类药性质
合集下载
第五章 类药性质

化合物
IC50 (μM/L)
pKa
4-OCH3 H
75
9.34
45
9.10
O2 H
H2N
SN
R3
4-Cl 4-I
35
8.56
25
8.17
R1 R2
2-Cl, 4-OCH3
19
8.81
3-CF3
15
7.98
2-Cl
14
8.18
4-COCH3 4-CN
11
7.52
7
7.36
4-NO2
7
2-OCH3, 4-NO2
生物系统
物理化学性质: 溶解度,渗透性,化学稳定性
生物化学性质: 代谢,转运体亲和,蛋白结合,靶标亲和
物理环境
蛋白质
结构特征: 分子量,氢键,亲脂性,极性表面积,pKa,形状,反应性
化合物的结构决定了其基本特征,基本特征决定了其物理化学和 生物化学性质,后者最终决定了它的药动学和毒性。
化合物性质影响药物发现研究质量
O
N Cl
效价=1 nmol/L, 口服吸收差
HN
氢键供体数=1
氢键受体数=6
分子量=591
Log p=7.3
PSA=50 可旋转键数=14 氢键总数=6
“Rule of 3”规则(Oprea,T.I.)
--类先导化合物(Lead like compound)规则
MW≤ 300 Clog P≤ 3 可自由旋转键≤ 3 氢键供体≤ 3 氢键受体≤ 3 极性表面积≤60Ų
OH
茚地那韦
IC50 = 0.41 nmol/L ClC95 = 24~100 nmol/L 人口服生物利用度为60%
磺胺类药物结构理化性质及临床应用 化学治疗药 药物化学课件

本身具有抗菌活性,与其它抗菌药合用可增强其它抗菌药 的抗菌活性,如甲氧苄胺嘧啶
本身不具有抗菌活性或抗菌活性很弱,与其它抗菌药合用 可增强其它抗菌药的抗菌活性,如棒酸
本身不具有抗菌活性,与其它抗菌药合用时通过影响其代 谢可增强其它抗菌药的抗菌活性,如丙磺舒
抗菌增效剂
TMP(甲氧苄啶):磺胺增效剂,使细菌生长受双重阻止作用; 丙磺舒:抑制有机酸排泄,增加药物在血中浓度,与β-内酰胺类 抗生 素合用有增效作用;
相关链接
临床常用抗菌增效剂的作用特点
棒酸(克拉维酸)本身抗菌活性很弱,但具有抑制β内酰胺酶的作用,可显著增强β-内酰胺类抗生素的作用, 如与头孢霉素、羟氨苄西林合用分别可增强其抗菌活性 2~8倍与130倍。
丙磺舒(增加尿酸排泄药)可抑制有机酸抗菌药物从 哺乳动物肾脏的排泄,因而可以抑制青霉素类、头孢菌 素类及对氨基水杨酸等有机酸类抗菌药物的排泄。如与 青霉素合用可降低青霉素的排泄速度,提高其在血中的 浓度而增强青霉素的抗菌作用。
吸电子基团可以是酰基,也可以是芳香杂环。N,N-双取代化合物一般丧失活性。
(4)苯环若被其他芳环或芳杂环取代,或在苯环上引入其他基团,抑菌活性降低或丧失。
第一节 磺胺类药物及抗菌增效剂
磺胺类药物的构效关系
其他芳环或引入其他基团 活性降低或丧失
单取代活性增加, 杂环取代更好
游离氨基或潜在的游离氨基 是活性关键
本代谢物竞争性或干扰基本代谢物的被利用,或掺入生物大分子的合成之中形成伪 生物大分子,导致致死合成(lethal synthesis),从而影响细胞的生长。抗代谢 物的设计多采用生物电子等排原理。代谢拮抗概念已广泛应用于抗菌、抗疟及抗癌 药物等设计中。
第一节 磺胺类药物及抗菌增效剂
本身不具有抗菌活性或抗菌活性很弱,与其它抗菌药合用 可增强其它抗菌药的抗菌活性,如棒酸
本身不具有抗菌活性,与其它抗菌药合用时通过影响其代 谢可增强其它抗菌药的抗菌活性,如丙磺舒
抗菌增效剂
TMP(甲氧苄啶):磺胺增效剂,使细菌生长受双重阻止作用; 丙磺舒:抑制有机酸排泄,增加药物在血中浓度,与β-内酰胺类 抗生 素合用有增效作用;
相关链接
临床常用抗菌增效剂的作用特点
棒酸(克拉维酸)本身抗菌活性很弱,但具有抑制β内酰胺酶的作用,可显著增强β-内酰胺类抗生素的作用, 如与头孢霉素、羟氨苄西林合用分别可增强其抗菌活性 2~8倍与130倍。
丙磺舒(增加尿酸排泄药)可抑制有机酸抗菌药物从 哺乳动物肾脏的排泄,因而可以抑制青霉素类、头孢菌 素类及对氨基水杨酸等有机酸类抗菌药物的排泄。如与 青霉素合用可降低青霉素的排泄速度,提高其在血中的 浓度而增强青霉素的抗菌作用。
