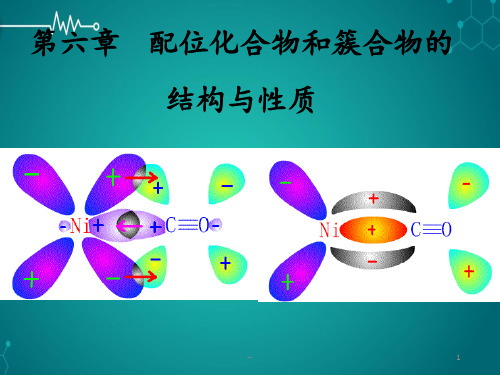
第六章配合物和簇合物的结构与性质
合集下载
配位化合物的结构和性质

2
3
sp
sp2
直线形
平面三角形
[Ag(NH3)2]+
[CuCl3]2-
4
sp3
d3s dsp2
四面体
四面体 平面正方形 三角双锥形 四方锥形
[Ni(NH3)4]2+
MnO4[Ni(CN)4]2Fe(CO)5 [TiF5]2-
5
dsp3 d4s
6
d2sp3
八面体
[Fe(CN)6]3-
6.2 价键理论
(3) 平行正方形场
在平行正方形配合物中,四个配体沿x,y轴正负方向与
中心离子接近。 在平行正方形配合物中,dx2-y2轨道的电子云极大值方向 指向配体,因此能级最高,高于Es能级;dxy 轨其也在xy平面上,所以
也要受到较大的排斥故能级也高于Es能级;dz2 轨道的能级 较低,低于Es能级;dyz和dxz轨道受到的排斥作用相同,是 简并的,能级最低。这样,在平面正方形场中,能级分裂为 四组。
由于△值通常从光谱确定,故称这个顺序为光谱化学序列。
分裂能和成对能
b) 当配体固定时,分裂能随中心离子的不同而不同,其
大小次序为:
Pt 4 Ir 3 Pd 4 Rh3 Mo3 Ru3 Co3 Cr 3 Fe 3 V 2 Co2 Ni 2 Mn2
中正负离子的静电作用;
中心离子在配体的静电作用下,使原来简并的d轨道分 裂成能级不同的几组轨道;
d电子在分裂的d轨道上重新排布,优先占据能量较低
的轨道,往往使体系的总能量有所降低,形成强场低自 旋、弱场高自旋的配合物。
二、d轨道在晶体场中的分裂
d原子轨道的角度分布图
原子簇化合物

原子簇化合物
第一节
定义和结构特点
一、定义:簇状配合物是指含有金属-金属键(M—M) 的多面体分子,它们的电子结构是以离域的多中心 键为特征的。
特点:这类配合物不是经典的配合物,也不是一般 的多核配合物。
例:
[Co(NH3)6]Cl3 经典配合物 O C Mn C O O C Mn C O
[(NH ) Co
三角形
四面体
三角双锥
四方锥
2) 簇的结构中心多数是“空”的,无中心金属 原子存在,只有少数例外。如Au11I3[P(p-ClC6H4)3]7 结构中,11个Au中,有一个在中心。
3) 簇的金属骨架结构中的边并不代表经典价键 理论中的双中心电子对键。骨架中的成键作用以离 域的多中心键为主要特征。 4) 占据骨架结构中顶点的不仅可以是同种或 异种过渡金属原子,也可以是主族金属原子,甚至 非金属原子C、B、P等。 5) 簇状配合物的结构绝大多数是三角形或以 三角形为基本结构单元的三角形多面体。
3 3
H O H O H O
Co(NH3)3
]
3+
多核配合物 CO CO CO
OC OC OC
原子簇配合物
二、M—M键的形成条件 能形成M—M键化合物的金属元素可分为两类: 一类是某些主族金属元素,它们生成无配体结合的 “裸露”金属原子簇离子。如:Ge92-、Sn94-、Pb94-等。 它们不属于配合物。 另一类是某些金属元素在形成M—M键的同时, 还与卤素、CO、RNC、膦等发生配位,即为簇状配 合物。
1、金属-金属多重键 M—M多重键的概念由美国学者F. A. Cotton首 先提出。研究的最充分的是:[Re2Cl8]2-和[Mo2Cl8]4-。
结构特点:M—M键极短:Re—Re为2.24 Å, Mo—Mo为2.14 Å。相应金属本身为:Re—Re为2.741 Å和Mo—Mo为2.725 Å。
第一节
定义和结构特点
一、定义:簇状配合物是指含有金属-金属键(M—M) 的多面体分子,它们的电子结构是以离域的多中心 键为特征的。
特点:这类配合物不是经典的配合物,也不是一般 的多核配合物。
例:
[Co(NH3)6]Cl3 经典配合物 O C Mn C O O C Mn C O
[(NH ) Co
三角形
四面体
三角双锥
四方锥
2) 簇的结构中心多数是“空”的,无中心金属 原子存在,只有少数例外。如Au11I3[P(p-ClC6H4)3]7 结构中,11个Au中,有一个在中心。
3) 簇的金属骨架结构中的边并不代表经典价键 理论中的双中心电子对键。骨架中的成键作用以离 域的多中心键为主要特征。 4) 占据骨架结构中顶点的不仅可以是同种或 异种过渡金属原子,也可以是主族金属原子,甚至 非金属原子C、B、P等。 5) 簇状配合物的结构绝大多数是三角形或以 三角形为基本结构单元的三角形多面体。
3 3
H O H O H O
Co(NH3)3
]
3+
