药动学概述
药动学
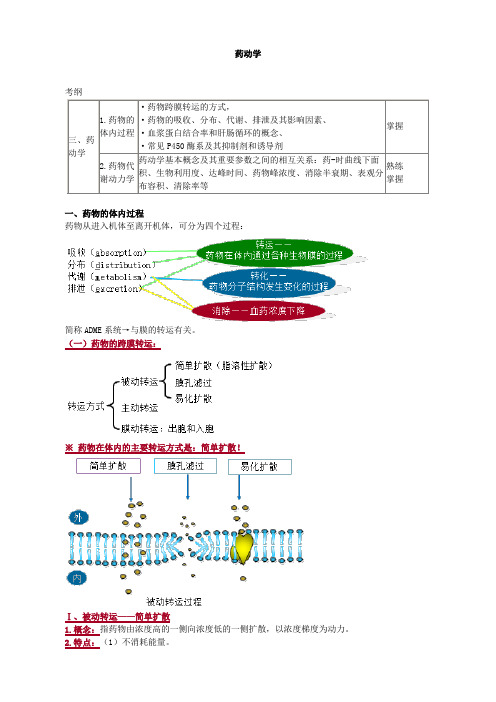
药动学考纲三、药动学1.药物的体内过程·药物跨膜转运的方式,·药物的吸收、分布、代谢、排泄及其影响因素、·血浆蛋白结合率和肝肠循环的概念、·常见P450酶系及其抑制剂和诱导剂掌握2.药物代谢动力学药动学基本概念及其重要参数之间的相互关系:药-时曲线下面积、生物利用度、达峰时间、药物峰浓度、消除半衰期、表观分布容积、清除率等熟练掌握一、药物的体内过程药物从进入机体至离开机体,可分为四个过程:简称ADME系统→与膜的转运有关。
(一)药物的跨膜转运:※药物在体内的主要转运方式是:简单扩散!Ⅰ、被动转运——简单扩散1.概念:指药物由浓度高的一侧向浓度低的一侧扩散,以浓度梯度为动力。
2.特点:(1)不消耗能量。
(2)不需要载体。
(3)转运时无饱和现象。
(4)不同药物同时转运时无竞争性抑制现象。
(5)当膜两侧浓度达到平衡时转运即停止。
3.影响简单扩散的药物理化性质(影响跨膜转运的因素)(1)分子量分子量小的药物易扩散。
(2)溶解性脂溶性大,极性小的物质易扩散。
(3)解离性非离子型药物可以自由穿透。
离子障是指离子型药物被限制在膜的一侧的现象。
※药物的解离程度受体液pH 值的影响离子型非离子型4.体液pH值对弱酸或弱碱药物的解离的影响:※pK a(解离常数)的含义:>>是指解离和不解离的药物相等时,即药物解离一半时,溶液的pH值。
>>每一种药物都有自己的pK a。
若为弱酸性药物,则:若为弱碱性药物,则:从公式可见,体液pH算数级的变化,会导致解离与不解离药物浓度差的指数级的变化,所以,pH值微小的变动将显著影响药物的解离和转运。
一个pK a=8.4的弱酸性药物在血浆中的解离度为A.10%B.40%C.50%D.60%E.90%『正确答案』A『正确解析』pH对弱酸性药物解离影响的公式为:即:解离度为107.4-8.4=10-1=0.1。
※四两拨千斤:体液pH值对药物解离度的影响规律:◇酸性药物——在酸性环境中解离少,容易跨膜转运。
药理学 第三章药动学 重点知识总结

第三章药动学药动学:机体对药物的作用。
药物自进入机体到离开机体历经吸收、分布、代谢及排泄过程,这是机体对药物的处置,这些处置可以概括为药物的转运(吸收、分布、排泄)和药物的转化(代谢)1、吸收(absorption ):是指药物自体外或给药部位经过细胞组成的屏障进入血液循环的过程.1.2、药物的转运方式:被动转运和主动转运被动转运:单纯扩散:(脂溶性物质直接溶于膜的类脂相而通过)、易化扩散:*需特异性载体*顺浓度梯度,不耗能、滤过扩散主动转运特点:耗能,逆浓度差,需载体参与影响药物吸收的因素:(1)、给药途径静脉>吸入>肌肉(im)>皮下(ih)>舌下>直肠>口服>经皮。
(2)口服给药对药物吸收的影响首关消除(第一关卡效应或首过消除):有些口服药物首次通过肝脏就发生转化,减少进入体循环量,(3)血液循环的状态也影响药物的吸收(4)生物利用度也影响药物的吸收2、分布:指吸入血液的药物被转运至组织器官的过程。
药物在体内的分布速率主要取决于药物的理化性质,各器官组织的血流量与对药物的通透性,以及药物在组织与血浆的分配比。