吸电子基团可以是酰基,也可以是芳香杂环。N,N-双取代化合物一般丧失活性。
(4)苯环若被其他芳环或芳杂环取代,或在苯环上引入其他基团,抑菌活性降低或丧失。
第一节 磺胺类药物及抗菌增效剂
磺胺类药物的构效关系
其他芳环或引入其他基团 活性降低或丧失
单取代活性增加, 杂环取代更好
游离氨基或潜在的游离氨基 是活性关键
本代谢物竞争性或干扰基本代谢物的被利用,或掺入生物大分子的合成之中形成伪 生物大分子,导致致死合成(lethal synthesis),从而影响细胞的生长。抗代谢 物的设计多采用生物电子等排原理。代谢拮抗概念已广泛应用于抗菌、抗疟及抗癌 药物等设计中。
第一节 磺胺类药物及抗菌增效剂
生物碱类药物的性质

H H HO
H3CO
O
H O
OCH3
酯键:易水解
OCH3
OCH3 OCH3
吲哚环上的氮(N2)由 于与芳环共轭,几乎无 碱性,不与酸成盐
2
N
O
H H
N1
H O
, HNO3
硝酸士的宁
生物碱类药物的性质
活泼氢:呈酸性,可溶于 碱的水溶液中
O H3C N
ON
CH3 N
N
, H2O
CH3
咖啡因
O
H3C N
H N
ON N CH3
茶碱
, H2O
本类药物结构中含有四个氮原子, 但受邻位羰基的影响,碱性极弱。 不易与酸成盐,以游离碱供药用
黄嘌呤结构呈紫脲 酸铵特征反应
液分别发生反应,可用于鉴别 ,亦可与铁氰化钾试液反应, 生成蓝绿色而与可待因区别
生物碱类药物的性质
利血平
士的宁分子中脂环叔氨氮(N1)碱性较 强,可与硝酸成盐;利血平分子中的脂 环叔胺氮(N1)受邻近基团空间位阻的 影响,碱性较弱,不能与酸结合成稳定 的盐,而以游离状态存在
H3CO
2
N HH
N1 H
N
喹啉环芳环氮:碱性弱,不 能与硫酸成盐
硫酸奎宁
2
荧光特性:硫酸奎宁和硫 酸奎尼丁在稀硫酸溶液中 显蓝色荧光
生物碱类药物的性质
(四)异喹啉类生物碱
酚羟基和叔胺基团:具有酸 碱两性,但碱性略强 H3C
N H
, HCl , 3H2O
HO
O
盐酸吗啡
H 吗啡生物碱的显色反应 盐酸 H OH 吗啡可与甲醛硫酸、钼硫酸试
H OH O
, H2SO4 , H2O
药品生产技术《3.巴比妥类药物的主要性质》

巴比妥类药物的主要性质
①弱酸性:巴比妥类药物分子结构中都有1,3-二酰亚氨基团,能使其分子发生酮式与烯醇式互变异构,在水溶液中可发生二级电离。
因此,本类药物的水溶液显弱酸性,可与强碱形成水溶性的盐类,常见为钠盐。
由弱酸与强碱形成的巴比妥钠盐,其水溶液呈碱性,加酸酸化后,那么析出结晶性的游离巴比妥类药物,可用有机溶剂将其提取出来。
②水解反响:本类药物的分子结构中含有酰亚胺结构,与碱溶液共沸即水解释放氨气,可使红色石蕊试纸变蓝。
该反响可用于鉴别该类药物。
在吸湿情况下,本类药物的钠盐也能水解为无效物质。
一般,在室温和pH10以下水解较慢;pH11以上随着碱度的增加水解速度加快。
③与重金属离子的反响:巴比妥类药物分子中因含有—CONHCONHCO—〔丙二酰脲〕基团,所以在适宜pH溶液中,可与某些重金属反响,生成有色或不溶性有色物质。
以此性质,可对本类药物进行鉴别和含量测定。
④与香草醛的反响:该类药物分子中具有活泼氢,可与香草醛在浓硫酸存在下发生缩合反响,产生棕红色产物。
⑤紫外吸收光谱特征:在碱性溶液中,巴比妥类药物电离为具有共轭体系的结构,而产生明显的紫外吸收,其吸收光谱随其
电离级数的不同而变化。
⑥薄层色谱行为特征:巴比妥类药物结构不同,其薄层色谱行为也自然不同,可进行鉴别。
⑦显微结晶:大局部巴比妥类药物本身或与某种试剂的反响产物,具有特殊的晶形,可根据结晶形状鉴别。
典型药物的分析 甾体激素类药物典型药物结构与性质

地塞米松杂质检查:外标法,按峰面积计算,限度0.5% 其他杂质: 不加校正因子的主成分自身稀释对照法,限度为0.5% 总杂质:除地塞米松为加校正因子的主成分自身稀释对照法外,其余杂质均为不加校正因子的 主成分自身稀释对照法,总杂质限度为1.0%
醋酸地塞米松及其制剂分析 原料药——【检查】
硒
杂质来源? 检查原因?
