2013-第六章 配合物的反应动力学解析
配位化学-中科院总结(4-6章)

(3) [Ni(en)3]2+ 和 [Fe(en)3]2+ 二者中心离子的d电子数不同,其CFSE不同。 [Ni(en)3]2+ : 3d8,电子排布为t2g6eg2,CFSE = -12Dq; [Fe(en)3]2+ : 3d6,电子排布为t2g6,CFSE = -24Dq。 所以, [Fe(en)3]2+ 更稳定。 (4) [Ni(H2O)6]2+ 和 [Ni(en)3]2+ en为螯合配体,其配合物具有螯合效应, 所以, [Ni(en)3]2+ 更稳定。
+ H2O
若为SN2机理: [Co(NH3)5 X]2+
v = k[Co(NH3)5X2+] + H2O 慢 [Co(NH3)5 X H2O]2+
[Co(NH3)5 X H2O]2+ 快 [Co(NH3)5 H2O]3+ + Xv = k[Co(NH3)5X][H2O]≈ k[Co(NH3)5X2+]
6.如何用晶体场理论判断配合物的活性和惰性。 比较活化配合物与反应物的CFSE确定。
7. [Co(NH3)5X]的水解反应机理和速率方程,如何验 证反应机理?
例如: [Co(NH3)5X]的酸式水解 若为SN1机理:[Co(NH3)5
[Co(NH3)5 ]2+ 慢 快
X]2+
[Co(NH3)5]3+ + X[Co(NH3)5H2O]3+
A5 A3 A2 A6 M B1 X4
A5
A5
-X
A2
A3
M B1 A6
4
+Y
A2
A3
M B1 A6
第五章配合物的反应动力学

CFAE≤0 所需活化能小,是活性配合物; 若: CFAE≤0,所需活化能小,是活性配合物; 所需活化能大,是惰性配合物. CFAE >0,所需活化能大,是惰性配合物. 构型配合物为例,计算其晶体场活化能. 以d6构型配合物为例,计算其晶体场活化能. d6构型配合物八面体的晶体场稳定化能: 构型配合物八面体的晶体场稳定化能: 低自旋(d : 低自旋 xz2dyz2dxy2): CFSE=6*(-4)= -24Dq 高自旋( 高自旋( dxz2dyz1dxy1dz21dx2-y21): CFSE=4*(-4)+2*6= -4Dq (a),按SN1反应机理,经过四方锥中间体. , 反应机理, 反应机理 经过四方锥中间体. 构型配合物四方锥体的晶体场稳定化能: d6构型配合物四方锥体的晶体场稳定化能:
ns
np
nd
解释:若按 机理反应, 解释:若按SN2机理反应,容易理解. 机理反应 容易理解. 外轨型配合物: 外轨型配合物: 因为nd轨道与 轨道与sp 轨道能量接近, 因为 轨道与 3d2轨道能量接近,插入取代配 体的一对电子,因此不需要消耗很大的活化能. 体的一对电子,因此不需要消耗很大的活化能. 内轨型配合物: 内轨型配合物: 有空轨道, 若 (n-1)d有空轨道, 则可接受第 个配体的电子 有空轨道 则可接受第7个配体的电子 因此是活性配合物. 对,因此是活性配合物. 若无(n-1)d空轨道 , 第 7个配体的电子对加入有 空轨道, 若无 空轨道 个配体的电子对加入有 三种可能: 三种可能: (1),加到 轨道上; 轨道上; ,加到nd轨道上 (2),使(n-1)d轨道的成单电子配对(d3,d4); 轨道的成单电子配对( , 轨道的成单电子配对 (3),(n-1)d轨道中的电子激发至较高级上,以空 轨道中的电子激发至较高级上, , 轨道中的电子激发至较高级上 轨道. 出(n-1)d轨道. 轨道 以上三种方式均需较高活化能,因此是惰性的. 以上三种方式均需较高活化能,因此是惰性的.