影响因素:(1)与血浆蛋白的结合率(2)体内屏障(3)与组织的亲和力(4)组织器官的血流量3、生物转化(代谢):指药物在体内发生的化学过程,这种变化主要是结构的变化,由于结构变化引起性质变化,以至作用强度的变化。
注意:有少数药不发生化学变化,原型作用,原型排泄,如色甘酸、链霉素等。
1、转化的场所:肝脏微粒体2、生物转化的类型第一步:为氧化、还原、水解。
这步反应多数药物灭活,但也有例外(可待因)。
第二步:为结合。
总使药物活性降低或灭活并使极性增加。
影响药物转化的因素肝脏的功能:肝脏的功能是药物代谢的主要器官,肝脏功能不全时可影响代谢。
药酶诱导剂:某些药物能使肝脏药酶的活性增加或加速其合成。
如:苯巴比妥、水合氯醛、保泰松等可加速其代谢,使药物作用减弱。
药酶抑制剂:凡能抑制药酶活性或减少药酶合成的药物。
药物动力学(PK)与药效动力学

评估药物的疗效和安全性,了解药物对生理功能的影响,确定药物的适应症和用法用量。源自新药临床试验中的PK/PD研究
VS
提供药物的吸收、分布、代谢和排泄数据,以及给药方案和剂量选择依据。
PD数据
提供药物的疗效和安全性数据,包括临床试验结果和不良反应监测数据,以支持新药的上市申请。
PK数据
新药上市申请中的PK/PD数据
药物吸收
研究药物在体内的吸收速率和程度,确定最佳给药途径和剂量。
药物分布
了解药物在体内的分布情况,预测药物在不同组织中的浓度和作用。
药物代谢
研究药物在体内的代谢过程,包括代谢产物的性质和作用。
药物排泄
研究药物从体内排出的途径和速率,评估药物的消除和半衰期。
新药临床前PK/PD研究
PK研究
通过临床试验,进一步了解药物在人体内的吸收、分布、代谢和排泄过程,为临床用药提供依据。
生物信息学
开发新型的生物传感器和检测方法,实现药物在体内实时动态监测,为个体化用药提供更精确的指导。
实时监测技术
利用体外实验模型模拟体内环境,提高PK/PD研究的可靠性和可重复性。
体外实验与体内实验相结合
PK/PD研究技术的发展
个体差异大,不同个体对药物的反应不同,需要更精确的预测和调整用药方案。
02
CHAPTER
药效动力学(PD)概述
药效动力学(PD)是研究药物如何产生预期效果的科学,主要关注药物在体内的效应和作用机制。
定义
通过药效动力学研究,了解药物如何与靶点结合并产生作用,预测药物在不同个体内的效果和安全性,为临床用药提供科学依据。
目标
定义与目标
药物与靶点的相互作用
药物通过与体内的靶点(如受体、酶或离子通道)相互作用,产生药理效应。
药物动力学在临床药学中的应用

药物动力学在临床药学中的应用药物动力学是指研究药物在体内的吸收、分布、代谢和排泄的过程。
对于临床药学而言,药物动力学是至关重要的,它能帮助临床药师更好地了解和预测药物在患者体内的表现,从而指导用药的合理性和安全性。
一、药物动力学概述1. 药物吸收药物吸收是指药物从给药部位进入到血液循环的过程。
它受到很多因素的影响,比如药物的理化性质、给药途径和个体差异等。
了解药物吸收的特点和规律可以帮助临床药师选择合适的给药途径,并根据患者的个体差异进行个体化用药。
2. 药物分布药物分布是指药物在体内组织和器官中的分布情况。
它受到血液循环、药物与蛋白结合、脂溶性等因素的影响。
临床药师需要了解药物分布的规律,从而确定药物的给药剂量和给药间隔,以及预测药物在靶组织的作用情况。