问题3:杂质限量是多少?如何计算?
精密量取供试品溶液与对照溶液各20μl,分别注入液相色谱仪,记录色谱图至供试品溶液主 成分峰保留时间的2倍。供试品溶液的色谱图中如有与对照溶液中地塞米松峰保留时间一致的杂 质峰,按外标法以峰面积计算,不得过0.5%;其他单个杂质峰面积不得大于对照溶液中醋酸地 塞米松峰面积的0.5倍(0.5%),各杂质峰面积(与地塞米松峰保留时间一致的杂质峰面积乘以 1.13)的和不得大于对照溶液中醋酸地塞米松峰面积(1.0%)。供试品溶液色谱图中小于对照 溶液中醋酸地塞米松峰面积0.01倍(0.01%)的峰忽略不计。
醋酸地塞米松供试品溶液(临用新制):0.5mg/ml 目的:检查杂质 地塞米松对照品溶液:0.5mg/ml 目的:地塞米松杂质对照
对照溶液: 醋酸地塞米松0.005 mg/ml、地塞米松:0.005mg/ml 目的:检测系统适用性、其他杂质对照
醋酸地塞米松及其制剂分析
原料药——【检查】
有关物质 问题2:本项检查应用了HPLC检查杂质中的哪种方法?
C17位醋酸酯: 易水解,可利用水解产物进行鉴别
C10、C13、C16位有角甲基 C11位为羟基
C9位氟(F):
△1,4–3–酮基
有机破坏后可与茜素氟蓝、硝酸亚铈反应
(1)紫外吸收
(2)与羰基试剂(异烟肼、2,4-二硝基苯肼、硫酸苯肼)反应呈色
醋酸地塞米松及其制剂分析 原料药——【检查】
硒
杂质来源? 检查原因?
问题3:杂质限量是多少?如何计算?
精密量取供试品溶液与对照溶液各20μl,分别注入液相色谱仪,记录色谱图至供试品溶液主 成分峰保留时间的2倍。供试品溶液的色谱图中如有与对照溶液中地塞米松峰保留时间一致的杂 质峰,按外标法以峰面积计算,不得过0.5%;其他单个杂质峰面积不得大于对照溶液中醋酸地 塞米松峰面积的0.5倍(0.5%),各杂质峰面积(与地塞米松峰保留时间一致的杂质峰面积乘以 1.13)的和不得大于对照溶液中醋酸地塞米松峰面积(1.0%)。供试品溶液色谱图中小于对照 溶液中醋酸地塞米松峰面积0.01倍(0.01%)的峰忽略不计。
醋酸地塞米松供试品溶液(临用新制):0.5mg/ml 目的:检查杂质 地塞米松对照品溶液:0.5mg/ml 目的:地塞米松杂质对照
对照溶液: 醋酸地塞米松0.005 mg/ml、地塞米松:0.005mg/ml 目的:检测系统适用性、其他杂质对照
醋酸地塞米松及其制剂分析
原料药——【检查】
有关物质 问题2:本项检查应用了HPLC检查杂质中的哪种方法?