多核配合物 CO CO CO
OC OC OC
原子簇配合物
二、M—M键的形成条件 能形成M—M键化合物的金属元素可分为两类: 一类是某些主族金属元素,它们生成无配体结合的 “裸露”金属原子簇离子。如:Ge92-、Sn94-、Pb94-等。 它们不属于配合物。 另一类是某些金属元素在形成M—M键的同时, 还与卤素、CO、RNC、膦等发生配位,即为簇状配 合物。
1、金属-金属多重键 M—M多重键的概念由美国学者F. A. Cotton首 先提出。研究的最充分的是:[Re2Cl8]2-和[Mo2Cl8]4-。
结构特点:M—M键极短:Re—Re为2.24 Å, Mo—Mo为2.14 Å。相应金属本身为:Re—Re为2.741 Å和Mo—Mo为2.725 Å。
簇状配合物

对于一个正常的共面八面体,M处于八面体中心, M—M距离为2.80埃。 Cr2Cl93M—M 磁性 3.12 Mo2Cl932.67 W2Cl932.41 反磁性
顺磁性三个未成 反磁性 对电子(d3)
成键情况 不存在 Cr—Cr键
有强 中等强度 Mo—Mo键 W—W键
稳定规律:同一族金属离子生成簇状配合物的稳定 性从上→下稳定性增大,即较重的元素容易生成 M—M键。
( 2)簇的金属骨架结构中的“边”并不代表经典
价键理论的双中心电子对,骨架中的成键作用以离域
的多中心键为主要特征。 ( 3 )占据骨架结构中心的顶点,可以是同种或异 种过渡金属原子,也可以是主族金属原子,甚至可为 非金属原子C,S,P等。
( 4 )簇的结构中心多数是“空”的,无中心金属
原子存在。
例如: Au11I3[P(ρ-ClC6H4)3]7 。 11 个 Au 中有一个在
中心。
簇状配合物的基本骨架结构
2. 为什么簇状配合物的结构绝大多数是三角形或 以三角形为基本结构单元的三角面多面体? 在一个三角形结构单元中,金属原子间的键合, 作用关不局限于边,而且也能通过三角形中心, 而三角形中心到顶点的距离比正方形中心到顶点 的距离要短。通过三角形中心,两个金属原子间 的距离短,成键强,故多数是以三角形为基本结 构单元的三角形多面体。
(2)若为非对称结构,则将其中一个中心原子及其配体 一起作为另一个中心原子的配体(词尾用“基”)来 命名。另一个作为主要的中心原子是元素符号的英文 字母在后的金属。 例如:[(C6H5)3AsAuMn(CO)5] 五羰基· [(三苯基胂)金基]合锰
2. 中心原子间既有桥联基团又有金属之间键
此类化合物应按桥联配合物来命名,并将金属- 金属键的元素符号在括号中缀在整个名称之后。 例如: (CO)3Co(CO)2Co(CO)3 二(μ- 羰基) · 二(三羰基合钴)(Co-Co) 3. 同种金属原子簇状配合物的命名 命名时在金属原子之前写明该金属原子簇的几何 形状(如三角、四方、四面等)。
第六章配合物和簇合物的结构与性质PPT课件
的点电荷与M的d轨道电子云产生排斥作用。
--
13
在晶体场中d 轨道能级分裂,引起电子排布及其他 一系列性质的变化,据此可解释配位化合物的各种 性质。如光谱、水合热及几何结构等特性。
--
14
六配位和四配位是过渡金属离子配合物的主要形式,
分别称为正八面体场(Oh)、正四面体场(Td)和正
方形场(D4h)。
D4h场
自由离子
Es
dx2-y2
ddxz2y dxz dyz
d轨道 球对称作用部分
x
y
四配位化合物的配位型式与d电子数的多少及配体的电
负性有关
--
21
立方体
四面体
t2g
t2
八面体 eg
平面正方形 dx2-y2
dxy
球对称
t2g
dz2
Td
Oh
eg Oh
配体对称性决定了d轨道能级的分裂
dxz, dyz
这与键的形成有关:dxy,dxz,dyz虽不能与配体L形 成键,但条件适合可形成键,从而影响的大小 ,这与配位场理论有关。
B:当配位体固定时, 随中心离子而改变:
对同一金属离子而言,电荷越高, 值越大;
如:Mn2+对H2O的值为7800cm-1,而Mn3+对H2O的
值为21000cm-1
--
26
对电子构型(d电子及价数)相同的不同金属离子,主 量子数越大(周期),则值越大; 5d 4d 3d
--
12
6.1.1 晶体场理论(CFT)
1.晶体场理论的内容: 把中心离子(M)和配体(L)的相互作用看作类似
离子晶体中正负离子的静电作用。
当L接近M时,M中的d轨道受到L负电荷的
高二化学配合物结构和性质PPT学习教案

(1)画出这两种固体分子的几何构型图
NH
Cl
Cl-P3 t-Cl
Cl-Pt-NH3
NH
NH
①淡3 黄色
②黄3 绿色