第讲配合物的热力学性质和动力学性质
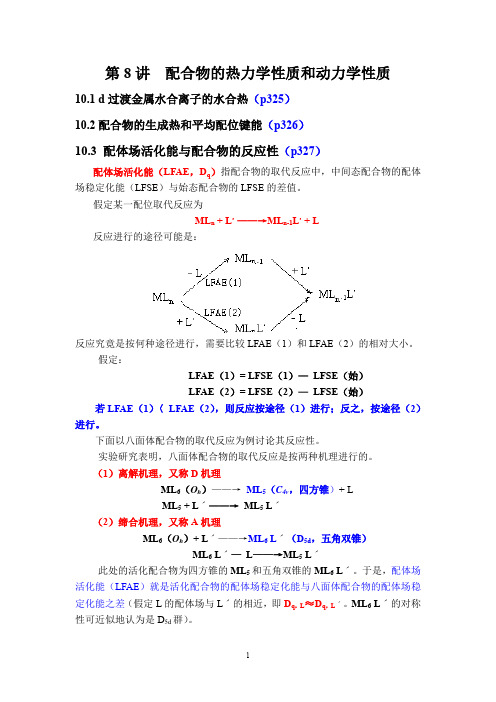
第8讲配合物的热力学性质和动力学性质10.1 d过渡金属水合离子的水合热(p325)10.2配合物的生成热和平均配位键能(p326)10.3 配体场活化能与配合物的反应性(p327)配体场活化能(LFAE,D q)指配合物的取代反应中,中间态配合物的配体场稳定化能(LFSE)与始态配合物的LFSE的差值。
假定某一配位取代反应为ML n + L,——→ML n-1L,+ L反应进行的途径可能是:反应究竟是按何种途径进行,需要比较LFAE(1)和LFAE(2)的相对大小。
假定:LFAE(1)= LFSE(1)—LFSE(始)LFAE(2)= LFSE(2)—LFSE(始)若LFAE(1)〈LFAE(2),则反应按途径(1)进行;反之,按途径(2)进行。
下面以八面体配合物的取代反应为例讨论其反应性。
实验研究表明,八面体配合物的取代反应是按两种机理进行的。
(1)离解机理,又称D机理ML6(O h)——→ML5(C4v,四方锥)+ LML5 + Lˊ——→ML5 Lˊ(2)缔合机理,又称A机理ML6(O h)+ Lˊ——→ML6 Lˊ(D5d,五角双锥)ML6 Lˊ—L——→ML5 Lˊ此处的活化配合物为四方锥的ML5和五角双锥的ML6 Lˊ。
于是,配体场活化能(LFAE)就是活化配合物的配体场稳定化能与八面体配合物的配体场稳定化能之差(假定L的配体场与Lˊ的相近,即D q,L ≈D q,Lˊ。
ML6 Lˊ的对称性可近似地认为是D5d群)。
表1 离解机理的配体场活化能(D q)离子构型强场弱场四方锥八面体LFAE 四方锥八面体LFAEd00 0 0 0 0 0 d1-4.57 -4.00 -0.57 -4.57 -4.00 -0.57 d2-9.14 -8.00 -1.14 -9.14 -8.00 -1.14 d3-10.00 -12.00 2.00 -10.00 -12.00 2.00 d4-14.57 -16.00 1.43 -9.14 -6.00 -3.14 d5-19.14 -20.00 0.86 0 0 0 d6-20.00 -24.00 4.00 -4.57 -4.00 -0.57 d7-19.14 -18.00 -1.14 -9.14 -8.00 -1.14 d8-10.00 -12.00 2.00 -10.00 -12.00 2.00 d9-9.14 -6.00 -3.14 -9.14 -6.00 -3.14 d100 0 0 0 0 0表2 缔合机理的配体场稳定化能(D q)离子构型强场弱场五角双锥八面体LFAE 五角双锥八面体LFAEd00 0 0 0 0 0 d1-5.28 -4.00 -1.28 -5.28 -4.00 -1.28 d2-10.56 -8.00 -2.56 -10.56 -8.00 -2.56 d3-15.84 -12.00 -3.84 -7.74 -12.00 4.16 d4-21.12 -16.00 -5.12 -4.93 -6.00 1.07 d5-18.30 -20.00 1.70 0 0 0 d6-15.48 -24.00 8.52 -5.28 -4.00 -1.28 d7-10.56 -18.00 7.45 -10.56 -8.00 -2.56 d8-7.74 -12.00 4.26 -7.74 -12.00 4.16 d9-4.92 -6.00 1.08 -4.92 -6.00 1.08 d100 0 0 0 0 0事实上,D 机理和A 机理仅是两种极端情况。
