铜铝合金价电子结构的计算
铜铝两种金属材质间的渗透

铜铝两种金属材质间的渗透甘 霖天津欧能电气有限公司 天津 300457摘 要:文章首先简述了铜资源概述,然后分析了铝资源概述,最后重点探讨了关于铜铝两种金属材质间的渗透关键词:铜金属;铝金属;渗透中图分类号:C35 文献标识码:A一、前言随着改革开放,我国的经济迅速发展,由于工业技术的不断革命和电子技术突飞猛进的迅速发展,鉴于铝/铜金属的优点与特性,为了能够更好地满足了当今工业生产的要求,铜铝两种金属材质间渗透成为了国内外研究的话题。
二、铜资源概述1、铜是人类最早发现和使用的金属之一紫红色,比重8.89,熔点1083.4℃。
铜及其合金由于导电率和热导率好,抗腐蚀能力强,易加工,抗拉强度和疲劳强度好而被广泛应用,在金属材料消费中仅次于钢铁和铝,成为国计民生和国防工程乃至高新技术领域中不可缺少的基础材料和战略物资,在电气工业、机械工业、化学工业、国防工业等部门具有广泛的用途。
铜按自然界中存在形态可分为:自然铜,铜含量在99%以上,但储量极少氧化铜矿,为数也不多硫化铜矿,世界上80%以上的铜是从硫化铜矿精炼出来的按生产过程分类的话,铜产品则可大致分为铜精矿、粗铜和纯铜。
铜精矿是低品位的含铜原矿石经过选矿工艺处理达到一定质量指标的精矿,可直接供冶炼厂炼铜。
粗铜是铜精矿冶炼后的产品,含铜量在95~98%。
纯铜是火炼或电解之后含量达99%以上的铜。
火炼可得99~99.9%的纯铜,电解则可以使铜的纯度达到99.95~99.99%。
按主要合金成分分类的话,铜合金主要有黄铜、青铜和白铜。
黄铜为铜锌合金。
青铜是铜锡等合金,除了锌镍外,加入其他元素的合金均称青铜。
白铜则为铜镍合金,藏银工艺品大多是这种成分。
按产品形态分类,则主要有铜管、铜棒、铜线、铜板、铜带、铜条、铜箔等。
2、铜矿产资源情况全球铜资源情况铜相对于铝、锌、铅、铁等其他基本金属较为稀有,在地壳中的含量仅有万分之零点五左右,远小于其他基本金属。
全世界探明的铜矿储量约6亿多吨,遍及六大洲,有150多个国家都有铜矿资源,部分国家可采年限达100年以上,智利铜储量居世界第一位,探明铜金属储量达1.9亿吨,约占世界储量的1/3,其次为秘鲁4%、澳大利亚4%、墨西哥、美国15,3%、中国、俄罗斯5%、印度尼西亚、波兰15%、赞比亚6%、哈萨克斯坦和加拿大4%,上述12个国家铜储量合计占世界总储量的近90%。
Al_Cu合金GP区的价电子结构

文章编号:1009-3818(2003)02-0019-03Al-Cu合金GP区的价电子结构*高英俊刘慧(广西大学物理系广西南宁530004)摘要:应用固体电子理论(EET),对Al-Cu合金G.P区的价电子结构进行计算,在价电子结构层次上,对Al-Cu合金的时效硬化过程进行了分析.关键词:Al-Cu合金;G.P区;价电子结构中图分类号:TG111.1文献标识码:AAl-Cu合金由于具有良好的室温和高温性能而得到广泛应用,特别是含4~5%wt.Cu的高强度铸造Al-Cu合金,其力学性能优异,完全可以替代钢铁材料,近年来得到了较快的发展[1~3].对Al中添加少量的Cu,形成Al-Cu固溶体可以增加合金的强度[4].在较低的温度下对Al-Cu合金进行人工时效,可以从过饱和固溶体A相中依次地析出G.P(I)区、亚稳相H d、H c和稳定相(Al2Cu),其中亚稳相H d、H c和H的界面与A存在一定的共格或半共格关系,而稳定相H则是与基体完全不共格[5].这些析出的G.P(I)区、H d、H c和H相在不同的时效状态下,对合金的强化作用贡献不同,有的起强化作用,有的起弱化作用.析出相对基体的强化和弱化作用,都与析出相的价电子结构有着密切的关系.基于价键理论[6]建立的固体电子理论(EE T)[7],提供了一个处理复杂体系价电子结构的简捷实用的经验方法)))键距差(B LD)法,该方法已在合金改性及设计上得到了广泛应用并取得了成功[8].本文应用EE T方法对Al-Cu合金中时效形成的G.P区的电子成键特征进行计算,尝试从价电子结构层次解释时效过程中析出的G.P区对合金强化的原因.收稿日期:2003-03-20第一作者:高英俊(1962-)男教授,材料学博士研究方向为金属物理*基金项目:国家自然科学基金项目[50061001];广西科学基金匹配项目[桂科配0135006];广西科学基金项目[0007020];广西/十百千人才工程0项目[2001207]1模型1.1A-Al的晶体结构图1AL-cu合金G、P区模型Fig.1G、P Zone of Al-Cu alloy图2G、P区的单位晶脆Fig.2Unit cell of G、P zone对纯金属Al和Cu,它们的晶体结构均为面心立方结构,Al和C u的晶格常数分别为a Al= 0.40496nm和a Cu=0.36147nm.在Al-C u固溶相中,文献[4]指出,铝的晶格常数随含铜量的增加成直线下降,在平衡溶解极限5.7%Cu时为0.403 8nm.以含4wt.%Cu的Al-Cu合金为例,此时合金的晶格常数为0.40416nm.时效前的淬火态,可以认为溶质Cu原子是杂乱分散在溶剂Al中,形成无序A固溶体.