(2)淡黄色固体在水中的溶解度小而黄绿色固体溶解度大
的
①中结构原对称,分子无极性因;②的分子有极性是
_,__据__相__似__相__溶__规__则__可__知__,__前__者__溶__解__度__小__而__后__者__大。
(Cu2+的半径小于Zn2+的半径)
答案:B
第28页/共30页
思考题:
向CoCl2溶液中滴加氨水,使生成的Co(OH)2沉 淀溶解生成[Co(NH3)6]2+。此时向溶液中通入空气 ,得到的产物中有一种其组成可用CoCl3·5NH3表 示。把分离出的CoCl3·5NH3溶于水后立即加硝酸 银溶液,则析出AgCl沉淀。经测定,每1mol CoCl3·5NH3只生成2mol AgCl。请写出表示此配合 物结构的化学式(钴的配位数为6):
高二化学配合物结构和性质
会计学
1
一、什么叫配合物
由提供孤电子对的配体与接受孤电 子对的中心原子以配位键结合形成的化 合物称为配合物。
请思考NH4Cl是否是配合物?
第1页/共30页
配合物的组成
[Cu(NH3)4] SO4
中心离子 配位体 配位数 外界离子
内界
外界
配合物
NH3中的N为配位原子
第2页/共30页
第12页/共30页
[Ag(NH3)2]+的成键情况和空间结构 Ag+空的5s轨道和5p轨道形成sp杂化轨 道,接受2个NH3分子提供的孤电子对, 形成直线形的[Ag(NH3)2]+。
原子簇化合物(1)

13
BnHn+6 BH7 B2H8 B3H9 B4H10 B5H11 B6H12 B7H13 B8H14
B9H15 B10H16 B11H17 B12H18 B13H19 B14H20 B15H20
BnHn+4 BH5 B2H6 B3H7 B4H8 B5H9 B6H10 B7H11 B8H12
B9H13 B10H14 B11H15 B12H16 B13H17 B14H28
6个B, 3×6=18e,6个H用6个e, 余12 + 2(电荷), 7 = n + 1 对
五角双锥 (7) D5h
22
B8H82-
B9H92-
十二面体 (8) D2d
三顶三棱柱 (9) D3h 十四面体
23
B10H102-
B11H112-
双帽四方反棱 (10) D4d 十六面体
十八面体 (11) C2v
39
③ 阴离子(硼酸根)
B12H2-12
闭式的通式
十二氢- 闭式 - 十二硼酸根(2-)离子
④ 硼烷及衍生物的编号法则
选最高次对成轴,由上 → 下
40
↓4
↓4
↓5
双帽四方反棱 D4d
20 面体 I h
选最高次对成轴,由上→ 下
41
B10H14 骨架硼原子的编号
42
←C ←C
闭式-1,2-C2B10H12 杂原子编号最低
(1). 结构类型 闭合(closo) 巢式(nido) 蜘蛛式(arachno) 敞网式(hypho) 稠合式(conjuncto)
20
封闭型硼烷阴离子 BnHn2- (n = 6~12)
B4H42-
B5H52-
BnHn+6 BH7 B2H8 B3H9 B4H10 B5H11 B6H12 B7H13 B8H14
B9H15 B10H16 B11H17 B12H18 B13H19 B14H20 B15H20
BnHn+4 BH5 B2H6 B3H7 B4H8 B5H9 B6H10 B7H11 B8H12
B9H13 B10H14 B11H15 B12H16 B13H17 B14H28
6个B, 3×6=18e,6个H用6个e, 余12 + 2(电荷), 7 = n + 1 对
五角双锥 (7) D5h
22
B8H82-
B9H92-
十二面体 (8) D2d
三顶三棱柱 (9) D3h 十四面体
23
B10H102-
B11H112-
双帽四方反棱 (10) D4d 十六面体
十八面体 (11) C2v
39
③ 阴离子(硼酸根)
B12H2-12
闭式的通式
十二氢- 闭式 - 十二硼酸根(2-)离子
④ 硼烷及衍生物的编号法则
选最高次对成轴,由上 → 下
40
↓4
↓4
↓5
双帽四方反棱 D4d
20 面体 I h
选最高次对成轴,由上→ 下
41
B10H14 骨架硼原子的编号
42
←C ←C
闭式-1,2-C2B10H12 杂原子编号最低
(1). 结构类型 闭合(closo) 巢式(nido) 蜘蛛式(arachno) 敞网式(hypho) 稠合式(conjuncto)
20
封闭型硼烷阴离子 BnHn2- (n = 6~12)
B4H42-
B5H52-
第六章 配位化合物和簇合物的结构与性质

