DNAman序列拼接-修正版.pdf
DNAman使用方法教学课件ppt

基因组浏览器
总结词
DnaMan具备可视化基因组浏览器,方便用户查看基因组结构和基因注释。
详细描述
DnaMan的基因组浏览器支持多种基因组数据可视化展示,包括基因注释、基因 转录本、SNP、重复序列等,可帮助用户深入分析基因组结构及功能。
分子进化分析
总结词
DnaMan支持分子进化分析,可探究物种间的亲缘关系和演 化历程。
详细描述
DnaMan的分子进化分析功能包括多种进化树构建方法,如 NJ树、MP树、ML树等,并提供了丰富的分子进化分析工具 ,如PAML、CODEML等。
04
DnaMan使用中的常见问题及解决 办法
序列导入问题
总结词
在导入序列时遇到的各种问题,如格式不正确、文件名限制等。
详细描述
DnaMan支持多种格式导入,如FASTA、GenBank等,但常因格式问题导致 导入失败;另外,序列文件名中不能含有特殊字符,否则无法导入。
DnaMan具备强大的序列编辑、可视化和注释功能,同时提 供了大量内置的分析工具和数据库,方便用户进行各种生物 信息学研究。
DnaMan的起源与发展
1
DnaMan起源于20世纪90年代末期,由美国一 家生物技术公司开发。
2
经过多年的发展和不断更新,DnaMan已经成 为了生物信息学领域中广受欢迎的工具之一。
插入视频
添加注释
在课件中插入DnaMan软件操作视频,以 便学生更好地了解软件操作流程和功能使用 方法。
对于一些复杂的操作和功能,可以添加注释 来帮助学生更好地理解和记忆。
演示教学课件的技巧
熟悉课件
在演示教学课件前,需要熟悉课 件的内容和排版,以便更好地讲 解。
互动教学
利用SeqMan进行序列拼接

利用SeqMan进行序列拼接利用SeqMan进行序列拼接Step1:打开Seqman软件Step2:加入你要拼接的序列点击Add sequences查找并选中要拼接的序列(可按住control键进行多选)点击Add按钮填加选择的序列填加完后点击done注:最好用测序的图谱尽量不要直接用测序得到的序列Step3:去除末端序列主要是去除序列末端测序质量差或是载体序列有两种方法可以用来去除这类末端序列其一:利用Seqman自带的去除工具自动去除(利用Trim ends 按钮进行)其二:手工去除个人感觉手工去除方法最有效,因此下边我们以后工去除为例进行演示手工去除侧翼序列双击要去除侧翼序列的目标序列将鼠标放到测序图谱左边的一个黑色的竖线上,此时鼠标会变成一个有两个箭头的水平线按住左键拖动黑竖线,那么你就会发现侧翼序列的颜色变浅,这部分变浅的序列则就被去除,不再参加后面的拼接此步请将测序不准确或认为是载体的序列用这种方法去除。
测序准确的峰形图峰形规则,一般在序列的中部,如下图所示测序不准确的峰形图峰形较乱,很难判断是哪个碱基,一般位于序列两端,如下图所示Step4:进行序列拼接点击Assemble按钮在新出现窗口处点击拼接好的contig1在出现的Alignment of contig1 窗口中点击左三角显示序列的测序图谱点击菜单contig->strategy view可以观察序列拼接的宏观图Step5:查找拼接错误find conflict 点击菜单Edit点击Find Previous或Find Next查找接接中出现的错误还可以通过Seqman左下角的快捷按钮查找错误的拼接查找错误的拼接错误的拼接的类型类型1:两条序列的测序结果不一致并明显一条测序质量好而另一条质量差处理:直接将该处修改为正确的碱基类型2:两条序列的测序结果不一致并两条测序质量都比较差处理:重新测序或用新的合适引物重新测定类型3:两条序列的测序结果不一致并明显两条测序质量都好处理:测序过程出现问题,重新测定Step6:导出拼接的序列可选择合适的格式,导出拼接好的序列通过以上几步我们就能很快将几个测序片段进行拼接,大家可以拿着自己的序列试试!当然如果两个测序片段的拼接片段太短可能利用默认的参数不能完成拼接,大家可以试着修改一下拼接参数试试!如降低Match size及Minimum Match Percentage的值!修改参数命令。
dnaman使用方法