由于固溶体A相与纯Al、Cu金属同属于面心立方结构,而且原子的点阵排列,也只是由Al或Cu原子之间相互替换混合而成,故可采用/平均原子0模型[7]描述原子的状态.1.2G.P区结构模型Al-Cu合金淬火后,在时效进行的初期,会在固溶体中形成与基体共格的G.P区.Al-Cu合金中G.P区的原子排列模型已由文献[9]给出,如图1所第15卷第2期常德师范学院学报(自然科学版)Vol.15No.2 2003年6月Journal of Changde Teachers University(Natural Science Edition)Jun.2003示.图中所表示的原子排列为(100)面,而(001)和(010)面则垂直于图面.当一层Cu原子集中在(001)面上时,由于Cu原子的半径较Al原子要小,将引起Cu附近的晶格发生畸变,影响范围在1~2层原子范围.按文献[9]给出的Cu原子半径为0.118nm,Al 原子的半径为0.124nm,因此,近似认为Cu原子引起的邻近两层Al原子向Cu原子收缩畸变约为5%.因此,依据图1的G.P区结构模型,可设想这个G.P区的单位晶胞结构如图2所示.这个晶胞结构与基体Al的晶胞结构(fcc)是完全共格的,只是(001)面上的Al原子被Cu原子所替代.由于在[001]方向,G.P区引起晶格畸变,C u原子层最近邻的两层Al原子层间距收缩大约为5%,次近邻各原子层间距亦将有不同程度的收缩,距铜原子层愈远,收缩也愈少.为简化起见,在一级近似情况下,只考虑最近邻的两层Al原子间距变化,忽略第二层以上的Al 层的弹性畸变.这样,G.P区的晶胞可以近似看成为L10型晶胞结构,晶胞的晶胞常数由实验给出为a= 0.404nm,c=0.380nm.2计算方法与结果按照EET[7]理论,原子的共价电子是分布在连接最近邻、次近邻以及A近邻原子的键上.各键上共价电子对数(即键级n s)由下列键距公式表示:D(n A)=R A+R B-B lgn A(1)这里R是单键半径,参数B按文献[6]中的取值选取,晶胞内的共价电子数可以写成下述关系式:k1n A c+k2n B c=6A I A n A(2)式中k1、k2分别为晶胞中A、B原子的个数,n A c、n B c 分别为A、B原子的共价电子数.I s为n s键级的等同键数,各等同键数的确定可依照文献[7]的作法.由于各晶胞的晶体结构已确定,晶格常数已有实验结果,因此,运用键距差(BLD)方法[7]建立n A 方程,参见文献[7,10]的求解步骤,求解(1)、(2)方程组,逐个计算各晶胞中原子的电子结构,计算结果分别列在表1和表2.表1给出了面心立方的无序A固溶体晶胞的价电子结构,它是根据/平均原子0模型[7]计算求得.按照图2简化的G.P区模型,计算得到的G.P区价电子结构结果列在表2.表1A-Al(无序Al-Cu)晶胞的价电子结构Tab.1The electronic structure of A-Al unit cella:0.40416nm B=0.710Al CuR Al:4n c:2.5296R(I):0.119n m R Cu:9n c:4.5504R(1):0.11481nm键名I A D n A(nm)D n A(nm)n A$D(nm) D n A S120.285770.285760.21160.00001 D n B S60.404160.404140.00460.00001表2G.P区单位晶胞(Al-Cu晶胞)的价电子结构T ab.2The electronic structure of G.P zone cella0:0.404n m B=0.710a Cu:0.380n mAl CuR Al:3n c:1.1734R(I):0.119n m R Cu:8n c:4.4006R(1):0.11492nm键名I A D n A(nm)D n A(nm)n A$D(n m) D n A Al-Cu320.277320.276760.24920.00056 D nB Al-Al80.285670.285120.21700.00056 D n C Cu-Cu80.285670.285120.16650.00056 D nD Al-Al20.380000.379440.01020.00056 D nE Al-Al100.404000.403440.00470.00056 D n F Cu-Cu80.404000.403440.00360.00056 20常德师范学院学报(自然科学版)2003年3讨论经淬火后的Al-Cu过饱和固溶体,由于基体残存空位等缺陷,在时效的初期铜原子容易通过这些空位缺陷扩散,在基体的(001)面上偏聚出Cu原子,逐渐形成G.P区.从表1和表2分别列出的对无序A固溶体Al-Cu晶胞和G.P区晶胞的价电子结构计算结果,可以解释G.P区形成对基体强化作用的结果:由表1可见基体A固溶体的最强键Al-Al键的共价电子对数为n A=0.2116,而在G.P区的单位晶胞中,表2给出的最强键为Al-Cu键,共价电子对数为n A=0.2492,次强的Al-Al键共价电子对数为n A=0.2170,均比基体A固溶体的最强键要强得多.因此,这些较强的Al-Cu键有规则的排列,易形成Cu原子偏聚的Al-Cu原子区域)))G.P区.由于G.P区与基体完全共格,当大量的G.P区脱溶沉淀出时,将起到提高合金整体键强的作用,因而使得合金的强度得到明显提高.参考文献1Shan Liu,R.E.Napoli tano,R.Trivedi.Measurement Of Anisotropy Of Crystal-Melt Interfacial Energy For A B-inary Al-Cu Alloy[J].Acta mater.2001,4:4271-4276.2J.M.Silcock,ments on the nucleati on and growth of in AlCu alloys.