C17位醋酸酯: 易水解,可利用水解产物进行鉴别
C10、C13、C16位有角甲基 C11位为羟基
C9位氟(F):
△1,4–3–酮基
有机破坏后可与茜素氟蓝、硝酸亚铈反应
(1)紫外吸收
(2)与羰基试剂(异烟肼、2,4-二硝基苯肼、硫酸苯肼)反应呈色
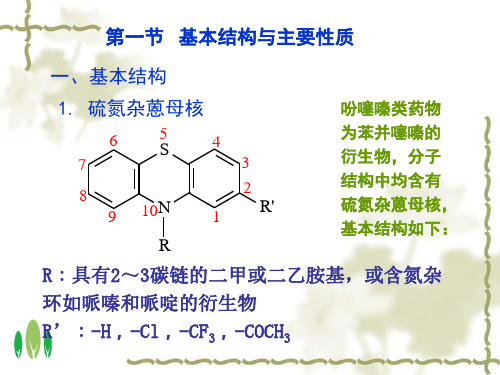
吩噻嗪类药物分析

本类药物母核的硫为二价,易氧化,其氧化产物为亚砜及砜,与未取代的吩噻嗪母核的吸收光谱有明显不同,它们具有四个峰值。因此,可以利用紫外吸收光谱的这些特征测定药物中杂质氧化物存在的量;同时也可在药物含量测定时对氧化产物的干扰进行校正。
吩噻嗪类药物取代基R和R‘的不同,产生不同的红外吸收光谱,国内外药典已用于本类药物较多品种的鉴别。
注意点:
①异丙嗪遇光不稳定,上述检查应在避光条件下操作;
②溶液应临用时配制,否则杂质斑点增多。
四、含量测定
(一)非水溶液滴定法
吩噻嗪类药物母核上10位取代基的烃胺-NR2、哌嗪基及哌啶基具有碱性,可在非水介质中以高氯酸-冰醋酸液滴定。
吩噻嗪类原料药物国内外药典多采用非水碱量法测定,所用溶剂除酸性溶剂如醋酸、醋酐外,也有采用中性或近中性溶剂的,如丙酮、二氧六环、乙腈等。.
2.易氧化呈色吩噻嗪类药物遇不同氧化剂例如硫酸、硝酸、三氯化铁试液及过氧化氢等,其母核易被氧化成自由基型产物和非离子型产物(砜、亚砜、3-羟基吩噻嗪)等不同产物,随着取代基的不同,而呈不同的颜色。(可用于鉴别)
3.与金属离子络合呈色 母核中未被氧化的S原子,可与金属离子(如Pd2+)形成有色络合物,其氧化产物砜和亚砜则无此反应。利用此性质可进行鉴别和含量测定,并具有专属性,可排除氧化产物的干扰。
(二)检查方法
取本品,加二氯甲烷制成每1ml中含10mg的溶液,作为供试品溶液;精密量取适量,加二氯考,试大收集整理甲烷制成每1ml中含0.15mg和0.05mg的溶液,作为对照溶液(1)和(2)。吸取上述三种溶液各10μl,分别点于同一硅胶GF254薄层板上,以己烷-丙酮-二乙胺(8.5:1:0.5)为展开剂,展开后,晾干,置紫外光灯(254nm)下检视,供试品溶液如显杂质斑点,不得多于3个;其杂质斑点与对照溶液(2)的主斑点比较,不得更深;如有一点超过,应不得深于对照溶液(1)的主斑点。
吩噻嗪类药物取代基R和R‘的不同,产生不同的红外吸收光谱,国内外药典已用于本类药物较多品种的鉴别。
注意点:
①异丙嗪遇光不稳定,上述检查应在避光条件下操作;
②溶液应临用时配制,否则杂质斑点增多。
四、含量测定
(一)非水溶液滴定法
吩噻嗪类药物母核上10位取代基的烃胺-NR2、哌嗪基及哌啶基具有碱性,可在非水介质中以高氯酸-冰醋酸液滴定。
吩噻嗪类原料药物国内外药典多采用非水碱量法测定,所用溶剂除酸性溶剂如醋酸、醋酐外,也有采用中性或近中性溶剂的,如丙酮、二氧六环、乙腈等。.