3. 药物代谢药物代谢是指药物在体内被生物转化成代谢产物的过程。
主要发生在肝脏中。
了解药物代谢的途径和速度可以帮助临床药师评估患者的肝功能,并指导用药剂量的调整。
4. 药物排泄药物排泄是指药物从体内排出的过程。
主要通过肾脏排泄和肠道排泄。
了解药物排泄的规律可以帮助临床药师评估患者的肾功能和肠道功能,并指导用药剂量的调整。
二、药物动力学在临床药学中的应用药物动力学在临床药学中有着广泛的应用。
它能帮助临床药师评估药物的疗效和毒副作用,从而指导用药方案的制定。
它能帮助临床药师了解患者的体内药物浓度的变化,从而指导用药剂量的调整。
另外,它还能帮助临床药师评估患者的肝肾功能和药物相互作用等情况,从而指导用药的安全性和合理性。
三、个人观点和理解作为一名临床药师,我认为药物动力学是非常重要的。
它能够帮助我们更好地了解药物在体内的表现,从而指导临床用药。
在未来,我希望能够进一步深入学习和掌握药物动力学的知识,不断提升自己的临床实践能力。
总结药物动力学在临床药学中扮演着重要的角色,它有助于临床药师更好地了解和预测药物在体内的表现,从而指导用药的合理性和安全性。
个体化用药和合理用药也是未来临床药学发展的重要方向,而药物动力学无疑将在这个过程中发挥重要作用。
临床药理学-药动学概述201008

给药途径
胃肠道pH环境
胃排空速率
首过效应等
影响口服药物吸收的生理因素
胃肠道pH值
胃排空速率
首过效应
食物的影响
胃肠道pH
消化道不同的pH下,药物的解离状态不同,影
响药物的吸收
弱酸药物 主要在胃吸收
弱碱药物 主要在小肠吸收
水和食物 餐后pH显著
疾病 十二指肠溃疡 pH
C
lgC
t
t
Cmax、tmax
口服给药形成的曲线是由迅速上升的以吸收为
主的吸收相和缓慢下降的以消除为主的消除相 两部分组成。 C
峰浓度 Cmax
tmax 达峰时间
t
药-时曲线下面积(AUC)
AUC0 指药物从零时间至所有
原形药物全部消除这一段时间的药-时
曲线下总面积,反映药物进入血循环的
k12 k21
中央室
k
K10:药物从中央室消除的一级消除速 率常数
生物半衰期(t1/2)
包括吸收半衰期(t1/2Ka) 、分布半衰期(t1/2α)
和消除半衰期(t1/2)
消除半衰期
体内药量或血药浓度下降一半所需要的时间 , 其长短可反映体内药物消除速度。
C C0
C0/2
C0/4 t1/2 t1/2
排泄(elimination)
药物的原形或其代谢产物通过排泄器官排出体
外的过程称为排泄。
药物的排泄途径 肾排泄
肾脏是最重要的排泄器官
胆汁排泄 其它途径
肾排泄 : • 肾小球滤过 • 肾小管
分子量小于两万
主动分泌(弱酸和弱碱)
两个系统均为非特异性,可发生竞争性抑制。 如丙磺舒(酸性药物)可能抑制其他酸性药物, 如青霉素
药动学概述

药动学研究的新进展
(1) 矩量法(statistical moment theory)
➢ 统计矩概念的基础是:当一定量的药物输入机体时,不论是在 给药部位或在整个机体内,各个药物分子滞留时间的长短,均 属随机变量。药物的吸收、分布及消除可视为这种随机变量所 相应的总体效应,因而药-时曲线是某种概率统计曲线。