[J].Scripta M ater.,2002,46:389-394.3S.P.Ringer,K.Hono.Microstructural Evolution and Age Hardening in Aluminiu m Alloys[J].Mater.Characteriza-tion,2000,44:101-131.4L.F.蒙多尔福编著.铝合金的组织与性能[M].北京:冶金工业出版社,1984,221-240.5P.哈森.物理金属学[M].北京:科学出版社,1984, 183.6L.鲍林著.化学键本质[M].上海:上海科学技术出版社,1966,393.7张瑞林.固体与分子经验电子理论[M].吉林:吉林科学技术出版社,1993,1-3008刘志林.合金价电子结构与成分设计[M].吉林:吉林科学技术出版社,1990,290.9K.Hono,T.Satoh,and K.Hi rano,Evidence of multi-layer GP zones in Al-Cu alloy[J].Philos.Mag.A,1986,53:493-504.10高英俊,钟夏平,刘慧.微量Sc对Al-Mg合金晶粒细化影响的电子结构分析[J].中国有色金属学报,2002(S.2):132-134.CALCULATION ON VALENC EELECTRON STRUC TURES OFAl-Cu ALLOY IN EA RLYAGING COND ITIONGAO Ying-jun LIU Hu i(Department of Physics,Guangxi Universi ty,Nanning,Guangxi530004,P.R.China)Abstract According to the/E mpirical Electronic Theory in Solid and Molecules0(EE T),the valence electron s tructures of the GP zones of Al-Cu alloy in aging condition were calculated and the process of strength in aging was investigated.Key words Al-Cu alloy;G.P zone;valence electron struc-ture(责任编校:熊振国)21第2期高英俊刘慧Al-Cu合金GP区的价电子结构。
关于Mn3Al块体合金的电子结构计算
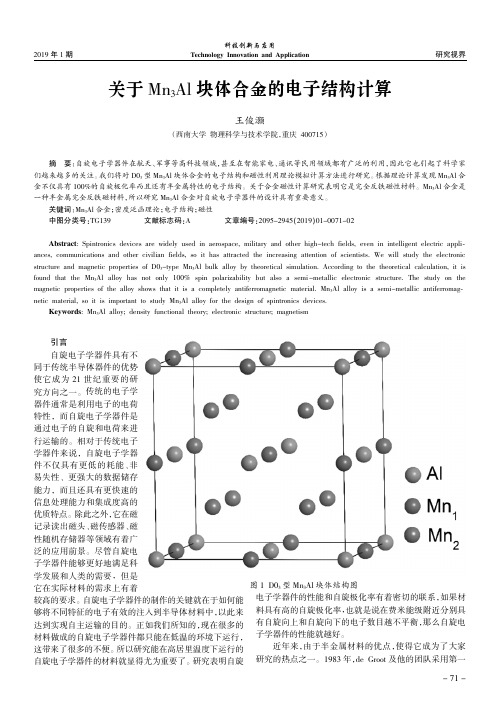
2019年1期研究视界科技创新与应用Technology Innovation and Application关于Mn 3Al 块体合金的电子结构计算王俊灏(西南大学物理科学与技术学院,重庆400715)引言自旋电子学器件具有不同于传统半导体器件的优势使它成为21世纪重要的研究方向之一。
传统的电子学器件通常是利用电子的电荷特性,而自旋电子学器件是通过电子的自旋和电荷来进行运输的。
相对于传统电子学器件来说,自旋电子学器件不仅具有更低的耗能、非易失性、更强大的数据储存能力,而且还具有更快速的信息处理能力和集成度高的优质特点。
除此之外,它在磁记录读出磁头、磁传感器、磁性随机存储器等领域有着广泛的应用前景。
尽管自旋电子学器件能够更好地满足科学发展和人类的需要,但是它在实际材料的需求上有着较高的要求。
自旋电子学器件的制作的关键就在于如何能够将不同特征的电子有效的注入到半导体材料中,以此来达到实现自主运输的目的。
正如我们所知的,现在很多的材料做成的自旋电子学器件都只能在低温的环境下运行,这带来了很多的不便。
所以研究能在高居里温度下运行的自旋电子学器件的材料就显得尤为重要了。
研究表明自旋电子学器件的性能和自旋极化率有着密切的联系,如果材料具有高的自旋极化率,也就是说在费米能级附近分别具有自旋向上和自旋向下的电子数目越不平衡,那么自旋电子学器件的性能就越好。
近年来,由于半金属材料的优点,使得它成为了大家研究的热点之一。
1983年,de Groot 及他的团队采用第一摘要:自旋电子学器件在航天、军事等高科技领域,甚至在智能家电、通讯等民用领域都有广泛的利用,因此它也引起了科学家们越来越多的关注。
我们将对D03型Mn 3Al 块体合金的电子结构和磁性利用理论模拟计算方法进行研究。
根据理论计算发现Mn 3Al 合金不仅具有100%的自旋极化率而且还有半金属特性的电子结构。
关于合金磁性计算研究表明它是完全反铁磁性材料。
铜单质价电子结构参数统计值的计算

铜单质价电子结构参数统计值的计算
李飞;赵侠
【期刊名称】《辽宁石油化工大学学报》
【年(卷),期】2010(030)002
【摘要】基于固体与分子经验电子理论,对Cu单质的价电子结构进行了分析,计算了价电子结构参数统计值n'A、E'A,给出了价电子结构参数统计值的计算方法,将统计值n'A、E'A与最可几值nA、EA进行了对比.计算结果表明,Cu原子处于甲种杂化第1~12杂阶时所处的状态为其实际中可能存在的;Cu的统计值n'A、E'A与相应的最可几值nA、EA的计算偏差分别为5.72%和6.86%,可以用n'A、E'A代替nA、EA来讨论铜的宏观性能.