dnaman使用方法使用DNAMAN的方法DNAMAN是一款功能强大的生物信息学软件,广泛应用于DNA 和蛋白质序列分析、比对、编辑和可视化等领域。
本文将介绍DNAMAN的使用方法,帮助读者快速上手并熟练运用该软件。
一、安装和启动DNAMAN1. 下载软件安装包:在DNAMAN官方网站上下载适用于您的操作系统的安装包,并保存到本地。
2. 安装软件:双击安装包,按照提示完成软件的安装过程。
3. 启动软件:安装完成后,双击桌面上的DNAMAN图标,即可启动软件。
二、导入和编辑序列1. 导入序列:在DNAMAN的主界面,点击"文件"菜单,选择"导入序列",然后选择您要导入的序列文件(支持FASTA、GenBank 等格式)。
2. 编辑序列:在序列编辑界面,您可以对序列进行添加、删除、替换等操作。
点击"编辑"菜单,选择相应的编辑功能,然后在弹出的对话框中进行操作。
三、序列比对和分析1. 序列比对:点击"工具"菜单,选择"序列比对",然后选择比对算法和参数设置,点击"开始比对"按钮即可进行序列比对。
2. 序列分析:DNAMAN提供了丰富的序列分析工具,如ORF预测、限制酶切位点分析、引物设计等。
点击"工具"菜单,选择相应的分析工具,按照提示进行操作。
四、序列可视化和输出1. 序列可视化:DNAMAN提供了多种序列可视化方式,如线性图、环形图、比对图等。
点击"视图"菜单,选择相应的可视化方式,即可查看序列的结构和特征。
2. 输出结果:完成序列分析后,您可以将结果导出为文本文件或图像文件。
点击"文件"菜单,选择"导出结果",然后选择输出格式和保存路径,点击"保存"按钮即可导出结果。
五、保存和管理项目1. 保存项目:在使用DNAMAN进行序列分析时,建议您保存项目以便后续操作。
4实验四 DNAMAN 软件的使用,多序列比对

实验目的
1.理解什么是多序列连配以及其目的。 2.学会使用DNAMAN软件。 3.用DNAMAN软件进行多序列连配、开放阅 读框寻找、氨基酸二级结构预测等。
实验材料
• 计算机,网络。 • DNAMAN。 • 基因核苷酸和氨基酸鸡、斑马鱼等物种的下 列基因中任何一个基因的核苷酸 和氨基酸序列:
• LPL, FAS, FABP, FTO, GDF11, ACC,
LEPTIN, MC4R, IGF1,IGF2,BMP2,HSL, PPARγ,STARS, ADD1, NHX1,SAMDC, DLX5,ENR, Lpin1
进行如下操作
• ORF寻找 • 不同物种氨基酸之间多序列比对 • 蛋白质二级结构预测 • 保存结果,并抓图
基因核苷酸和氨基酸序列基因核苷酸和氨基酸序列实验过程实验过程从从ncbincbi下载人小鼠大鼠下载人小鼠大鼠猪羊鸡斑马鱼等物种的下猪羊鸡斑马鱼等物种的下列基因中任何一个基因的核苷酸列基因中任何一个基因的核苷酸和氨基酸序列
生物信息学实验课件
邢晋祎 生命科学学院 Copyright
实验四
DNAMAN软件的使用、 多序列联配
DNAMAN的使用方法

文件菜单中选择“打开”或通过快捷键Ctrl+O,打开存储在计算机中的DNA序列文件。
保存文件
文件菜单中选择“保存”或通过快捷键Ctrl+S,将更改后的DNA序列存储到硬盘或其他存储设 备中。
文件格式要求
只支持常见的序列文件格式如FASTA、GenBank等,可以选择单个文件或批量导入。
D N A 序列的导入和编辑
分析序列统计学
序列长度分布
序列特征统计图
DNAMAN可以为所有序列生成序 列长度分布图,从而确定序列长 度最常见的地方,并且甚至可以 根据自己的喜好来更改分布参数。
这是用于比较DNA序列中各类特 征的常用工具。统计图通常是以 直方图的形式出现,幸运的是 DNAMAN可以自动生成这种统计 图并轻松进行定位和分析。
D N A 序列编辑
D N A 序列导入
D N A 序列统计信息
DNAMAN提供多种序列编辑工具, 例如添加碱基、删除碱基、反转 序列和互补序列等。
DNAMAN支持多种序列格式导入, 例如FASTA、GenBank等。
在编辑界面右侧的信息面板中, DNAMAN会自动生成序列的碱基 组成、止旋镇性能力等信息。
多样性分析
发现多样性和图形化分组模型对 于了解疾病的分布和传播至关重 要,DNAMAN通过比对分析大量 的DNA序列,可以进行多样性分 析并演示图形化分组模型。
D N A M A N 在植物和动物遗传学中的应用
BA C分析
通过资料库查询和选择BAC、 BIBAC、Cosmid等载体中的 DNA,进行序列分析和匹配 以获得目标DNA序列。
常见问题解答
1 D N A M A N 支持哪些文件格式?
便携式数据格式(PDF)、HTML网页、Microsoft Word和图像文件(PNG、JPEG、GIF)等。
利用SeqMan进行序列拼接