配位体: 配位体: 具有孤对电子或π 具有孤对电子或 π 键电子并能与金属离子进行配位的原子 或分子 原子主要是: Cl, 原子主要是: N,O,C,P,S,Cl,F
配位体分子分为: 配位体分子分为: 单啮配位体:只有一个配位点的配位体, 单啮配位体:只有一个配位点的配位体, 如NH 3 非螯合多啮配位体:一个配位体有多个配位点, 多个金属 非螯合多啮配位体: 一个配位体有多个配位点, 与多个金属 离子配位,但不能直接与同一金属离子配位。 离子配位,但不能直接与同一金属离子配位。 螯合配位体:有多个配位点,且能直接与同一金属 螯合配位体:有多个配位点,且能直接与同一金属 同一 离子配位,形成螯合配位化合物,如乙二胺, 离子配位,形成螯合配位化合物,如乙二胺, 三联吡啶 芳香烃, π配位体:含有π电子的烯、炔、芳香烃,与过渡金属形成配 配位体:含有π电子的烯、 位化合物
③ ∆ 随配位原子半径的减小而增大: 如 I < Br < Cl < S < F < O < N < C p :电子成对能。使体系能量升高。 2、 d轨道中d电子的排布:要从 ∆ 和 p 综合考虑。 ① ∆< p 配体是弱场,∆ 较小 d电子尽量采取高自旋态。 ② 如: Fe2+
∆> p
配体是强场,∆ 较大 d电子尽量采取低自旋态。
按微扰理论, d轨道的平均能量不变,并令Es=0
2 Eeg + 3Et2 g = 0 ∴ Eeg − Et2 g = 10 Dg
那
Eeg = 6 Dg Et2 g = −4 Dg
3、正四面体场的作用:配体从四面体的四个顶点接近中心离子
L
L
一种 d xy , d yz , d zx
簇状配合物.

②桥基:与两个金属原子结合的配体称为“线桥基” (M—L—M)或简称“桥基”。以μ2—L表示。
③面桥基:与多面体中同一面的几个金属原子相连 的配体称为“面桥基”以μx—L表示。 由于簇状配合物的结构多以三角面为基本结构单元, 故面桥基以μ3—L最为常见。
例如:
[Fe4(µ3-CO)(µ2-CO)3(CO)9]2- M4(µ2-CO)3(CO)9(M=Co,Rh)
3. 键能 一般规定M—M键能达到20Kcal∕mol附近即认
为金属键存在。
注意:不同金属M-M键能的比较一般须在同 类配合物及同种测定方法的情况下进行比较判断。
三、结构特点及表示形式 1. 结构特点
(1)簇状配合物的结构是以成簇的金属原子所 构成的金属骨架为特征,骨架中的金属原子以一种 多角形或多面体排列着。
在簇状配合物中三种键合状态的羰基——端羰基、桥 基、面桥基可由红外光谱的羰基伸缩频率来区别。
C-O基类型 C-O键长(埃) 频率(cm-1)
端基
1.12-1.19
2150-1950
桥基
1.165-1.20
1900-1750
面基
1.19-1.22
1800-1700
由端基→桥基→面基红外光谱的羰基伸缩频率逐 渐减小。
Re3X9L3类三原子簇中M—M成键分子轨道
在Re3X9L3中1个Re3+为5d4,3个Re3+离子共有 12个电子,这12个电子全部填充在成键轨道上, 因此成键轨道全部占满。具有最大的M—M键合 为双键,成键电子都配对,化合物是反磁性的。
若从电子配对法来看,由于有6对电子配给Re3三 角形的3个Re—Re边,每个边可分到2对电子。所 以每个Re—Re键是一个双键,组成一个σ键和一个 垂直于Re3平面的π键。
③面桥基:与多面体中同一面的几个金属原子相连 的配体称为“面桥基”以μx—L表示。 由于簇状配合物的结构多以三角面为基本结构单元, 故面桥基以μ3—L最为常见。
例如:
[Fe4(µ3-CO)(µ2-CO)3(CO)9]2- M4(µ2-CO)3(CO)9(M=Co,Rh)
3. 键能 一般规定M—M键能达到20Kcal∕mol附近即认
为金属键存在。
注意:不同金属M-M键能的比较一般须在同 类配合物及同种测定方法的情况下进行比较判断。
三、结构特点及表示形式 1. 结构特点
(1)簇状配合物的结构是以成簇的金属原子所 构成的金属骨架为特征,骨架中的金属原子以一种 多角形或多面体排列着。
在簇状配合物中三种键合状态的羰基——端羰基、桥 基、面桥基可由红外光谱的羰基伸缩频率来区别。
C-O基类型 C-O键长(埃) 频率(cm-1)
端基
1.12-1.19
2150-1950
桥基
1.165-1.20
1900-1750
面基
1.19-1.22
1800-1700
由端基→桥基→面基红外光谱的羰基伸缩频率逐 渐减小。
Re3X9L3类三原子簇中M—M成键分子轨道