【总页数】5页(P11-14,22)
【作者】李飞;赵侠
【作者单位】辽宁石油化工大学职业技术学院,辽宁抚顺,113001;辽宁石油化工大学职业技术学院,辽宁抚顺,113001
【正文语种】中文
【中图分类】TG111.1
【相关文献】
1.Cu、Ag、Au价电子结构参数统计值及熔点的理论研究 [J], 李飞;赵侠;李志武
2.钨铌合金价电子结构参数统计值计算及性能研究 [J], 陈天宝;刘宁;刘爱军
3.铜铝合金价电子结构的计算 [J], 许万马;彭金璋;石长柏;廖劢鑫
4.铝的价电子结构参数统计值的计算 [J], 李飞;赵侠
5.云环境下多方保密计算最大值、最小值及其统计学应用 [J], 李占利;陈立朝;陈振华;刘娅茹
因版权原因,仅展示原文概要,查看原文内容请购买。
Mg—Sc合金的价电子结构分析

素的密度更低 ; S 镁合金可以提高镁固溶体的 含 c 熔 点 ;c S 的熔点 ( 4 ℃ ) 于其 它 稀 土 元 素 , 15 1 高 在
镁合 金 中具有更 低 的扩 散 速 度 ; c 另外 的元 素 S与 组 合能形 成 Mg S 化合 物强化 相 , 、c 改善 合金 的室 温 和高温 性能 。最 近研究 表 明 , 土元 素 S 能 够 稀 c 将 MgR 系 合 金 服 役 温 度 提 高 到 3 0 以 —E 0℃
收 稿 日期 :0 01—3 2 1 —01 作 者 简 介 : 中柱 (9 5 , , 西原 平人 , 原 大 学机 电 工 程 系助教 , 要 研 究 方 向 : 型镁 合 金 材 料 的研 究 与 开发 。 谢 17 一) 男 山 太 主 新
15 ・ 3
・
杂 化状态 如表 1 所示 。
t 6 。 p l= 1 mt 2 n = 0 r = 1 Rt : s6 2 t : t t
共 价 电子数 :
=
∞
(
+ ^ ^ C +( , + f f f ( ) +n )^ r f + )f 4 l
(^ ^ z) +(f f fC 1+ +, C ^ 1十 + )f () 5
总 电子数 :
=
研
单键 半距 :
RdI R“1C +R, )缸 ( ) )h d ( C 1 () 6
各 杂 阶原子状 态 特征参 量 由以下公 式确 定 : 杂 阶 的 h态和 t 的组 分为 : 态
C +是 C 一 t= 1 2 h =1 a 一 a 一 ( ‘ 2 )
晶格 电子数 :
:
( 一r )h +( 一r)t 1 hl C 1 ,lf c
铝的价电子结构参数统计值的计算

[] 8 的思 想 , 算 了铝 的价 电子 结 构参 数 统计 值 , 计 从 而 为铝 及 铝合 金 的 成 分设 计 和 开 发 性 应 用 提 供 了
基 础数 据 。
在 A l的 价 电 子 结 构 计 算 中 , 足 A < 满 D 0 05r .0 i m条件 的有 2组解 , 两 种解 便 视 为 晶胞 中 这 A 原 子最 可 能存 在 的 状 态 。根 据 价 电子 结 构 参 数 l
1 9期
李
飞, : 等 铝的价 电子结构参数统计值 的计算
4 8 57
∑, r= 1.5 9 卢 = 0 0 1n 2 148 .7 m
N=2
P “ ’
式 ( ) , 为 实 际 晶胞 中可 能 存 在 的原 子 状 1中 态 数 目; n 为处 于 第 i 可 能存 在 的原 子 状 态 时 该 种 晶胞 内最强 键上 的共 用 电子 对 数 ; 为处 于第 i 种 可 能存 在 的原 子 状 态 时 该 晶胞 内 最 强 键 上 的 键 能
于实 验与 测 试 的角 度 ’ , 少 深 人 到 电 子结 构 层 3很 J 次 。众所 周 知 , 料 的组 织 、 能 是 由微 观结 构 来 材 性
胞 内的原 子排 列及 键 络 分 布 见 图 1 。按 文献 [ ] 7 的 计算 方法 给 出了铝 的价 电子结 构 , 算结 果见表 1 计 。
Ⅳ {
组态 i 和 时 , 晶胞 的 (k) h1晶面 上 的共 价 电子 密 度 。 将 表 1中所 有 的 P “ 、r 代人式 ( )则有 ¨ o 值 2,
一
^
=
∑ C= 25 +156 = t ÷(.39 36 ) 500 .78
Al—Cu—Mg合金界面电子结构计算及其对合金性能的影响

[ 谰] 1 uMg合金 ;界 面 电子绪 构 GP Z 关键 A一 - C B
_
- -
[ 中圄分类号】Q T
【 文献标识褐] A l
【 文章编号]0716(000 —040 10 —8 52 1)40 3—3
Ca c l to fI e f c e t o t uc ur nd I lue eo o ri so lu a i nso nt ra eElc r n S r t ea nf nc n Pr pe te f AICu - g Alo - - - M ly
i a ig rc si r c l l e s gte m i c l l t nT e r E T i S l n l ue , e eut sg et a te oa b n i t s yo n gn o es gwee ac a d i pr a Ee r h oy( E ) n oi a dMoe ls t s l g s dt th t o d gi e i f p n ut un hE i co d c h r su e h t l n n n t S”
Al_Mg_Si合金的价电子结构分析

文章编号: 1672-6146(2003)04-0009-03Al-Mg-Si合金的价电子结构分析3高英俊 李云雯(广西大学物理学院 广西南宁 530004)摘 要: 用经验电子理论(EET)对Al-Mg-S i合金中主要成分(如α-Al、镁、硅,以及强化相(β-Mg2S i)的价电子结构进行计算,计算结果表明各成分晶胞最强键上的共价电子对数与其密度、熔点、硬度、电负性有对应关系;Mg、S i原子上次强键的共价电子分布有利于β相的形成.关键词: Al-Mg-S i合金;Mg2S i;价电子结构中图分类号:TG111.1 文献标识码:AAl-Mg-Si合金由于具有优良的屈服强度、伸长率、电阻率、冲击韧性、耐腐蚀等性能[1-2],在航空航天、汽车、电子工业等领域得到广泛应用[3].随着对航空航天材料性能要求的提高,从微观结构层次对含Si的铝合金进行改性研究和开发具有高性能的铝合金材料,已成为当前材料科学家对Al-Mg合金关注的重点[4].