类型3错误拼接的类型13 2• 为了区分修整过和没有修整 过的数据,我们给修整过的 数据加一个有颜色的背景。 选择菜单 Project→Parameters→Editing Color打开下面的对话框。确 定use consensus match color 和use other color已被选中。
• 修整完毕后 Alignment View 中在序列的左边会有一个黑色的垂直棒, 右边有一个小的黑三角形。
• 要找回修整去掉的序列末端,只需把垂直棒向序列的两端拖动即可, 以前修整去掉的序列有明亮的黄色背景。
Pre-Assembly Options 操作及序列装配
• 在拼接前面,可以将所要拼接的片段中清除载体和污染序列,优化 装配顺序,设定片段末端和标记重复序列
查看修整序列前后的跟踪数据
• 右键选择6 号样本,然后Show Original Trace Data,打开Trace:Sample 6.abi 窗口
• 从 5’末端起变淡的部分是载体序列,将不会用于序列装配,故被清除。 • 垂直的黑棒出现于修整和未修整的序列之间,根据需要拖动垂直黑棒,可以调
整用于装配的序列末端。
利用SeqMan进行序列拼接
Step2:加入你要拼接的序列
点击Add sequences
查找并选中要拼接的 序列
点击Add按钮
填加完后点击done
注:最好用测序的图谱(*.abi)尽量不要直接用测序得到的序列 (.seq)
1 点击Assemble按钮 2 点击拼接好的co
Alignment of contig1 窗口中点击 左三角显示序列的测 序图谱
1. 两条序列的测序结果 不一致并明显一条测 序质量好而另一条质 量差
处理:直接将该处修改为 正确的碱基
序列分析软件DNAMAN 的使用方法简介