在Re3X9L3中1个Re3+为5d4,3个Re3+离子共有 12个电子,这12个电子全部填充在成键轨道上, 因此成键轨道全部占满。具有最大的M—M键合 为双键,成键电子都配对,化合物是反磁性的。
若从电子配对法来看,由于有6对电子配给Re3三 角形的3个Re—Re边,每个边可分到2对电子。所 以每个Re—Re键是一个双键,组成一个σ键和一个 垂直于Re3平面的π键。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
z
z
y
y
z
x
x dxy
dxz
dyz
z
y
y
x
x dz2
dx2-y2
x
d轨道分裂 为:
eg (dz2 , dx2- y2 ) t2g (dxy, dxz, dyz )
eg t2g
d
=晶体场分裂能
八面体场(Oh)
Es 球对称作用部分
eg t2g
自由离子 d轨道 0=10Dq
Eeg= 6 Dq Et2g= -4Dq
Werner提出副价概念,藉以补充当时不完善的 原子价理论,这是他的重要贡献之一。正是为 了满足副价的要求,主价已经饱和的分子、离 子可以进一步反应生成配合物。
内界、外界的概念说明了配合物的结构和物理、 化学性能。而Werner创造性地把有机化学的空 间结构理论扩展到无机化合物领域,奠定了配 合物立体化学的基础,这是他的又一重大贡献。 由于Werner对配位学说的杰出贡献,1913年获 得Nobel化学奖,成为获得此项科学奖金的第 一位无机化学家。
物。
★ 有时中心原子和配位体直接结合成不带电的中性
配位化合物分子。
●单核配位化合物:一个配位化合物分子(或离子)中 只含有一个中心原子。
●多核配位化合物:含两个或两个以上中心原子。
●金属原子簇化合物:在多核配位化合物中, 若M—M之间有键合称为金属原子簇化合物。
目前,研究配位场的理论是晶体场理论(CFT)
• 对经典化合价理论提出了尖锐挑战:化合价已经饱 和的CoCl3和NH3为什么还能相互结合生成很稳定的 “复杂化合物”?
配位化学的奠基人—维尔纳
• 苏黎世大学年仅25岁的A.Werner从1891年起 发表了“对于无机化合物的结构贡献“等一 系列论文,并于1893年在“关于无机化合物 的结构问题”的论文中,将其结构观点系统 化、理论化。这就是在当今称之为“配位学 说”的伟大发现。
2Eeg+3Et2g=0
Eeg - Et2g=10Dq
② 四面体场 中心离子位于立方体的中心,在立方体的八个角上每隔 一个角(上下错开)放一个配体。
dz2
dx2-y2
dxz, dyz, dxy
2 的角度极大值指向立方体的 y
与八面体场相反,dz2,dx2-
面心; 而dxy,dxz,dyz的极大值指向立方体四个边线的中 心。前者离配体较远,斥力小,轨道能量上升少;后者与 配体的斥力大,轨道能量上升多。
8.7
9
Mn4+
Mo3+
23
24.6 27.0
12.0 Rh3+
尿素
HAc 乙醇
0.92
0.94 0.97
NH3
en CN-
1.25
1.28 1.7
Fe3+
Cr3+ Co3+
14.0 Tc4+
17.4 18.2 Ir3+ Pt4+
30
32 36
② 成对能(P)
如果迫使本来自旋平行分占两个轨道的电子挤到同一 轨道上所需要提供的能量称为电子的成对能,用 P表 示。 电子在 d 轨道的排布与 和 P 的相对大小有关。若 P>时,为弱场,则高自旋排布稳定,为弱场高自旋 (HS);若P<时,为强场,则低自旋排布稳定,为 强场低自旋(LS)。
6.1.1
晶体场理论(CFT)
1.晶体场理论的内容:
把中心离子(M)和配体(L)的相互作用看作类 似离子晶体中正负离子的静电作用。 当L接近M时,M中的d轨道受到L负电荷的 静电微扰作用,使原来能级简并的d轨道发生 分裂。按微扰理论可计算分裂能的大小,因计 算较繁,定性地将配体看作按一定对称性排布 的点电荷与M的d轨道电子云产生排斥作用。
值为21000cm-1
对电子构型(d电子及价数)相同的不同金属离子,主 量子数越大(周期),则值越大; 5d 4d 3d 如:当周期数增大时,同族同价的第二系列过渡金属 离子比第一系列过渡金属离子的 值增大约 40~50% , 而第三系列比第二系列的值又增大约20~25%。
C:值为配位体的贡献 (f)和中心离子的贡献 (g)的乘积 ,即
第六章
配位化合物和簇合物的
结构与性质
第六章目录
6.1 配位场理论简介 6.1.1 晶体场理论 6.1.2 配位场理论简介 6.2 CO和N2配位化合物的结构与性质 6.2.1 羰基配合物 6.2.2 N2的配位化合物与固氮
Contents