2013-第三章--配合物的化学键理论解析

与羰基配合物成键过程相似,CN-配体中C上的 孤电子对向金属的杂化空轨道配位,形成σ配键,金 属的d电子向CN- π* 轨道配位,形成d-pπ配键。
(3) 烯烃配合物
1827年,Zeise合成了K[ PtCl3(C2H4) ]·H2O,这是第 一个有机金属化合物,但其结构直到120多年后才确定。 乙烯的成键π电子向铂的杂化轨道配位,按成键的对称 性应为σ配键;金属d轨道的电子向乙烯的 π* 轨道配位, 形成d-pπ配键。
z
y x
1. d轨道的分裂
dz2
dyz
dxz
dx2-y2
z
y
x
dxy
d 轨道分裂情况 八面体场中:
dz2 , dx2-y2, 轨道能量升高 (eg 或 dγ) dxy, dyz, dxz 轨道能量降低 (t2g 或 dε) 四面体场中:
dz2 , dx2-y2, 轨道能量降低 (eg) dxy, dyz, dxz 轨道能量升高 (t2g)
dx2-y2
x y
极大值指向面心
dxy
x
y
极大值指向棱的中点
1. d轨道的分裂 ( 在Oh场中的分裂 )
分裂能 o = 10 Dq 场强参数Dq: D—中心离子的
极化度 q:配体电荷
Dq具有能量单位
重心守恒原理: 分裂前后五个d轨 道的总能量相等
没有不成对电子
稳定性:内轨型配合物 > 外轨型配合物
根据实验测得的有效磁矩,判断下列各种离子分
别有多少个未成对电子?哪个是外轨?哪个是内轨?
① Fe (e n22)
5.5 B.M.
配合物的反应动力学及反应机理

SN1速度
减小
增加
(zēngjiā)
不影响
不影响
减小
增加 增加 增加
SN2速度
增加
增加
增加 减小 减小
减小 减小 减小
* 近似地适用于半满或全满d壳层情况,不适用于具有共价键或键配合物
共八十四页
2. 配体的取代(qǔdài)反应
2.3 八面体配合(pèihé)物的取代反应的速率方程
1. 离解(D)机理
共八十四页
1. 基本概念
1.6 势能(shìnéng)曲线 活化(自由)能
过渡态:瞬间存在的不稳定物种;
中间体:相对稳定的过渡(guòdù)化合物,在慢反应中能检测其存在;
共八十四页
1. 基本概念
1.7 活化(huóhuà)参数
通常反应机理(jī lǐ)不能够完全由速率方程决定,还需从实验获得的活化参数予以证实。
例:[Fe(H2O)6]3+ + Y 比 [Fe(H2O)5(OH)]2+ + Y 反应速率大2个数量级
反应速率只与水合金属离子溶度有关,与外来配体L无关。
反应机理:
r k[M(H2O)x ]
[M(H2O)x ]n slow[M(H2O)x1]n H2O [M(H2O)x1]n Lm fast[M(H2O)x1L]nm
键的断裂是反应速率的决定步骤
共八十四页
2. 配体的取代(qǔdài)反应
(2)缔合(置换(zhìhuàn))机理(A机理,SN2机理)
1. 中心原子的电子结构
d0-d2电子:有空的d轨道,易接受外来配体的电子,生成 配位数为7 的活化配合物。
Cr3 (d3 ) :
d3-d6电子:惰性。(需要较高活化能)
2013-第六章 配合物的反应动力学汇总

过渡态
Ea
反应物
CFSE(反应物)
CFSE(过渡态) 若CFAE≤0,所需活化能减小, 是活性配合物;
Ea'
产物 反应坐标
CFAE >0,所需活化能增加, 是惰性配合物。
由于CFAE与过渡态构型有关,因此可用来判定 SN1还是SN2机理(数据见表,P107)。