序列分析软件DNAMAN 的使用方法简介DNAMAN 是一种常用的核酸序列分析软件。
由于它功能强大,使用方便,已成为一种普遍使用的DNA 序列分析工具。
本文以DNAMAN 5.2.9 Demo version 为例,简单介绍其使用方法。
打开DNAMAN,可以看到如下界面:第一栏为主菜单栏。
除了帮助菜单外,有十个常用主菜单,第二栏为工具栏:第三栏为浏览器栏:在浏览器栏下方的工作区左侧,可见Channel 工具条,DNAMAN 提供20 个Channel,(如左所示:)点击Channel 工具条上相应的数字,即可击活相应的Channel。
每个Channel 可以装入一个序列。
将要分析的序列(DNA 序列或氨基酸序列)放入Channel 中可以节约存取序列时间,加快分析速度。
此版本DNAMAN 提供自动载入功能,用户只需激活某个Channel,然后打开一个序列文件,则打开的序列自动载入被激活的Channel 中。
本文以具体使用DNAMAN 的过程为例来说明如何使用DNAMAN 分析序列。
1.将待分析序列装入Channel(1)通过File Open 命令打开待分析序列文件,则打开的序列自动装入默认Channel。
(初始为channel1)可以通过激活不同的channel (例如:channel5)来改变序列装入的Channel。
(2)通过Sequence/Load Sequence 菜单的子菜单打开文件或将选定的部分序列装入Channel 。
通过Sequence/Current Sequence/Analysis Defination 命令打开一个对话框,通过此对话框可以设定序列的性质(DNA 或蛋白质),名称,要分析的片段等参数。
2.以不同形式显示序列通过Sequence//Display Sequence 命令打开对话框,如下图所示:根据不同的需要,可以选择显示不同的序列转换形式。
对话框选项说明如下:Sequence &Composition 显示序列和成分Reverse Complement Sequence 显示待分析序列的反向互补序列Reverse Sequence 显示待分析序列的反向序列Complement Sequence 显示待分析序列的互补序列Double Stranded Sequence 显示待分析序列的双链序列RNA Sequence 显示待分析序列的对应RNA 序列3.DNA 序列的限制性酶切位点分析将待分析的序列装入Channel,点击要分析的Channel,然后通过Restriction/Analysis 命令打开对话框,如下所示:参数说明如下:Results 分析结果显示其中包括:Show summary(显示概要) Show sites on sequence(在结果中显示酶切位点)Draw restriction map(显示限制性酶切图)Draw restriction pattern(显示限制性酶切模式图)Ignore enzymes with more than(忽略大于某设定值的酶切位点)Ignore enzymes with less than(忽略小于某设定值的酶切位点)Target DNA (目标DNA 特性)circular(环型DNA),dam/dcm methylation(dam/dcm 甲基化)all DNA in Sequence Channel(选择此项,在Sequence Channel 中的所有序列将被分析,如果选择了Draw restriction pattern,那么当所有的channel 中共有两条DNA 时,则只能选择两个酶分析,如果共有三个以上DNA 时,则只能用一个酶分析。
分子生物学专业软件DNAMAN使用方法介绍
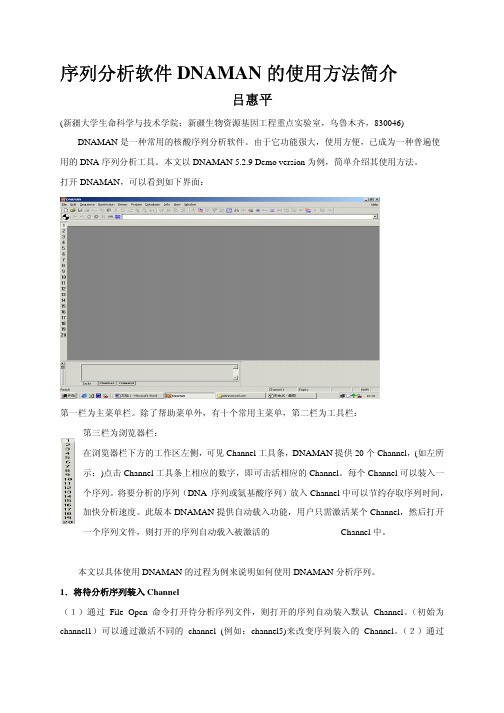
序列分析软件DNAMAN的使用方法简介吕惠平(新疆大学生命科学与技术学院;新疆生物资源基因工程重点实验室,乌鲁木齐,830046) DNAMAN是一种常用的核酸序列分析软件。
由于它功能强大,使用方便,已成为一种普遍使用的DNA序列分析工具。
本文以DNAMAN 5.2.9 Demo version为例,简单介绍其使用方法。
打开DNAMAN,可以看到如下界面:第一栏为主菜单栏。
除了帮助菜单外,有十个常用主菜单,第二栏为工具栏:第三栏为浏览器栏:在浏览器栏下方的工作区左侧,可见Channel工具条,DNAMAN提供20个Channel,(如左所示:)点击Channel工具条上相应的数字,即可击活相应的Channel。
每个Channel可以装入一个序列。
将要分析的序列(DNA 序列或氨基酸序列)放入Channel中可以节约存取序列时间,加快分析速度。
此版本DNAMAN提供自动载入功能,用户只需激活某个Channel,然后打开一个序列文件,则打开的序列自动载入被激活的 Channel中。
本文以具体使用DNAMAN的过程为例来说明如何使用DNAMAN分析序列。
1.将待分析序列装入Channel(1)通过File Open命令打开待分析序列文件,则打开的序列自动装入默认Channel。
(初始为channel1)可以通过激活不同的channel (例如:channel5)来改变序列装入的Channel。
(2)通过Sequence/Load Sequence菜单的子菜单打开文件或将选定的部分序列装入Channel。
通过Sequence/Current Sequence/Analysis Defination命令打开一个对话框,通过此对话框可以设定序列的性质(DNA 或蛋白质),名称,要分析的片段等参数。
2.以不同形式显示序列通过Sequence//Display Sequence命令打开对话框,如下图所示:根据不同的需要,可以选择显示不同的序列转换形式。