第六章目录
6.3 有机金属配合物的结构与性质 6.3.1 蔡斯(Zeise)盐 6.3.2 夹心式配合物 6.4 原子簇化合物的结构与性质 6.4.1 过渡金属簇合物 6.4.2 碳笼烯
• 配位化学目前已经发展成为无机化学、有 机化学、物理化学等学科的重要交叉学科。 当代配位化学已经突破了纯无机化学的范 畴,它渗入有机合成、高分子化学和生物 化学,形成了许多崭新的富有生命力的边 缘学科,成为当代化学学科中最活跃的领 域之一。 • 目前配位化学的结构理论有: (1)价键理论;(2)晶体场理论;(3) 分子轨道理论;(4)配位场理论
③ 八面体及四面体配合物中d轨道上电子的排布 在八面体场中,不论是强场还是弱场, d1,d2,d3,d8
,d9 和 d10 的排布方式都是一样的,未成对电子数相同
,没有高低自旋之分;而 d4,d5,d6,d7 则不同:当
P>时,为高自旋;当P<时,为低自旋。
如配位离子[Fe(H2O)6]2+和[Fe(CN)6]4-的中心金属均为 Fe2+ (d6)组态,但前者显强顺磁性而后者显反磁性,说 明配体H2O对Fe2+形成的Oh场为弱场,而配体CN-对 Fe2+形成的Oh场为强场。
e Td eg Oh
球对称性下降, d轨道分裂的组数增加。
其它晶体场的能级分裂情况见表6.1
3 d轨道中电子的排布——高自旋态和低自旋态 与价键理论一样,CFT中中心离子d轨道上的电子
排布也分为高自旋态和低自旋态。其排布情况与
电子的分裂能()和成对能(P)的相对大小有关。
配位学说
大多数元素具有两种原子价一主价(相当于现 在的氧化数)和副价(相当于后来的配位数)。 每一元素倾向于既满足它的主价又满足它的 副价。 在配合物中部分分子或离子与中心离子较紧 密地结合在一起,组成在溶液中能够稳定存 在的整体,称为“内界”。与中心离子结合 不够紧密的离子则处于“外界”。在溶液中 外界离子易于离解,内界分子或离子则难于 离解。 副价指向空间的确定位置,配合物有确定的 几何构型。
也称配位场强度序列)。
若只看配位体中直接配位的单个原子,则随原子序数的 增大而减小: C N O F S Cl Br I
问题:CFT无法解释为什么不带电荷的中性分子CO是
强场配体,而带电荷的卤素离子却是弱场配体? 这与 键的形成有关: dxy,dxz,dyz 虽不能与配体 L 形成 键,但条件适合可形成 键,从而影响 的大 小,这与配位场理论有关。 B:当配位体固定时, 随中心离子而改变: 对同一金属离子而言,电荷越高, 值越大; 如:Mn2+对H2O的值为7800cm-1,而Mn3+对H2O的
验规律: A: 当中心离子M固定时, 值随配体而改变
CO≈CN–NO2– 邻 蒽 菲 联 吡 啶 SO32– 乙 二 胺
(en)NH3 吡啶EDTA H2O F–OH– Cl– Br–I–
大者为强场配位体, 小者为弱场配位体。由于通
常由光谱实验确定,故称这个顺序为光谱化学序列(
为四面体排布; d1和d6一般仍采用四面体型,如VCl4,
FeCl42-。
对于 d8 的四配位化合物,应为平面正方形,因为这 种构型获得的 LFSE较多,这时配位化合物自旋成对
,呈反磁性 ,第二,第三长周期过渡元素确是如此,
如 PtCl42-, PdCl42- ,Au2Cl6等。 而第一长周期过渡元素,因金属离子较小,碰到电 负性高、体积大的配体时,则需考虑排斥作用, Ni(CN)42-为平面正方形,而 NiX42-(X=Cl-,Br-,I-)为四
d轨道分裂为:
t2 (dxy, dxz, dyz ) t2 e (dz2 , dx2- y2 ) e
t
d
t=4/9 0
t t2 e
四面体场(Td) 自由离子 d轨道 Ee= -2.67 Dq Et2= 1.78Dq Es 球对称作用部分
③ 平面正方形场 四个配体分别沿 x,y 四个方向向中心离子接近,其
Contents
6.1
配位场理论简介
配位化合物:又称络合物,是一类含有中心金属原子
(M)和若干配位体(L)的化合物(MLn)。
★ 中心原子M通常是过渡金属元素的原子 (或离
子),具有空的价轨道。
★ 配位体L则有一对或一对以上孤对电子。
★ M和L之间通过配位键结合,成为带电的配位离子,
配位离子与荷异性电荷的离子结合,形成配位化合
Δ= f (配体) × g ( 中心离子)
八面体场的 f 值 和 g 值
f 值 Br- 0.72 C2O42- 0.99 g 值(单位:1000cm-1 ) Mn2+ 8.0 Ru2+ 20
SCN-
Cl- F-
0.73
0.78 0.9