基于价键理论和能带理论建立的固体电子理论(EET)[5],提供了一个处理复杂体系价电子结构的计算方法———键距差(BLD)法,并成功地用于合金的电子结构计算与合金相变研究,使得研究合金的宏观性能可以追溯到合金原子的价电子结构层次,为合金改性设计提供了深层次的理论指导[6-7].本文则是运用EET理论对Al-Mg-Si合金的若干相的价电子结构进行计算,为设计和开发性能更好的Al-Mg-Si合金提供电子层次的键信息.1 模型铝晶胞为面心立方晶体结构,晶胞内含有4个Al原子,晶格常数为a=0.40496nm,晶胞结构如图1(a)所示;镁的晶体结构为六角密集结构,一个镁晶胞中含有4个镁原子,构成一个六面体结构,如图1(b)所示,镁晶胞的晶格常数为a=0.32094nm,c =0.52056nm.硅晶胞的空间结构为金刚石结构,一个硅晶胞中含有8个硅原子,构成一个四面体,如图1(c)所示,硅晶胞的晶格常数为a=0.54307nm. 由文献[8]知Al-Mg-Si合金中主要的析出相收稿日期:2003-09-203基金项目:国家自然科学基金项目[50061001],广西科学基金项目桂科配[0135006]、桂科自[0007020]、桂科基[0342004-1],广西“十百千人才工程”项目[2001207].第一作者:高英俊(1962-)男教授,材料学博士研究方向合金的微观结构与物理性能为Mg2Si相—β相,它在合金中起着强化的作用.一个Mg2Si晶胞中含有12个原子,为CaF2型结构,晶格常数为a=0.639nm,如图2所示.由于成分存在范围很窄,与α-Al基呈伪共晶关系,它的生成过程[8]可用如下表达式表示:S olute clusters(溶解团)→G PⅠZ ones(spherical,球状)→G PⅡ→Z ones/β″(针状)→β′(棒状)→β(面第15卷第4期湖南文理学院学报(自然科学版)V ol.15N o.4 2003年12月Journal of H unan U niversity of Arts and Science(N atural Science Edition)Dec.2003心立方).上述变化过程在Al-Mg-Si合金中,最终生成的相是β相,本文重点是对Mg2Si晶胞的键结构进行分析.2 计算方法与结果表1 纯Al晶胞的电子结构T ab.1 The electronic structure of Al键名D Al-Al IαD nα nm D′nα nm nαn A120.286350.286330.20857n B60.404960.404940.00445注:σ=4;a=0.40496nm;β=0.710;n c=2.5296;R Al(1)=0.1190nm;ΔD nα=0.000017nm表2 纯Mg晶胞的电子结构T ab.2 The electronic structure of Mg键名D Mg-Mg IαD nα nm D′nα nm nαn A60.319500.319590.11025n B60.320940.321030.10521n C60.452860.452950.00145n D20.520560.520650.00016n E60.555890.555980.00005注:σ=3,β=0.710;n c=1.3022;R Mg(1)=0.12580nm;ΔDnα=0.00009nm表3 纯S i晶胞的电子结构T ab.3 The electronic structure of S i键名D Si-Si IαD nα nm D′nα nm nαn A40.235160.234910.97105n B120.384010.383760.00778n C120.450290.450040.00091n D240.470310.470060.00047n E60.543070.542820.00004注:σ=6;β=0.710;n c=4;R Si(1)=0.1170nm;ΔD nα=0.00025nm.表4 Mg2S i的电子结构T ab.4 The electronic structure of Mg2S i键名IαD nα nm D′nα nm nαD Mg-Si(n A)160.276690.272110.32466D Mg-Mg(n B)120.319500.314920.08804D Si-Si(n C)120.451840.447260.00027D Mg-Mg(n D)240.451840.447260.00055D Mg-Si(n E)480.528310.525250.00002注:β=0.600;ΔD nα=0.004580nm;σ=3;R Mg(1)=0.12580nm;n c= 1.3022;σ=4;R Si(1)=0.1170nm;n c=3.664. 按照EET[5]理论,原子的共价电子是分布在连接最近邻、次近邻,以及s近邻原子的键上.各键上共价电子对数(即键级n s)由下列键距公式表示:D(n s)=R A+R B-βlg n s(1)这里R是单键半径,β按文献[5]中的取值选取,晶胞内的共价电子数可以写成下述方程:k1n A c+k2n B c=6s I s n s(2)式中k1、k2分别为晶胞中A、B原子的个数,n A c、n B c 分别为A、B原子的共价电子数.I s为n s键级的等同键数,各等同键数的选取可依照文献[5]的作法来确定.由于各晶胞的结构已确定,晶格常数已有实验结果,因此,运用键距差(BLD)方法[5]建立n A方程,参见文献[5,9]的求解步骤,联立(1)、(2)等方程组,逐个计算各晶胞中原子的价电子结构.计算结果如表1至4所示.3 分析和讨论从表1至表4给出的计算结果可见,铝晶胞的最强键上分布的电子对数为n A=0.20857,镁晶胞的最强键上分布的共价电子对数为n A=0.110251,硅晶胞的最强键上分布的共价电子对数为n A =0.97105,而Mg2Si晶胞的最强键上分布的共价电子对数为:n A=0.32799,介于镁与硅的最强键之间.从物理特性上看,硅的熔点为1414℃,密度为2.32g/cm3;镁的熔点为649℃,密度为1.74g/cm3;铝的熔点为660.3℃,密度为2.7g/cm3,而Mg2Si晶体具有较高的熔点(1085℃)、较低的密度(1.9g/ cm3)和热膨胀系数(7.5×10-6K-1)[11].显然Mg2Si 的密度、熔点也介于镁与硅的密度、熔点之间,与本文计算得到的最强共价键上分布的电子对数的情况相对应.其次,电负性是表征元素的原子在分子内吸引电子的能力,电负性越大,吸引电子的能力也就越强.