2.易氧化呈色吩噻嗪类药物遇不同氧化剂例如硫酸、硝酸、三氯化铁试液及过氧化氢等,其母核易被氧化成自由基型产物和非离子型产物(砜、亚砜、3-羟基吩噻嗪)等不同产物,随着取代基的不同,而呈不同的颜色。(可用于鉴别)
3.与金属离子络合呈色 母核中未被氧化的S原子,可与金属离子(如Pd2+)形成有色络合物,其氧化产物砜和亚砜则无此反应。利用此性质可进行鉴别和含量测定,并具有专属性,可排除氧化产物的干扰。
(二)检查方法
取本品,加二氯甲烷制成每1ml中含10mg的溶液,作为供试品溶液;精密量取适量,加二氯考,试大收集整理甲烷制成每1ml中含0.15mg和0.05mg的溶液,作为对照溶液(1)和(2)。吸取上述三种溶液各10μl,分别点于同一硅胶GF254薄层板上,以己烷-丙酮-二乙胺(8.5:1:0.5)为展开剂,展开后,晾干,置紫外光灯(254nm)下检视,供试品溶液如显杂质斑点,不得多于3个;其杂质斑点与对照溶液(2)的主斑点比较,不得更深;如有一点超过,应不得深于对照溶液(1)的主斑点。
吩噻嗪类药物分析
等 度,的即 差值足够大,A。b而1 待A测b2 组分在这两个波长处吸收
(2)定量依据
样品在二波长下吸收度差值(A):
A
Aab 1
Aab 2
(
Aa 1
Ab 1
)
(
Aa 2
Ab 2
)
Aa 1
Aa 2
( 1
2 )Cal
Ab 1
Ab 2
2位上取代基(R′)不同,会引起吸收峰发生位移。结 构中2价硫,易氧化,产物砜及亚砜有四个吸收峰。
5.红外吸收光谱特性:由于取代基R和R’的不同,具有 不同的红外光谱,已被药典用于不同品种鉴别。
第二节 鉴别试验
一、化学法
(一)与生物碱沉淀试剂反应 吩噻嗪类药物10位的含氮取代基有碱性,可与生物碱
【 ChP盐酸氯丙嗪含量测定】 取本品约0.2g.精
密称定.加冰醋酸10ml与醋酐30ml溶解后,照电位 滴定法,用高氯酸 (0.1mol/L)滴定液滴定,并将滴 定结果用空白试验校正。
每1ml高氯酸滴定液(0.1mol/L) 相当于35.53mg盐酸 氯丙嗪。
注意:
滴定剂的稳定性 非水溶液滴定法所用的溶剂为醋
注:避光操作;溶液临用新配。
LOGO
第四节 含量测定
酸碱滴定法 非水溶液滴定法 乙醇-水溶液中的氢氧化钠滴定法
紫外分光光度法 高效液相色谱法
一、酸碱滴定法
非水溶液滴定法
吩噻嗪类原料药物含量测定大多利用其侧链脂 肪胺碱性,国内外药典多采用非水滴定法测定本类药 物及其盐酸盐原料药的含量。
氯丙嗪的碱性较弱,醋酐解离生成 醋酐合乙酰氧离子比醋酸合质子的 酸性强,更利于增加氯丙嗪的碱性
(2)定量依据
样品在二波长下吸收度差值(A):
A
Aab 1
Aab 2
(
Aa 1
Ab 1
)
(
Aa 2
Ab 2
)
Aa 1
Aa 2
( 1
2 )Cal
Ab 1
Ab 2
2位上取代基(R′)不同,会引起吸收峰发生位移。结 构中2价硫,易氧化,产物砜及亚砜有四个吸收峰。
5.红外吸收光谱特性:由于取代基R和R’的不同,具有 不同的红外光谱,已被药典用于不同品种鉴别。
第二节 鉴别试验
一、化学法
(一)与生物碱沉淀试剂反应 吩噻嗪类药物10位的含氮取代基有碱性,可与生物碱
【 ChP盐酸氯丙嗪含量测定】 取本品约0.2g.精
密称定.加冰醋酸10ml与醋酐30ml溶解后,照电位 滴定法,用高氯酸 (0.1mol/L)滴定液滴定,并将滴 定结果用空白试验校正。
每1ml高氯酸滴定液(0.1mol/L) 相当于35.53mg盐酸 氯丙嗪。
注意:
滴定剂的稳定性 非水溶液滴定法所用的溶剂为醋
注:避光操作;溶液临用新配。
LOGO
第四节 含量测定
酸碱滴定法 非水溶液滴定法 乙醇-水溶液中的氢氧化钠滴定法
紫外分光光度法 高效液相色谱法
一、酸碱滴定法
非水溶液滴定法
吩噻嗪类原料药物含量测定大多利用其侧链脂 肪胺碱性,国内外药典多采用非水滴定法测定本类药 物及其盐酸盐原料药的含量。
氯丙嗪的碱性较弱,醋酐解离生成 醋酐合乙酰氧离子比醋酸合质子的 酸性强,更利于增加氯丙嗪的碱性
类药性

亲脂性
测量方法:高通量摇瓶法, 测量方法:高通量摇瓶法,毛细管 电泳法和反相色谱法 预测方法: 预测方法:A logP,ACD logP, ClogP, ,
MlogP,X logP ,
(二)pKa
pKa决定水溶性和透膜性的主要因素之 决定水溶性和透膜性的主要因素之 一 酸性或碱性较强的分子, 酸性或碱性较强的分子,在体内以较多 解离形式存在, 解离形式存在,导致透膜性和生物利用 度差,改善pKa可提高生物利用度 度差,改善 可提高生物利用度 胺类化合物: 胺类化合物: pKa 10~11 ~ 酸类化合物: 酸类化合物:pKa 3~5 ~
类药性评价方法
基于经验判断的类药性评价 基于经验判断的类药性评价 经验 基于理化性质的类药性评价 基于理化性质的类药性评价 理化性质 基于ADMET性质的类药性评价 性质的类药性评价 基于 性质
一、基于经验判断的类药性评价