A、d0、d1、d2、d10 及弱场下的 d5、d6、d7 八面体配合物的 CFAE≤0,是活性配合物。
事实上, 这两类配合物之间并不存在明显的界限。 Taube建议, 在室温下, 0.1mol·L-1的溶液中1分钟内 能反应完全的配合物, 叫活性配合物, 大于1分钟的则是 惰性配合物。
(2) 与热力学稳定常数的关系 活性与惰性是动力学上的概念,不可与稳定性混为
一谈。惰性配合物也可能是热力学不稳定的配合物。
个电子), 则这样的配合物就是惰性的。
这是因为当中心金属没有空 (n-1)d 轨道时, 要么 进入的配体的孤电子对必须填入 nd 轨道, 要么 (n- 1)d 轨道上的电子必须被激发到更高能级轨道, 以便腾 出一条空 (n-1)d 轨道, 而这两种情况都需要较高的活 化能之故。
↑ ↑ ↑ ↑↓ ↑↓
离解机理
交换机理 反应机理示意图
缔合机理
(a), (c) 有中间产物存在, (b) 无中间产物
a. 加到nd轨道上; b. 使 (n-1) d轨道的成单电子配对(d3,d4); c. (n-1) d轨道中的电子激发至较高级上,以空出轨道. 这三种方式均需较高活化能,因此是惰性的。
价键理论之不足之处: 只能作定性划分; 认为d8组态Ni2+八面体配合物(外轨型)为活性,
实验表明为惰性; 无法解释Cr3+(d3)和Co3+(d6)八面体配合物比
配合物的结构及异构现象
[Ni(CN)4]2- (d8) , [PdCl4]2- (d8), [Pt(NH3)4]2+ (d8), [Cu(NH3)4]2+ (d9)
一般地,当4个配体与不含有d8电子构型的过渡金属离子或原子配位时可形成四面体构型配合物。
而d8组态的过渡金属离子或原子一般是形成平面正方形配合物, 但具有d8组态的金属若因原子太小, 或配位体原子太大, 以致不可能形成平面正方形时, 也可能形成四面体的构型。
双帽四方反棱柱体 双帽12面体
配位数为10的配位多面体是复杂的, 通常遇到的有双帽四方反棱柱体和双帽12面体。
十一配位的化合物极少, 理论上计算表明, 配位数为11的配合物很难具有某个理想的配位多面体。 可能为单帽五角棱柱体或 单帽五角反棱柱体, 常见于 大环配位体和体积很小的 双齿硝酸根组成的配合物中。
早在1893年维尔纳(瑞士)建立配位理论时,就已经提出了使中心离子周围配体之间的静电斥力最小,配合物最稳定,即配体间应尽力远离,从而采取对称性分布,而实际测定结构的结果证实了这种设想。
配合物的配位数与其空间结构有一定的联系,但配位数相同时,由于配体的不同,与中心离子的作用不同,而空间结构也会不同。
配位数3 (D3h)三角形
配位数5 (D3h,T4v )主要为三角双锥和四方锥
配位数8 (D4d , D2d )四方反棱柱和十二面体
配合物的配位数与几何构形
价键理论顺利地解释了配合物的分子构型。
显然, 分子构型决定于杂化轨道的类型:
配 位 数 2 3 4 4 杂化轨道 sp sp2 sp3 dsp2 分子构型 直线 三角形 正四面体 正方形 配 位 数 5 5 6 杂化轨道 sp3d d2sp2, d4s sp3d2, d2sp3 分子构型 三角双锥 四方锥 正八面体
配位化学第六章分析
Coordination Chemistry
[L5M-X] + Y→ [L5M-X ···Y] → [MLn-1Y] + L
离去配体和进入配体在同一步中形成活化配 合物并发生交换, 并无真正的中间体, 而是形成 L─M--Y 过渡态
三、平面正方形配合物的取代反应