Pharmacokinetics,“PK” 药动学 应用动力学的原理与数学处理方法,定量地描述药物通过各种 途径进入体内吸收、分布、代谢、排泄过程的“量时”变化或 “血药浓度经时”变化动态变化规律的一门科学。
Pharmacodynamics ,“PD” 药效学
研究药物对机体的药理作用和作用机理,包括观测生理机能的改 变、生化指标的变化和组织形态学变化等内容。
杂的体内过程; ➢ 求出模型的特解与通解,推证与建立包括药物输入与输
出、体内与体外、药动与药效的关系、特殊个体动力学 参数与群体动力学参数的关系在内的一系列重要公式。 ➢ 理论上取得的重大突破,必将为药动学的实验设计及具 体应用开辟广阔前景。
二、药动学的基本研究任务
在实验上求参数
根据机体给药后观测到的样本药物浓度的经时数据,选择合适的模型, 并求出这种模型中有关的具体参数,使该模型与实验值能紧密地“嵌 合”起来,“模型嵌合”70年代前多采用残数法,后来多用计算机进 行编程处理。 准确的获得不同时间点体内药物的量:在线检测技术、固相微萃取技 术、液质联用技术等 用适宜的数据处理方法揭示药物在体内的动态变化规律: Laplace变 换法、输入函数与配置函数法、流线图法、残数法及人工神经网络等。
优点:
能更精细表征任何器官或组织中药物浓度的经时过程。 生理模型各参数采用真实解剖值,故机体生理病理的 改变而引起药物处置动力学的变化,能通过某些参数 的改变来估计。 种属之间的数据可以互相推算。
生物药剂学与药动学——药动学概述

生物药剂学与药动学——药动学概述一、药动学定义药动学是应用动力学的原理和数学处理方法,研究药物在体内的吸收、分布、代谢和排泄过程(即ADME 过程)的量变规律的学科,即药动学是研究药物体内过程动态变化规律的一门学科。
二、血药浓度与药物效应(一)治疗浓度范围治疗浓度范围即治疗窗,是指给药后产生药效的最低有效浓度和产生毒性的最低中毒浓度之间的浓度范围。
治疗窗窄的药物,其治疗浓度相对较难控制,易发生治疗失败或不良反应,常需进行治疗药物监测。
(二)血药浓度与药物效应的关系对于大多数药物及其制剂,药物进入体内后,血中的药物浓度与药物作用靶位的实际浓度呈正相关,从而间接反映药物的临床效应,包括治疗效果及不良反应。
药动学中常以血液中的药物总浓度作为观察指标。
三、药动学的基本概念和主要参数(一)血药浓度-时间曲线药动学的研究中,将药物制剂通过适当的方式给予受试者,然后按照适当的时间间隔抽取血样,检测血样中的药物浓度,每一个取血时间点有一个对应的药物浓度,由此就得到一系列的血药浓度相对于时间的实验数据,简称为药-时数据。
将其用坐标图表示,称为血药浓度-时间曲线,简称药-时曲线。
血管内给药的药-时曲线通常为曲线,而血管外给药的药-时曲线一般为拋物线。
根据研究的需要,常将药-时曲线的不同时间段用吸收相、平衡相和消除相来表示,表明该时间段(时相)体内过程的主要影响。
(二)血药浓度-时间曲线下面积血药浓度-时间曲线图中,药-时曲线与时间轴共同围成的面积称为血药浓度-时间曲线下面积,简称药-时曲线下面积,用AUC表示。
其与药物吸收的总量成正比,能够反映药物吸收的程度。
AUC越大,表明制剂中的药物被生物体吸收越完全。
血药浓度-时间曲线下面积是评价制剂生物利用度和生物等效性的重要参数。