硅的电负性为1.90,镁为1.31,铝为1.61,由表1至表4可见Al、Mg和Si的电负性与其最强键分布的电子对数成正比关系,电负性越大,最强键分布的电子对数越多.再次,发现最强共价键分布的电子对数也与其硬度成正比的关系:最强键分布的共价电子对数越多,硬度越大,铝的莫氏硬度为2.9,镁的莫氏硬度为2.0;硅的莫氏硬度为7.0.Mg2Si的莫氏硬度4.6介于Mg和Si的莫氏硬度之间.文献[11]的实验中:在β″相形成前期,晶胞中仍含有铝原子,在时效过程中,铝逐渐被硅所代替,因为最终生成的Mg2Si最强键上分布的共价电子对数比铝要相对多;Mg和Si的百分比含量比Al相对少得多,但这两种元素的次强键(Mg的次强共价键上分布的电子对数为n B=0.1052,Si的次强键上分布的共价电子对数为n B=0.0078)比铝(次强键上分布的电子对数为n B=0.004453)要大,因此形成01 湖南文理学院学报(自然科学版)2003年Mg 2Si 相后,结构更为稳定,其空间结构为十六面体,均匀分布在合金中.4 结论(1)各成分的熔点和密度与其对应晶胞最强键上的电子对数成正比关系,即最强键上的电子对数多,则其熔点、硬度和密度也相应地高.(2)晶胞中原子的电负性和晶胞的硬度也与其所对应的晶胞最强键上电子对数成正比关系.(3)在形成Mg 2Si 相时,Mg 和Si 原子的次强键上分布的共价电子对数起了一定的作用.参 考 文 献1 Ф.H.科瓦索夫主编.韩秉诚译.工业铝合金[M].北京:冶金工业出版社,1987:50-64.2 L.F.M ondolfo.S tructrue and Properties of Aluminum Alloy[M].London :Butterw orth &C o (Publishers )Ltd ,1976:566.3 R Chen ,Z Hang.M icrostructure of Al -Mg -S i Alloy[J ].Mater.Sci.Eng.,2000,A280:146.4 K Matsude ,Y Uetuni ,T Sato.Metastable phase in an Al -Mg -S i alloy[J ].Metal.Mater.T rans.,2001,A 32:1293.5 张瑞林.固体与分子经验电子理论[M].长春:吉林科学技术出版社,1993,1-90.6 刘志林.合金价电子结构与成分设计[M].长春:吉林科学技术出版社,1990,1-29.7 刘志林,孙振国,李志林.余氏理论和程氏理论在合金研究中的应用[J ].自然科学进展,1998,8(2):150.8 C D Mariora ,S J Andersen.Atomic M odel for G P -Z onesIn Al -Mg -S i Alloy[J ].Acta Mater.,2001,(49):321.9 高英俊,钟夏平,刘慧,等.微量Sc 对Al -Mg 合金晶粒细化影响的电子结构分析[J ].中国有色金属学报,2002,12(S2):132.10 Carbonneau Y,C outure A ,Van Neste.The observation ofa new ternary AlMgS i phase in Mg -S i alloys[J ].Metal.Mater.T rans.,1998,A 29:1759.11 A K Cupta ,D J Uloyd.Precipitation hardening processesin an Al -Mg -S i alloy [J ].Mater.Sci.Eng.,2001,A301:140.ANALYSIS OF VALENCE E LECTR ONIC STRUCTURES OF Al -Mg -Si ALLOYG AO Y ing -jun LI Y un -wen(Physics college of G uangxi University ,Nanning ,G uangxi 530004) Abstract Based on the em pirical electron theory (EET )thevalence electron structure of α-Al ,Mg ,S i and equilibrium (β-Mg 2S i )phase in Al -Mg -S i alloy was calculated.The calculated results show that there are represent the correspondence between the co -valence electrons which distribute in the strongest bonds with the density ,melting point ,hardness and electronegativity ,and the co -valence electrons which distribute in the second neighbor -bonds of Magnesium and S ilicon are of benefit to the appearance of βphase. K ey w ords Al -Mg -S i alloy ;Mg 2S i ;the valence electronstructure(责任编校:江 河) 我刊被评为湖南省一级期刊由湖南省科技厅、湖南省新闻出版局共同组织的2002年度湖南省科技期刊审读和等级评定工作日前结束。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第32卷第1期吉首大学学报(自然科学版)Vol.32No .12011年1月Journ al of Ji shou Universit y (Nat ural Science Edit ion)J an.2011文章编号:1007-2985(2011)01-0052-04铜铝合金价电子结构的计算*许万马,彭金璋,石长柏,廖劢鑫(吉首大学物理科学与信息工程学院,湖南吉首416000)摘要:采用余氏理论计算了铜铝合金在不同时效作用下析出相价电子结构,通过对键强的分析,从而发现相析出过程中合金强度逐渐得到强化.关键词:余氏理论;铜铝合金;价电子结构中图分类号:O469文献标志码:A1理论分析在室温和高温条件下铜铝合金具有良好的物理和化学性能,尤其含铜量为4~5%的高强度铸造铜铝合金,其力学性能较优异,完全可替代部分钢,近年得到迅速应用[1-5].