997)Predicting protein structure using hidden Markov models.Proteins,(Suppl1), 134–139.Laskowski,R.A.,Moss,D.S.and Thornton,J.M.(1993)Main-chain bond lengths and bond angles in protein structures.J.Mol.Biol., 231,1049–1067.Luthy,R.,Bowie,J.U.and Eisenberg,D.(1992)Assessment of protein models with three-dimensional profiles.Nature,356,83–85.Martin,A.C.,MacArthur,M.W.and Thornton,J.M.(1997)Assess-ment of comparative modeling in CASP2.Proteins,(Suppl1), 14–28.Melo,F.and Feytmans,E.(1997)Novel Knowledge-based Mean Force Potential at Atomic Level.J.Mol.Biol.,267,207–222. Morgenstern,B.(1999)DIALIGN2:improvement of the segment-to-segment approach to multiple sequence alignment.Bioinfor-matics,15,211–218.Mosimann,S.,Meleshko,R.and James,M.N.(1995)A critical assessment of comparative molecular modeling of tertiary structures of proteins.Proteins,23,301–317.Neuwald,A.F.,Liu,J.S.and Lawrence,C.E.(1995)Gibbs motif sampling:detection of bacterial outer membrane protein repeats.Protein Sci.,4,1618–1632.Rost,B.and O’Donoghue,S.(1997)Sisyphus and prediction of protein put.Appl.Biosci.,13,345–356.Sali,A.and Blundell,T.L.(1993)Comparative protein modelling by satisfaction of spatial restraints.J.Mol.Biol.,234,779–815. Sali,A.,Sanchez,R.and Badretdinov,A.(1997)MODELLER:A Program for Protein Structure Modeling Release4. Sanchez,R.and Sali,A.(1997)Advances in comparative protein-structure modeling.Curr.Opin.Struct.Biol.,7,206–214. Thompson,J.D.,Higgins,D.G.and Gibson,T.J.(1994)CLUSTALw: improving the sensitivity of progressive multiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice.Nucleic Acids Res.,22, 4673–4680.Thompson,J.D.,Plewniak,F.and Poch,O.(1999)A comprehensive comparison of multiple sequence alignment programs.Nucleic Acids Res.,27,2682–2690.Tramontano,A.(1998)Homology modeling with low sequence identity.Methods,14,293–300.Venclovas,C.,Zemla,A.,Fidelis,K.and Moult,J.(1999)Some measures of comparative performance in the three CASPs.Proteins,(Suppl3),231–237.Yang,A.S.and Honig,B.(1999)Sequence to structure alignment in comparative modeling using PrISM.Proteins,(Suppl3),66–72. Zemla,A.(2000)LGA program:A Method for Finding3D Similar-ities in Protein Structures.Accessed at http://PredictionCenter./local/lga.1256。