d8电子组态的过渡金属离子易生成平面正方 形配合物,如:Rh+, Ir+, Ni2+, Pd2+, Pt2+, Au3+等。 Pt(II)配合物构型稳定,反应速度适中,研 究的较多
Coordination Chemistry
1、平面正方形配合物的取代反应机理
d8:dsp2, 有空的p轨道,可接受外来配体,生 成配位数为5的配合物
Coordination Chemistry
第六章 配合物的反应动力学
Coordination Chemistry
配合物反应种类
配体取代反应 电子转移反应(氧化还原反应) 分子重排反应(异构化反应) 配合物的加成与消除反应 配体上进行的化学反应……
Coordination Chemistry
配合物反应机理
(a) MLn + Y → MLn-1Y + L 配体Y取代配体L
亲核取代反应 SN (nucleophilic substitution reaction)
(b) MLn + M´ → M´Ln + M 金属离子M´取代M
亲电取代反应 SE (electrophilic substitution reaction)
Ea
反应物
无机化学 第六章 化学动力学.
3. 实验测得aA + bB →cC + dD的动力学方程式为 r=kca(A)cb(B) ,下列说法错误的是( )
A. 该反应是基元反应 B. 反应级数为a+b C. 对反应物A是a级 D.对反应物 B是b级
4. 已知某反应是放热反应,如果升高温度,则( )
A. k增加,Kθ减小
B. k,Kθ均增加
6. 催化剂可以使反应的( )
A. Kθ增大
B. 反应自由能增大
C. 活化能改变
D. 反应焓减小
7. 反应2NO(g) + Br2(g) →2NOBr(g)的反应历程是: (1) NO(g) + Br2(g) →2NOBr2(g) 快 (2) NO(g) + NOBr2(g) →2NOBr2(g) 慢,则该反应对NO的反应 级数为( )
三、速率常数
速率方程中的 k 称为速率常数,物理意义是速率方程式 中各种物质浓度均为1 mol/L时的反应速率。
速率常数的量纲由速率方程确定,总反应级数不同,单位 不同。
速率常数越大,表明反应进行得越快。但应注意,两个反 应级数不同的反应,对比速率常数的大小是毫无意义的。
速率常数的大小取决于反应物的本质,而与浓度无关。 速率常数是温度的函数,当其他条件相同时,T 升高,则 k 增大。
第6章 化学动力学 6-1 化学反应速率
1. 平均速率: 指反应进程中某时间间隔(t)内反应物浓度的减少或生成
物浓度的增加来表示: r ≡ ︱cB / t︱ 或:r ≡ (1/B )cB / t
2. 瞬时速率
指某一瞬时(时刻)的速率,即时间间隔无限小,t→0时的 平均速率。
r ≡ ︱dcB / dt︱或 r ≡ (1/B )dcB / dt
配位化学多媒体课件 第六章 配合物的反应动力学和反应机理
反应中物种数增加, △S 也=是一个较大正值。
根据A机理, 由MLn+Y→MLnY可判断形成了新键, 一般要 放出能量, 因而总反应△H 表=现为一个较小的正值,
反应中物种数减小, △S 为=负值。
事实上, 这两类配合物之间并不存在明显的界限。
TaubemolL-1的溶液中一分钟内能反响完全的配合物, 叫活 性配合物, 大于一分钟的那么是惰性配合物。
活化配合物
Ea
反应物 △ H
产物
配合物的活性和惰性与热力学的稳 定性不应混淆。
由左边的图可以看出, 配合物的热力 学稳定性取决于反应物和产物的能量差, 而配合物的活性、惰性则决定于反应物 与活化配合物之间的能量差。