向纯铝金属加入适量铜经淬火后,形成含铜量为4%的过饱和固溶体,再经时效作用可提高合金强度[4].过饱和固溶体在较低温度下经不同人工时效作用,可依次析出GP 区、亚稳相、和稳定相(Al 2Cu),其中亚稳相、界面与基体基本上共格和半共格,稳定相界面与基体完全不共格[5],这些脱溶析出的合金相在不同温度下对合金基体作用不同,有的起强化作用,有的起弱化作用,而强化与弱化作用均与析出相电子结构有关,特别是与析出相中原子周围局域电子成键特征有着紧密关系[6-7].上世纪70年代,基于价键理论[8]和能带理论建立起来的固体与分子经验电子理论(EET),又称余氏理论.在该理论基础上,刘志林等提供了键距差法(BLD),使得合金宏观性能的研究可实现追溯到价电子结构层次,从而为合金良性设计提供深层次理论指导.在固体与分子经验电子理论中采用了以下4个基本假设:假设1在固体与分子中,每个原子一般由2个原子状态杂化而成,这2个状态称为始态和末态,即h态和t 态,其中至少有一个态在基态或靠近基态的激发态.2个状态都有它们各自的共价电子数n c 、晶格电子数n l 和单键半距R(1).晶格电子是在由多个原子组成的固体体系内,处于有3,4个甚至6个以上的原子围绕的空间内价电子.假设2一般情况下,状态杂化是不连续的,若以C t 表示t 态成分,那么在多数结构中C t 近似地由下式给出:k =l +m +n l +m+nl +m +n l +m+nl 3m 5n l3m5n,C t =1k 2.(1)*收稿日期:2010-11-20基金项目:湖南省自然科学基金资助项目(10JJ6068)作者简介许万马(),男,安徽安庆人,吉首大学物理科学与信息工程学院硕士生,主要从事凝聚态物理研究通讯作者彭金璋(63),男(土家族),湖南桑植人,吉首大学物理科学与信息工程学院教授,主要从事功能材料的物理特性研究:1980-:19-.其中:l,m,n,l ,m ,n 分别为h 态和t 态的s,p,d 轨道价电子.当s 轨道价电子是共价电子时,=1;当晶格电子时,=0,也同样如此.当h 态完全为晶格电子时,(1)式不成立,此时应为k =l +m +nll +m +nl 3m 5n.(2)若将1对确定的h 态和t 态构成的所有原子杂化状态称为原子的一种杂化状态的话,则把某种杂化状态下所具有的各种不同的稳定杂化的原子杂化称为杂化台阶,简称为杂阶,用表示,所有不同k 值的数目称为杂阶数.在EET 中,一个杂阶表示1个确定的原子状态,描述原子状态的特征参数n ,n c ,n l 以及R(1)在杂阶的数值可由下列各式给出:n =n h C h +n t C t =(l +m +n)C h +(l +m +n )C t ,n l =n l h C h +n l t C t =(1-)C h +(1-)C t ,n c =n c h C h +n c t C t =(l +m +n)C h +(l +m +n )C t ,R(1)=R(1)h C h +R(1)t C t .其中:C h 和C t 为h 态和t 态成分;C h =1-C 1,R(1)h 和R(1)t 为h 态和t 态单键半径距.假设3除特殊情况外,在结构中2个原子u 和v 之间总存在共价电子对,该共价电子对数用n 表示,而2原子间距将称为共价键距,用D uv (n )表示.根据鲍林研究[8],则D uv (n )=R u (1)+R v (1)-lg n .(3)其中,代表不同的键,有=A,B,,N ,它们代表结构中所有不可忽略的键.所谓不可忽略是指由这样长的键距,根据(3)式计算出的n 和结构中最大n M的误差相比还是不可忽略,而最大n M的可能误差当然决定于实验键距的可能误差值.的选取一律按照下列条件规定:=0.710nm n M <0.25或n M >0.75,0.0600n m 0.300nM0.700,0.0710-0.22n M =0.250+或n M =0.750-.其中0<0.250.假设4对于包括过渡元素在内的B 族元素以及镓、铟、铊,在固体和分子中的原子,它的外壳d 电子有一部分在空间扩散得远,使得它们对相邻原子的共价键距等效于更外层壳的s 和p 电子.对于Cu,Ag,Au 在晶格空间不同晶胞中p 价电子的取向是如此凌乱,使得其平均效果等效于s 电子.然而这些等效电子的相角分布和结合能计算的贡献仍然保持原来性质.来源于d 的等效s 、p 电子用(s )、(p )表示,而来源于p 的等效s 价电子用(s )表示.键距差方法是用N -1个键距差的实验值和由原子状态杂化表所选择的1个恰当杂化的参数n c 和R(1)组成N -1个lg r 方程和1个推导出的经验理论n A 方程共N 个方程求解出N 个未知数n ,然后将R(1)和n 代入鲍林研究存在的关系式求得N 个经验D n 值,并将它们与N 个相应的实验D n 比较看是否彼此接近.lg r 方程可以用lg r =lgnn A表达.对于一个分子式UV 的原子总数来说,一方面从价电子总数来看,则n c =n uc +n vc ;另一方面就共价电子在各条共价键上形成的共价电子对数的分布来看,则n c =I n =n AI r ,其中I 是等同键数,且n A =n c /I r .2晶体结构模型和价电子结构计算结果铜铝合金固溶体经淬火后,在时效初期会形成基体共格的GP 区,其原子排列模型由文献[9]给出.(001)面上的铝原子被铜原子所替代.按照文献[8]给出的GP 区铜原子半径为0.118nm,铝原子半径为,近似认为铜原子引起的近邻两层铝原子向铜原子收缩畸变约为5%,次近邻各原子层间距亦有不同程度的收缩,距铜原子层愈远,收缩愈少为简单起见,采取一级近似,即只考虑最近邻的两层铝原子间距变化,忽略第层以上的铝层畸变和铜原子层侧面端界畸变近似计算所得的晶格常数=53第1期许万马,等:铜铝合金价电子结构的计算0.124nm .2.:a 0.404图1GP 区的单位晶胞nm,c=0.380nm.GP 区单位晶胞见图1所示.随着时效时间的增加,亚稳相可以在基体中形成,也可以由GP 区直接演变产生.相的结构与GP 区相似,由若干铜原子层规则排列而成[2].相与基体共格,具有{100}//{100}基体的位相关系,其单位晶胞如图2所示,呈正方体结构[4],其晶格常数a=0.404nm,c=0.768nm.稳定相(Al 2Cu)晶胞含有12个原子,其中铝原子含8个,铜原子含4个,其晶格常数a=0.6066nm,c=0.4847nm,单位晶胞结构如图3所示.