在已知药物数据库中提取与类药性 相关联的理化性质和拓扑结构特征 相关联的理化性质和拓扑结构特征 的基础上形成的经验规律 用途:用于化学库(组合化学物库 用途:用于化学库( 或虚拟化合物库) 或虚拟化合物库)的早期设计和评 价 缺点:不能区别单个化合物是“ 缺点:不能区别单个化合物是“类 还是“非药” 药”还是“非药”
分子量在160~480之间,平均 ~ 之间, 分子量在 之间 平均357 原子数在20 之间, 原子数在 ~ 70之间,平均 之间 平均48 ALogP在-0.4 ~ 5.6之间,平均 之间, 在 之间 平均2.52 摩尔折射率在40 之间, 摩尔折射率在 ~ 130之间,平均 之间 平均97
(三) Verber法 法
血浆稳定性低: 血浆稳定性低:高清除率和较短的半衰 期
提高血浆稳定性
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
生物系统
物理化学性质: 溶解度,渗透性,化学稳定性
生物化学性质: 代谢,转运体亲和,蛋白结合,靶标亲和
物理环境
蛋白质
结构特征: 分子量,氢键,亲脂性,极性表面积,pKa,形状,反应性
化合物的结构决定了其基本特征,基本特征决定了其物理化学和 生物化学性质,后者最终决定了它的药动学和毒性。
化合物性质影响药物发现研究质量
理的消除和低毒性。
胃
小肠
门静脉 肝
血液 肾
器官
细胞
靶标
溶解度 pH 3~8
稳定性
∙代谢
稳定性
∙一相 ∙二相
蛋白结合
肾单位
∙渗透性 ∙尿排泄
分布 (全身)
细胞内分配 和代谢
稳定性
∙pH 2 ∙酶
稳定性
∙pH 3~8 ∙酶
上皮渗透性
∙被动 ∙pH 3~8 ∙Pgp外排
胆汁排泄
稳定性
∙酶
血-器官特殊渗透性
Log D: 某一特定pH(x)条件下,化合物部分以离 子形式、部分以中性分子形式存在,此pH条件 下其分配系数的对数
∙被动 ∙Pgp外排
渗透性
∙被动 ∙Pgp外排
药物经体内屏障输送到靶器官的概述
二、物理化学性质
1.从结构快速描述特征的规则
Lipinski规则(类药性5原则)
分子量 < 500 Clog P < 5 氢键给予体 < 5 (NH, OH数目的总和) 氢键接受体 < 10 (N, O数目的总和)
氢键供体
1(OH) 1(OH) 0 2(NH2) 1(NH) 0 0 0 0 0
氢键受体
1(O) 2(2O) 2(N, O) 1(N) 1(N) 1(O) 2(O) 1(O) 1(N) 1(N)
Veber规则 良好口服生物利用度(大鼠)应符合;
≤10个可旋转化学键 ≤140Ų极性表面积,或≤12个氢键总数(
例:
NH N
F3C
N
S
三氟拉嗪 碱类 pka=7.81
CNS+
HOOC O
Cl O
吲哚美辛 酸类 pka=4.18
CNS-
2. 亲脂性
亲脂性是许多ADME/Tox性质的主要决定因 素之一
Log P: 化合物的所有分子均以中性形式存在的 pH条件下,其分配系数的对数
Log P = Log ([anic]/[Compd.aqueous])
50 40 30 20 10
0
好的药物
性质
活性优化
好的配体
活性
改变候选药物策略,从专注于活性到平衡地关注活性和性 质。
新颖 性
药动性
溶解 性
稳定性 选择性
安全性
活性
药物发现的成功需要同时平衡不同的变量
生命系统中药物暴露的屏障
屏障包括生物膜、PH、代谢酶和转运体 生物屏障使给予的药物到达靶标的量减少 类药性佳,良好的吸收和分布、低代谢、合
溶解度低: 体外生物活性低或不一致 试验介质中不稳定: 引起生物活性低 渗透性差:酶或受体活性与细胞活性(大幅下降)不
一致 血脑屏障通透性差:CNS药物体内效价低 PK差、生物利用度低或血中不稳定:体内药效差。
源于各方面的失败率(%)
从1991年到2000年,由于药动学(PK)和生物利用度导致的 开发失败 率大幅降低。毒性、临床安全性和剂型仍然是突出的类药性质问题。
一、Introduction
类药化合物: 具有足以可接受的吸收、分布 、代谢、排泄和安全性(ADME/Tox), 能 在人体一期临床试验后生存下来的化合物。
类药性质是分子的内在性质,药物化学家的 责任不仅在于优化这些分子的药理性质,而 且还在于优化它们的类药性质。
类药性质是药物发现的组成部分之一
氢键供体数=7 可旋转键数=11
分子量=543
PSA=206
ClogP=-1.7
氢键总数=19
氢键受体数=12
根据计算,其口服生物利用度约5%
例: 神经肽YY1拮抗剂
N
O
N Cl
效价=2μmol/L 氢键供体数=0 氢键受体数=3 分子量=369 Log p=5.7 PSA=17 可旋转键数=6
N N
结构特征决定了化合物在体内的药动学和毒性 性质良好的化合物,可减少低效的研究、失败
率和费用 评估优化ADME/Tox性质是药物发现的重要
方面 最佳候选药物具活性和性质上的平衡
药物发现中应关注的诸多性质
结构特征
氢键 亲脂性 分子量 极性表面积 形状 反应性
受体加供体) 分子柔性、极性表面积(PSA)、氢键数目是决
定口服生物利用度的重要因素
Veber. D.F. J. Med. Chem. 2002, 45; 2615-2623.