(三)峰浓度和达峰时间血管外给药的药-时曲线一般为拋物线,其中有两项特征性参数,即血药峰浓度和达峰时间。
血药峰浓度即药-时数据中的最大浓度,用C max表示,C max的大小能够反映药物的疗效情况和毒性水平。
药动学的概念

药动学的概念药动学是研究药物在人体内的吸收、分布、代谢和排泄过程的科学。
通过对药物在体内的药物浓度和时间的改变进行定量分析,药动学可以提供关于药物疗效和安全性的重要信息。
下面是关于药动学的一些相关参考内容。
1. 药物吸收:药物吸收是指药物从给药部位进入血液循环的过程。
吸收的速度和程度受到多种因素的影响,如药物的解离性、溶解性、药物形式(片剂、胶囊、注射剂等)和给药途径(口服、静脉注射、皮下注射等)。
通过了解药物的吸收动力学特征,可以优化给药方式和剂量,提高药物的生物利用度。
2. 药物分布:药物分布是指药物在体内的分布情况,包括药物在组织器官间的分布均匀性和药物在组织器官内部的分布。
药物分布受到多种因素的影响,如药物亲脂性和亲水性、蛋白结合率、组织通透性等。
药物分布的特点决定了药物在靶组织中的浓度,进而影响药效和毒性。
3. 药物代谢:药物代谢是指药物在体内发生化学转化的过程。
在肝脏中的细胞中,酶系统将药物转化成代谢产物,以便药物的排泄。
药物代谢可以分为两个阶段:相对较快的药物转换为活性中间代谢物(相1)和相对较慢的活性中间代谢物被进一步转化为可溶性产物,以便在尿液或粪便中排泄(相2)。
药物代谢的速度和途径受到遗传因素、年龄和环境等因素的影响。
4. 药物排泄:药物排泄是指药物从体内被转运至体外的过程。
主要依靠肾脏、肝脏、肺和肠道等排泄器官完成。
药物排泄的速度和途径由药物的性质,如极性、分子量、药物结构和酸碱特性等决定。
通过了解药物的排泄动力学特征,可以调整药物剂量和给药频率,确保药物在体内的平衡。
5. 药物浓度-时间曲线:药物浓度-时间曲线是描述药物在体内浓度随时间变化的曲线。
药物浓度-时间曲线可以提供药物动力学参数,如峰值浓度(Cmax)、时间到达峰值浓度(Tmax)、消除半衰期(t1/2)和生物利用度(AUC)等信息。
通过分析药物浓度-时间曲线,可以评估药物的吸收、分布、代谢和排泄特征,为合理用药和药物治疗提供指导。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药动学概述
一、药动学定义
药动学是应用动力学的原理和数学处理方法,研究药物在体内的吸收、分布、代谢和排泄过程(即ADME 过程)的量变规律的学科,即药动学是研究药物体内过程动态变化规律的一门学科。
二、血药浓度与药物效应
(一)治疗浓度范围
治疗浓度范围即治疗窗,是指给药后产生药效的最低有效浓度和产生毒性的最低中毒浓度之间的浓度范围。
治疗窗窄的药物,其治疗浓度相对较难控制,易发生治疗失败或不良反应,常需进行治疗药物监测。
(二)血药浓度与药物效应的关系
对于大多数药物及其制剂,药物进入体内后,血中的药物浓度与药物作用靶位的实际浓度呈正相关,从而间接反映药物的临床效应,包括治疗效果及不良反应。
药动学中常以血液中的药物总浓度作为观察指标。
三、药动学的基本概念和主要参数
(一)血药浓度-时间曲线
药动学的研究中,将药物制剂通过适当的方式给予受试者,然后按照适当的时间间隔抽取血样,检测血样中的药物浓度,每一个取血时间点有一个对应的药物浓度,由此就得到一系列的血药浓度相对于时间的实验数据,简称为药-时数据。