H2 O
NCS- py
1.00
1.02 1.23
Ni2+
Co2+ V2+
作用下受到不同的影响,简并的能级发生不同的分裂。
z
-
z
-
y
z
y
x
y
-
x
- -
x
x dx2-y2
z
y
y
z
x
x dxy
dxz
dyz
z
y
y
x
x dz2
dx2-y2
x
d轨道分裂 为:
eg (dz2 , dx2- y2 ) t2g (dxy, dxz, dyz )
eg t2g
d
=晶体场分裂能
八面体场(Oh)
Es 球对称作用部分
eg t2g
自由离子 d轨道 0=10Dq
Eeg= 6 Dq Et2g= -4Dq
Werner提出副价概念,藉以补充当时不完善的 原子价理论,这是他的重要贡献之一。正是为 了满足副价的要求,主价已经饱和的分子、离 子可以进一步反应生成配合物。
内界、外界的概念说明了配合物的结构和物理、 化学性能。而Werner创造性地把有机化学的空 间结构理论扩展到无机化合物领域,奠定了配 合物立体化学的基础,这是他的又一重大贡献。 由于Werner对配位学说的杰出贡献,1913年获 得Nobel化学奖,成为获得此项科学奖金的第 一位无机化学家。
物。
★ 有时中心原子和配位体直接结合成不带电的中性
配位化合物分子。
●单核配位化合物:一个配位化合物分子(或离子)中 只含有一个中心原子。
●多核配位化合物:含两个或两个以上中心原子。
●金属原子簇化合物:在多核配位化合物中, 若M—M之间有键合称为金属原子簇化合物。
目前,研究配位场的理论是晶体场理论(CFT)
• 对经典化合价理论提出了尖锐挑战:化合价已经饱 和的CoCl3和NH3为什么还能相互结合生成很稳定的 “复杂化合物”?
配位化学的奠基人—维尔纳
• 苏黎世大学年仅25岁的A.Werner从1891年起 发表了“对于无机化合物的结构贡献“等一 系列论文,并于1893年在“关于无机化合物 的结构问题”的论文中,将其结构观点系统 化、理论化。这就是在当今称之为“配位学 说”的伟大发现。
2Eeg+3Et2g=0
Eeg - Et2g=10Dq
② 四面体场 中心离子位于立方体的中心,在立方体的八个角上每隔 一个角(上下错开)放一个配体。
dz2
dx2-y2
dxz, dyz, dxy
2 的角度极大值指向立方体的 y
与八面体场相反,dz2,dx2-
面心; 而dxy,dxz,dyz的极大值指向立方体四个边线的中 心。前者离配体较远,斥力小,轨道能量上升少;后者与 配体的斥力大,轨道能量上升多。
8.7
9
Mn4+
Mo3+
23
24.6 27.0
12.0 Rh3+
尿素
HAc 乙醇
0.92
0.94 0.97
NH3
en CN-
1.25
1.28 1.7
Fe3+
Cr3+ Co3+
14.0 Tc4+
17.4 18.2 Ir3+ Pt4+
30
32 36
② 成对能(P)
如果迫使本来自旋平行分占两个轨道的电子挤到同一 轨道上所需要提供的能量称为电子的成对能,用 P表 示。 电子在 d 轨道的排布与 和 P 的相对大小有关。若 P>时,为弱场,则高自旋排布稳定,为弱场高自旋 (HS);若P<时,为强场,则低自旋排布稳定,为 强场低自旋(LS)。
6.1.1
晶体场理论(CFT)
1.晶体场理论的内容:
把中心离子(M)和配体(L)的相互作用看作类 似离子晶体中正负离子的静电作用。 当L接近M时,M中的d轨道受到L负电荷的 静电微扰作用,使原来能级简并的d轨道发生 分裂。按微扰理论可计算分裂能的大小,因计 算较繁,定性地将配体看作按一定对称性排布 的点电荷与M的d轨道电子云产生排斥作用。
值为21000cm-1
对电子构型(d电子及价数)相同的不同金属离子,主 量子数越大(周期),则值越大; 5d 4d 3d 如:当周期数增大时,同族同价的第二系列过渡金属 离子比第一系列过渡金属离子的 值增大约 40~50% , 而第三系列比第二系列的值又增大约20~25%。
C:值为配位体的贡献 (f)和中心离子的贡献 (g)的乘积 ,即
第六章
配位化合物和簇合物的
结构与性质
第六章目录
6.1 配位场理论简介 6.1.1 晶体场理论 6.1.2 配位场理论简介 6.2 CO和N2配位化合物的结构与性质 6.2.1 羰基配合物 6.2.2 N2的配位化合物与固氮
Contents
第六章目录
6.3 有机金属配合物的结构与性质 6.3.1 蔡斯(Zeise)盐 6.3.2 夹心式配合物 6.4 原子簇化合物的结构与性质 6.4.1 过渡金属簇合物 6.4.2 碳笼烯