例:
O OH
O
OH
D C B A OH
OCH3O OH O
H3C
O
OH NH2
多柔比星
Lipinski规则
Veber规则
----------------- -----------------
Hale Waihona Puke 用于中枢的化合物结构特征 血脑屏障理化性质规则(Pardridge) 氢键总数<8~10 分子量<400~500 非酸类化合物
Clark和Lobell规则 N+O<6 PSA< 60~70Ų 分子量< 450 LogD = 1~3 ClogP - (N+O)>0
口服吸收好的化合物中,90%符合此规则; 同时违 背上述两项的化合物, 成药的概率较小。
(Lipinski, CA et al. Adv Drug Delivery Rev. 1997, 23: 3)
Lipinski 规则计算氢键数目实例
官能团
羟基 羧酸 —C(O)—N—R2 伯胺 仲胺 醛 酯 醚 腈 吡啶
物理化学性质
溶解度 渗透性 化学稳定性
生物化学性质
代谢(一相、二相) 蛋白结合和组织结合 转运(摄取、外排)
药动学(PK)和毒性
清除率 t1/2 生物利用度 药物-药物相互作用 LD50
药动学和毒性: 清除率,半衰期,生物利用度,LD50
O
N Cl
效价=1 nmol/L, 口服吸收差
HN
氢键供体数=1
氢键受体数=6
分子量=591
Log p=7.3
PSA=50 可旋转键数=14 氢键总数=6
“Rule of 3”规则(Oprea,T.I.)
--类先导化合物(Lead like compound)规则
MW≤ 300 Clog P≤ 3 可自由旋转键≤ 3 氢键供体≤ 3 氢键受体≤ 3 极性表面积≤60Ų
物理化学性质: 溶解度,渗透性,化学稳定性
生物化学性质: 代谢,转运体亲和,蛋白结合,靶标亲和
物理环境
蛋白质
结构特征: 分子量,氢键,亲脂性,极性表面积,pKa,形状,反应性
化合物的结构决定了其基本特征,基本特征决定了其物理化学和 生物化学性质,后者最终决定了它的药动学和毒性。
化合物性质影响药物发现研究质量
理的消除和低毒性。
胃
小肠
门静脉 肝
血液 肾
器官
细胞
靶标
溶解度 pH 3~8
稳定性
∙代谢
稳定性
∙一相 ∙二相
蛋白结合
肾单位
∙渗透性 ∙尿排泄
分布 (全身)
细胞内分配 和代谢
稳定性
∙pH 2 ∙酶
稳定性
∙pH 3~8 ∙酶
上皮渗透性
∙被动 ∙pH 3~8 ∙Pgp外排
胆汁排泄
稳定性
∙酶
血-器官特殊渗透性
Log D: 某一特定pH(x)条件下,化合物部分以离 子形式、部分以中性分子形式存在,此pH条件 下其分配系数的对数
∙被动 ∙Pgp外排
渗透性
∙被动 ∙Pgp外排
药物经体内屏障输送到靶器官的概述
二、物理化学性质
1.从结构快速描述特征的规则
Lipinski规则(类药性5原则)
分子量 < 500 Clog P < 5 氢键给予体 < 5 (NH, OH数目的总和) 氢键接受体 < 10 (N, O数目的总和)
氢键供体
1(OH) 1(OH) 0 2(NH2) 1(NH) 0 0 0 0 0
氢键受体
1(O) 2(2O) 2(N, O) 1(N) 1(N) 1(O) 2(O) 1(O) 1(N) 1(N)
Veber规则 良好口服生物利用度(大鼠)应符合;
≤10个可旋转化学键 ≤140Ų极性表面积,或≤12个氢键总数(
例:
NH N
F3C
N
S
三氟拉嗪 碱类 pka=7.81
CNS+
HOOC O
Cl O
吲哚美辛 酸类 pka=4.18
CNS-
2. 亲脂性
亲脂性是许多ADME/Tox性质的主要决定因 素之一
Log P: 化合物的所有分子均以中性形式存在的 pH条件下,其分配系数的对数
Log P = Log ([anic]/[Compd.aqueous])
50 40 30 20 10