将其用坐标图表示,称为血药浓度-时间曲线,简称药-时曲线。
血管内给药的药-时曲线通常为曲线,而血管外给药的药-时曲线一般为拋物线。
根据研究的需要,常将药-时曲线的不同时间段用吸收相、平衡相和消除相来表示,表明该时间段(时相)体内过程的主要影响。
(二)血药浓度-时间曲线下面积
血药浓度-时间曲线图中,药-时曲线与时间轴共同围成的面积称为血药浓度-时间曲线下面积,简称药-时曲线下面积,用AUC表示。
其与药物吸收的总量成正比,能够反映药物吸收的程度。
AUC越大,表明制剂中的药物被生物体吸收越完全。
血药浓度-时间曲线下面积是评价制剂生物利用度和生物等效性的重要参数。
(三)峰浓度和达峰时间
血管外给药的药-时曲线一般为拋物线,其中有两项特征性参数,即血药峰浓度和达峰时间。
血药峰浓度即药-时数据中的最大浓度,用C max表示,C max的大小能够反映药物的疗效情况和毒性水平。
与C max相对应的时间称为达峰时间,用t max表示,它能够反映药物吸收的快慢,t max越小,药物的吸收越快。
峰浓度和达峰时间是评价制剂生物利用度和生物等效性的重要参数。
单次血管外给药后血药浓度-时间曲线
(四)速率过程
与生物体药动学相关的速率过程主要有3种:一级速率过程、零级速率过程与非线性速率过程。
1.一级速率过程一级速率表示药物在体内某一部位的变化速率与该部位的药量或血药浓度的一次方
成正比,多数药物在常规给药剂量时的体内过程通常符合一级速率过程。
2.零级速率过程零级速率表示体内药物的变化速率与该部位的药量或血药浓度的零次方成正比,恒速静脉滴注的给药速率、理想的控释制剂的释药速率都符合零级速率过程。
3.非线性速率过程当体内的酶被饱和时的速率过程,称为受酶活力限制的速率过程,非线性速率过程。
符合非线性药动学特征的药物的速率过程通常以米氏方程描述。
(五)速率常数
药物在体内变化的速率过程的快慢由速度(率)常数k决定,k越大,体内药物的量或浓度变化得越快。
速率常数有多种,如吸收速率常数、分布速率常数、消除速率常数等,其中消除速率常数是最主要的一种。
如肾脏排泄、胆汁排泄、肺消除和生物转化的速率常数分别用k e、k bi、k lu和k b表示。
消除速率常数具有加和性,体内总的消除速率常数k是各种途径消除的速率常数之和。
k=k e+k bi+k lu+k b式(3-2-1)
消除速率常数的单位是时间的倒数,如h-1、min-1等。
符合线性药动学特征的药物的消除速率常数在健康人体内是一个常数,它只依赖于药物本身的性质,而与剂型及给药方法无关。
但消除速率常数与患者的肝、肾功能等病理因素有很大关系。
(六)生物半衰期
生物半衰期又称消除半衰期,是指药物在体内的量或血药浓度下降一半所需要的时间,以t1/2表示。
生物半衰期是衡量药物从体内消除快慢的指标。
根据体内过程不同,半衰期还有吸收半衰期[t1/2(α)]、分布半衰期[t1/2(α)]和消除半衰期[t1/2(β)]。
大多数药物在一定的剂量范围内符合一级消除,它的消除半衰期与消除速率常数有如下关系:t1/2=O.693/k 式(3-2-2)