由此可判断反应究竟是属于D机理还是A机理。
也可以根据形成过渡态时体积的变化△V 来进行判断, 显然
D机理 △V 0。 =A机理 △V 0
=
6.3 配合物的活性与惰性
配离子发生配位体交换反响的能力, 是用动力学稳定性的概 念来描述的, 配体交换反响进行得很快的配合物称为活性的配合 物, 而那些交换反响进行得很慢或实际上观察不到交换的配合物 那么称为惰性配合物。
配合物热力学稳定性与动力学活泼性处于矛盾情况的两个 典型例子是Co(NH3)63+和[Ni(CN)4]2-:
六氨合钴(III)离子在室温酸性水溶液中的H2O取代NH3的反 应, 在数日内无显著变化, 说明配合物是惰性的。但反应
Co(NH3)63++6H3O+ Co(H2O)63++6NH3+ 的平衡常数 K=1025, 说明配离子在热力学上是不稳定的。
如具有d3、d8电子构型的配合物 按离解机理进行取代时(中间体为四方锥),
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
于离解机理反应, 意味着配合物的惰性越大。
对于缔合机理(A)反应:
若进入配体的半径越小, 负电荷越大, 越有利于缔
合机理反应, 意味着配合物的活性越大。 但中心离子的电荷的增加有着相反的影响: 一方面使M-L键不容易断裂, 另一方面又使M-Y 键更易形成。究竟怎样影响要看两因素的相对大小。 一般来说, 中心离子半径和电荷的增加使得缔合机 理反应易于进行。进入配体半径很大时, 由于空间位阻, 缔合机理反应不易进行。
5)
2. 缔合机理(SN2机理,或A机理 )
慢 L5M-X+Y == L5MXY(配位数升高6
L5MXY == L5M-Y + X 反应速率:d[L5M-Y]/dt = k[L5M-X][Y] 动力学上属于二级反应。
7)
SN1和SN2是两种极限情況,大多数反应都是按照 这两种极限情况的中间机理进行的。
这是因为, 在八面体配合物中, 如果配合物中含有
一个空的 (n-1)d 轨道, 那么一个进来的配体 (即取代
配体) 就可以从空轨道的四个叶瓣所对应的方向以较小 的静电排斥去接近配合物, 并进而发生取代反应。这样 的配合物显然是比较活性的。
② 内轨型配合物的取代反应速率
一般取决于中心离子的 (n-1)d轨道中的电子分布.
当 (n-1)d 轨道中至少有一条轨道是空的配合物
就是活性配合物。
同样是因为当中心金属含有空轨道时,进来的配
体可以从空轨道的波瓣所对应的方向以较小的静电排
斥去接近配合物(从而配体上的孤电子对就可以填入
空轨道),进而发生取代反应。
↑↓ ↑↓
↑↓
↑↓
↑↓
↑↓
活性:d0
↑ ↑↓ ↑↓ ↑↓ ↑↓ ↑↓ ↑↓
二、活性与惰性配合物及理论解释
1. 活性与惰性配合物
配离子发生配位体交换反应的能力, 是用动力学稳 定性的概念来描述的。 配体交换反应进行的很快的配合物称为活性配合 物, 而那些交换反应进行得很慢或实际上观察不到交换 的配合物则称为惰性配合物。 事实上, 这两类配合物之间并不存在明显的界限。 Taube建议, 在室温下, 0.1mol· L-1的溶液中1分钟内 能反应完全的配合物, 叫活性配合物, 大于1分钟的则是 惰性配合物。
进入的配体的孤电子对必须填入 nd 轨道, 要么 (n-
1)d 轨道上的电子必须被激发到更高能级轨道, 以便腾
第一章 配位化学基础知识 第二章 配合物的结构及异构现象 第三章 配合物的化学键理论 第四章 配合物的电子光谱 第五章 配合物在溶液中的稳定性 第六章 配合物反应动力学 第七章 新型配合物