图2亚稳相的单位晶胞图3稳定相的单位晶胞GP 区、亚稳相、稳定相(Al 2Cu)的价电子结构计算结果见表1所示.表1价电子结构的计算结果GP zonesa0.404nm Al 2R(1)0.119nmn 3n c 1.0330n l 1.9670CuA8R(1)0.11492nm n 5.4006n 4.4006n l 1c0.380nm0.071nm I A 32I B 8I C 8I D 4I E 8I F 8n A 0.2429n B0.2115n C 0.1623n D 0.0099n E 0.0046n F 0.0035phase a0.404nm Al 3R(1)0.119nmn 3n c 1.1734n l 1.8266Cu B8R(1)0.11835nm n 6.2858n c 5.2858n l 1c 0.768nm 0.0643nm I A 32I B 8I C 8I D 32I E 8I F 2I G 8n A0.2804n B 0.1828n C 0.1828n D0.1828n E0.0165n F 0.0051n G 0.0026phase a0.6066nm Al 4R(1)0.119nmn 3n c 2.5296n l 0.4704CuA1R(1)0.11520nm n 5n c 4n l 1c 0.4874nm 0.060nm I A 2I B 16I C 2I D 4I E 8I F 4IG 4n A0.6019n B0.3874n C0.2818n D0.1345n E0.0631n F0.0124n G0.00013结论经固溶体淬火后的铜铝合金过饱和固溶体,由于基体残存空位等缺陷,在时效初期半径较小的铜原子容易通过这些空位缺陷扩散,在基体的(001)面上积聚铜原子,逐渐形成GP 区.由于GP 区与基体完全共格,当大量的GP 区脱溶沉淀时,较强的Al-Cu 键有规则的排列,形成铜原子偏聚的铜铝原子区域GP 区,将起到提高合金整体键强的作用,因而使得合金的强度得到明显提高.随着时效时间的延长,铜原子不断聚集,基体内的G 区逐渐长大,为相的形成提供了浓度组分条件,而且相一旦开始形核,由于铜原子半径较小,溶质铜原子会通过空位通道不断向晶核聚集由表可以看出,相的最强键共价电子对数为=,较G 区最强键共价电子对数=大,从而使得合金的键强进一步提高,合金强54吉首大学学报(自然科学版)第32卷P .1n A 0.2804P n A 0.2429度也进一步提高.随着时效时间的继续延长,铜原子通过空位通道继续向晶核聚集,铝原子逐渐呈现出无序化排列,逐渐形成与基体完全不共格的原子排列的相.相的最强键共价电子对数为n A =0.6019,达到了GP 区和相的最强键共价电子对数的2倍多,进而使合金的键强得到大大提高,合金强度得到显著提高.参考文献:[1]SILCOCK J M,FLOWER H ments on the Nucleation and Gr owth of in Al Cu Alloys [J].Scr ipta Mater,2002,46(3):389-394.[2]RINGER S P ,H ONO K.Micr ostructural Evolut ion and Age H ardening in Aluminium Alloys [J].M ater Character iza tion,2000,44(1):101-131.[3]MONDOLFO L F.Aluminum Alloys:Structure and P ropert y [M ].New York:Butterwor ths P ress,1976:260-320.[4]H AASON P,XIAO Ji mei.Physical Metallurgy [M ].Beijing:Science press,1984:183-230.[5]ANDO Y,MI HAMA K.Growth of GP Zones and P hase in Al Cu Based Alloys [J].J.Cr ystal Growt h.1974(24):581-584.[6]KAR LI K M,JOWF FR EY B.Study of GP Zones in Al Cu Based Alloys [J].Acta Mater ,1997,45(8):3251-3263.[7]TAKEDA M,MAEDA Y.Disscontiniuty of GP Zones and P hase in Al Cu Based Alloys [J].Scripta Mater ,1999,41(6):643-649.[8]P AULING L.The Natur e of the Chemical Bond [M].San Simeon:Cor rnell Univer sity P ress,1960:300-400.[9]H ONO K,SAT OH T,HIR ANO K.Evidence of Mult i Layer GP Zones in Al Cu Alloy [J].Philos M ag A,1986,53(4):493-504.Calculation of Electronic Structures of Al Cu AlloysXU Wan ma,PENG Jin zhang,SH I Chang bo,LIAO Mai xin(College of Physics Science and Infor mation Engineer ing,Jishou University,Jishou 416000,H unan China)Abstr act:Using empirical electron theory of solids and molecules (EET ),the electr onic structur es of the precipitates of Al Cu alloys,which pr ecipitates in differ ent aging condition,wer e calculated.It is conclu ded that the strength of the alloys is consolidated successively by analysing the covalence bond str ength.Key words:EET ;Al Cu alloys;electronic structur es(责任编辑陈炳权)55第1期许万马,等:铜铝合金价电子结构的计算。