和消除速率常数一样,消除半衰期也是药物的固有性质,符合线性动力学特征的药物的消除半衰期对于健康人来说也是个常数,而与剂量、剂型和给药途径无关。
(七)表观分布容积
表观分布容积表示给药剂量若按照所测得的血药浓度来分布而求算得到的体积数,是体内药量与血药浓度间的一个比例常数,用V表示,没有直接的生理学意义。
对于一室模型药物的静脉注射,给药剂量或体内药量X0与血药初浓度C0之间存在如下关系:
V= X0/C0 式(3-2-3)
表观分布容积是药物的一个特征性参数,能够反映药物在组织器官中分布的大致情况,表观分布容积大,表明药物的体内分布广泛。
表观分布容积的单位为L或者L/kg(体重)。
药物的组织分布与药物的理化性质有关,一般来说,亲脂性药物的组织亲和性大,体内分布广泛,表观分布容积相对也较大,它们的数值往往大于体液总体积,如地高辛的表观分布容积约为580L;而亲水性药物主要分布在水性的细胞外液中,因此表观分布容积较小,如利福平的表观分布容积约为65L。
表观分布容积还与人的体型和病理状态有关。
肥胖者体内的脂肪组织多,亲脂性药物在其中的分布也多,血药浓度相对降低,因此亲脂性药物在肥胖者体内的V值就会高于一般体型的人。
某些疾病如白蛋白血症,对于血浆蛋白结合力高的药物,血中的药物浓度升高,V值下降;而水肿患者因体液总量增加,所以药物的V值也会发生变化。
(八)清除率
清除率是指单位时间内机体能将相当于多少体积血液中的药物完全清除,即单位时间内从体内清除的药物的表观分布容积,常用“Cl”表示。
可以用消除速率常数和消除半衰期来表示,计算公式为:Cl=k·V 式(3-2-4)
清除率的单位为L/h或L/(h·kg)。
和消除速率常数一样,各种途径的清除率也具有加和性,体内的总清除率(TBCL)是各种途径的清除率之和。
在临床药物动力学中,总清除率是个非常重要的参数,它是制订和调整肝、肾功能不全患者的给药方案的依据。
(九)隔室模型
药物通过各种途径进入血液循环,然后向体内的各个组织分布,药物的性质不同,其分布速度和分布部位也可能存在差异。
可分为单(一)室模型、双(二)室模型和多室模型。
1.单室模型整个机体可视为一个隔室,称为单室模型或一室模型。
2.双室模型机体给药后药物首先迅速分布于血流比较丰富的中央室,并且迅速达到动态平衡,然后再分布于血流不太丰富的外周(又称外室、周边室),此类体内过程称为双室模型或二室模型。
3.多室模型
隔室的划分是按药物转运速率划分的,并不具有解剖学意义。
(十)线性与非线性药动学
药动学按给药剂量与动力学参数的线性关系不同可以分为线性和非线性药动学两大类。
临床应用的大部分药物符合线性药动学特征,它们在体内的转运和消除速率常数与给药剂量或药物浓度不相关。
而对于具有非线性药动学特征的药物,尤其是治疗窗窄的药物,在临床应用时要特别谨慎,因为剂量的少许增加可能会引起血药浓度的急剧上升,从而导致药物中毒。
(十一)统计矩
统计矩源于概率统计理论,具有相同化学结构的各个药物分子,其体内的转运是一个随机过程,具有概率性。
药动学的统计矩分析中,通常将血药浓度-时间曲线下面积(AUC)定义为零阶矩,而将时间与血药浓度的乘积-时间曲线下面积(AUMC)定义为一阶矩。
一阶矩与零阶矩的比值即为药物在体内的平均滞留时间(MRT),表示消除给药剂量的63.2%所需要的时间,与消除半衰期的意义类似。