第六章 配合物反应动力学
反应机理是对整个反应中所发生的涉及分子、 原子、自由基或离子的各步反应过程的详细说明。 反应机理的研究在于说明新的化学反应的变化过程, 预测化合物的反应性,指导新化合物的合成 方面有着重要的实际和理论意义。 研究范围:取代、氧化还原、异构化、加成与消除、 配体上进行的反应
2. 活性与惰性配合物的理论解释
迄今, 对于配合物动力学稳定性的差异还没有完
全定量的理论,但有一些经验理论:
(1) 简单静电理论
(2) 价键理论
(3) 晶体场理论
(1) 简单静电理论
该理论认为, 取代反应的主要的影响因素是中心离
子和进入配体及离去配体的电荷和半径的大小。
对于离解机理(D)反应:
中心离子或离去配体的半径越小, 电荷越高, 则金
(2) 价键理论:
Taube认为, 过渡金属配合物的反应活性或惰性与
金属离子的d电子构型有关。
① 外轨型配合物是活性的(如sp3d2杂化配合物); ② 内轨型配合物,如(n-1)d轨道中有空轨道,则是 活性的,否则是惰性的。
① 外轨型配合物的取代反应速率
含有空(n-1)d轨道的外轨型配合物一般是活性的.
本章只讲述:取代反应和氧化还原反应
6-1 取代反应动力学 一、取代的反应机理 二、活性与惰性配合物及理论解释 三、 八面体配合物的取代反应 四、 平面四方形配合物 6-2 配合物的氧化还原反应机理
两类机理
配体取代 (交换反应) 电子转移反应 (氧化还原反应)
反应机理的研究 研究对象: Co3+ Cr3+ Ni2+ Pt2+等中心离子, 简单配体
Co(NH3)63+ + 6H3O+ == Co(H2O)63++ 6 NH4+
K≈1025( Co(NH3)63+热力学不稳定)
数日内无显著变化(惰性) Ni(CN)42- + 6H2O == Ni(H2O)62++ 4CNK≈10-22( Ni(CN)42-热力学稳定) 取代速度快(活性) 平衡常数 K=10-22, 说明[Ni(CN)4]2-是热力学稳定的。 然而研究表明, [Ni(CN)4]2-在Байду номын сангаас力学上却是活泼的。
(2) 与热力学稳定常数的关系 活性与惰性是动力学上的概念,不可与稳定性混为 一谈。惰性配合物也可能是热力学不稳定的配合物。 (a)活性与惰性是配合物的动力学性质,配合物的稳 定性和不稳定性是热力学性质;
活化配合物
活性由活化能决定, 稳定性由生成焓决定
Ea 反应物
△H
产物
(b)热力学稳定性与动力学稳定性无必然联系。
Sc3+, Y3+, La3+ 活性:d1 Ti3+ 活性:d2 V3+
↑
↑
↑↓
↑↓
↑↓
↑↓
↑↓
↑ ↓
当 (n-1) d 轨道中至少有一条轨道是空的配合物
就是活性配合物。
内轨型配合物
如果没有空的(n-1)d 轨道 (每条轨道中至少有一
个电子), 则这样的配合物就是惰性的。
这是因为当中心金属没有空 (n-1)d 轨道时, 要么
研究的时标(time scale )范围:
100s (惰性化合物) 配体交换
ms (活性化合物)
s (电荷迁移和电子转移)
6-1 取代反应动力学 取代反应:配离子中一个配体被另一个自由配体取
代的反应。
例:L5M-X + Y L5M-Y+X 一、取代的反应机理 1. 离解机理(SN1机理,或D机理 ) 慢 L5M-X == L5M+X(配位数下降6 L5M+Y== L5M-Y 速率方程:d[L5M-Y]/dt = k [L5M-X] 速率与Y的浓度无关,是对[L5M-X]的一级反应。