0
好的药物
性质
活性优化
好的配体
活性
改变候选药物策略,从专注于活性到平衡地关注活性和性 质。
新颖 性
药动性
溶解 性
稳定性 选择性
安全性
活性
药物发现的成功需要同时平衡不同的变量
生命系统中药物暴露的屏障
屏障包括生物膜、PH、代谢酶和转运体 生物屏障使给予的药物到达靶标的量减少 类药性佳,良好的吸收和分布、低代谢、合
溶解度低: 体外生物活性低或不一致 试验介质中不稳定: 引起生物活性低 渗透性差:酶或受体活性与细胞活性(大幅下降)不
一致 血脑屏障通透性差:CNS药物体内效价低 PK差、生物利用度低或血中不稳定:体内药效差。
源于各方面的失败率(%)
从1991年到2000年,由于药动学(PK)和生物利用度导致的 开发失败 率大幅降低。毒性、临床安全性和剂型仍然是突出的类药性质问题。
一、Introduction
类药化合物: 具有足以可接受的吸收、分布 、代谢、排泄和安全性(ADME/Tox), 能 在人体一期临床试验后生存下来的化合物。
类药性质是分子的内在性质,药物化学家的 责任不仅在于优化这些分子的药理性质,而 且还在于优化它们的类药性质。
类药性质是药物发现的组成部分之一
氢键供体数=7 可旋转键数=11
分子量=543
PSA=206
ClogP=-1.7
氢键总数=19
氢键受体数=12
根据计算,其口服生物利用度约5%
例: 神经肽YY1拮抗剂
N
O
N Cl
效价=2μmol/L 氢键供体数=0 氢键受体数=3 分子量=369 Log p=5.7 PSA=17 可旋转键数=6
N N
结构特征决定了化合物在体内的药动学和毒性 性质良好的化合物,可减少低效的研究、失败
率和费用 评估优化ADME/Tox性质是药物发现的重要
方面 最佳候选药物具活性和性质上的平衡
药物发现中应关注的诸多性质
结构特征
氢键 亲脂性 分子量 极性表面积 形状 反应性
受体加供体) 分子柔性、极性表面积(PSA)、氢键数目是决
定口服生物利用度的重要因素
Veber. D.F. J. Med. Chem. 2002, 45; 2615-2623.
例:
O OH
O
OH
D C B A OH
OCH3O OH O
H3C
O
OH NH2
多柔比星
Lipinski规则
Veber规则
----------------- -----------------
Hale Waihona Puke 用于中枢的化合物结构特征 血脑屏障理化性质规则(Pardridge) 氢键总数<8~10 分子量<400~500 非酸类化合物
Clark和Lobell规则 N+O<6 PSA< 60~70Ų 分子量< 450 LogD = 1~3 ClogP - (N+O)>0
口服吸收好的化合物中,90%符合此规则; 同时违 背上述两项的化合物, 成药的概率较小。
(Lipinski, CA et al. Adv Drug Delivery Rev. 1997, 23: 3)
Lipinski 规则计算氢键数目实例
官能团
羟基 羧酸 —C(O)—N—R2 伯胺 仲胺 醛 酯 醚 腈 吡啶
物理化学性质
溶解度 渗透性 化学稳定性
生物化学性质
代谢(一相、二相) 蛋白结合和组织结合 转运(摄取、外排)
药动学(PK)和毒性
清除率 t1/2 生物利用度 药物-药物相互作用 LD50
药动学和毒性: 清除率,半衰期,生物利用度,LD50
O
N Cl
效价=1 nmol/L, 口服吸收差
HN
氢键供体数=1
氢键受体数=6
分子量=591
Log p=7.3
PSA=50 可旋转键数=14 氢键总数=6
“Rule of 3”规则(Oprea,T.I.)
--类先导化合物(Lead like compound)规则
MW≤ 300 Clog P≤ 3 可自由旋转键≤ 3 氢键供体≤ 3 氢键受体≤ 3 极性表面积≤60Ų