• 配位化学目前已经发展成为无机化学、有 机化学、物理化学等学科的重要交叉学科。 当代配位化学已经突破了纯无机化学的范 畴,它渗入有机合成、高分子化学和生物 化学,形成了许多崭新的富有生命力的边 缘学科,成为当代化学学科中最活跃的领 域之一。 • 目前配位化学的结构理论有: (1)价键理论;(2)晶体场理论;(3) 分子轨道理论;(4)配位场理论
③ 八面体及四面体配合物中d轨道上电子的排布 在八面体场中,不论是强场还是弱场, d1,d2,d3,d8
,d9 和 d10 的排布方式都是一样的,未成对电子数相同
,没有高低自旋之分;而 d4,d5,d6,d7 则不同:当
P>时,为高自旋;当P<时,为低自旋。
如配位离子[Fe(H2O)6]2+和[Fe(CN)6]4-的中心金属均为 Fe2+ (d6)组态,但前者显强顺磁性而后者显反磁性,说 明配体H2O对Fe2+形成的Oh场为弱场,而配体CN-对 Fe2+形成的Oh场为强场。
e Td eg Oh
球对称性下降, d轨道分裂的组数增加。
其它晶体场的能级分裂情况见表6.1
3 d轨道中电子的排布——高自旋态和低自旋态 与价键理论一样,CFT中中心离子d轨道上的电子
排布也分为高自旋态和低自旋态。其排布情况与
电子的分裂能()和成对能(P)的相对大小有关。
配位学说
大多数元素具有两种原子价一主价(相当于现 在的氧化数)和副价(相当于后来的配位数)。 每一元素倾向于既满足它的主价又满足它的 副价。 在配合物中部分分子或离子与中心离子较紧 密地结合在一起,组成在溶液中能够稳定存 在的整体,称为“内界”。与中心离子结合 不够紧密的离子则处于“外界”。在溶液中 外界离子易于离解,内界分子或离子则难于 离解。 副价指向空间的确定位置,配合物有确定的 几何构型。
也称配位场强度序列)。
若只看配位体中直接配位的单个原子,则随原子序数的 增大而减小: C N O F S Cl Br I
问题:CFT无法解释为什么不带电荷的中性分子CO是
强场配体,而带电荷的卤素离子却是弱场配体? 这与 键的形成有关: dxy,dxz,dyz 虽不能与配体 L 形成 键,但条件适合可形成 键,从而影响 的大 小,这与配位场理论有关。 B:当配位体固定时, 随中心离子而改变: 对同一金属离子而言,电荷越高, 值越大; 如:Mn2+对H2O的值为7800cm-1,而Mn3+对H2O的
验规律: A: 当中心离子M固定时, 值随配体而改变
CO≈CN–NO2– 邻 蒽 菲 联 吡 啶 SO32– 乙 二 胺
(en)NH3 吡啶EDTA H2O F–OH– Cl– Br–I–
大者为强场配位体, 小者为弱场配位体。由于通
常由光谱实验确定,故称这个顺序为光谱化学序列(
为四面体排布; d1和d6一般仍采用四面体型,如VCl4,
FeCl42-。
对于 d8 的四配位化合物,应为平面正方形,因为这 种构型获得的 LFSE较多,这时配位化合物自旋成对
,呈反磁性 ,第二,第三长周期过渡元素确是如此,
如 PtCl42-, PdCl42- ,Au2Cl6等。 而第一长周期过渡元素,因金属离子较小,碰到电 负性高、体积大的配体时,则需考虑排斥作用, Ni(CN)42-为平面正方形,而 NiX42-(X=Cl-,Br-,I-)为四
d轨道分裂为:
t2 (dxy, dxz, dyz ) t2 e (dz2 , dx2- y2 ) e
t
d
t=4/9 0
t t2 e
四面体场(Td) 自由离子 d轨道 Ee= -2.67 Dq Et2= 1.78Dq Es 球对称作用部分
③ 平面正方形场 四个配体分别沿 x,y 四个方向向中心离子接近,其
Contents
6.1
配位场理论简介
配位化合物:又称络合物,是一类含有中心金属原子
(M)和若干配位体(L)的化合物(MLn)。
★ 中心原子M通常是过渡金属元素的原子 (或离
子),具有空的价轨道。
★ 配位体L则有一对或一对以上孤对电子。
★ M和L之间通过配位键结合,成为带电的配位离子,
配位离子与荷异性电荷的离子结合,形成配位化合
Δ= f (配体) × g ( 中心离子)
八面体场的 f 值 和 g 值
f 值 Br- 0.72 C2O42- 0.99 g 值(单位:1000cm-1 ) Mn2+ 8.0 Ru2+ 20
SCN-
Cl- F-
0.73
0.78 0.9
H2 O
NCS- py
1.00
1.02 1.23
Ni2+
Co2+ V2+
作用下受到不同的影响,简并的能级发生不同的分裂。
z
-
z
-
y
z
y
x
y
-
x
- -
x
x dx2-y2