催化臭氧化_一种有前景的水处理高级氧化技术_李来胜
臭氧催化氧化技术

臭氧催化氧化技术1. 臭氧催化氧化技术的原理:臭氧催化氧化技术是一种利用臭氧氧化剂将有机物和无机物氧化的技术。
臭氧氧化剂是一种具有臭氧催化作用的物质,其作用是将臭氧分解成活性氧,从而发生氧化反应。
臭氧氧化剂可以有效地将有机物、无机物和溶剂等物质氧化,从而达到净化空气的目的。
2. 臭氧催化氧化技术的应用臭氧催化氧化技术可以用于去除污染物,如挥发性有机物(VOCs)、氨氮、硫化氢、氯气等,以及臭气、有害气体等。
它可以用于处理各种类型的废气,如工业废气、汽车尾气、医疗废气、垃圾焚烧废气等,以及处理空气污染物。
此外,臭氧催化氧化技术还可以用于处理水污染物,如氨氮、硫化物、氯化物等。
它还可以用于处理固体废物,如有机废料、植物秸秆、废旧电子产品等。
3. 臭氧催化氧化技术的优缺点优点:臭氧催化氧化技术可以有效地去除水中的有机物,污染物和病原体;具有较高的处理效率,可以在短时间内实现高浓度污染物的去除;操作简单,易于控制和维护;可以有效地去除水中的挥发性有机物;可以有效地降低水中的氨氮含量。
缺点:臭氧催化氧化技术的成本较高;臭氧的使用可能会产生有害的副产物,如臭氧氧化物;臭氧催化氧化技术只能有效地去除水中的有机物,而无法有效地去除水中的无机物;臭氧催化氧化技术的处理效率受污染物浓度、温度、pH值等因素的影响。
4. 臭氧催化氧化技术的发展趋势臭氧催化氧化技术的发展趋势主要有以下几点:首先,臭氧催化氧化技术的应用范围将不断扩大,将更多的污染物纳入治理范围;其次,技术的发展将更加精细化,将更加精确地控制臭氧催化氧化技术的反应条件;第三,将更多的研究和开发投入到臭氧催化氧化技术中,以提高臭氧催化氧化技术的效率和稳定性;最后,臭氧催化氧化技术的成本将逐渐降低,以便更多的污染物得到有效的治理。
臭氧催化氧化技术是一种利用臭氧氧化剂氧化有机物的技术,它可以有效地去除污染物,减少对环境的污染。
近年来,臭氧催化氧化技术受到了越来越多的关注,因为它在环境保护方面有着重要的作用。
催化臭氧化技术在水处理中的应用研究进展

催化臭氧化技术在水处理中的应用研究进展作者:石角来源:《科技风》2019年第16期摘要:所谓催化臭氧化就是在反应过程中产生的强氧化性烃基自由基对水中有机物进行氧化分解,在常温常压条件下对难以被臭氧单独氧化的对有机物实现氧化降解。
和单一的臭氧化相比,此种技术具备更为经济和安全的优势,能在很大程度上提升臭氧利用率,基于催化臭氧技术的特征,本文就将对其在水处理中的应用情况进行研究,希望对这项工作的开展提供一定理论帮助和指导作用。
关键词:催化臭氧化技术;水处理;应用研究催化臭氧化技术通常分为两种类型:借助溶液中的金属离子对固态金属、金属氧化物等物质进行均相催化臭氧化,或是金属氧化物的非均相催化臭氧化。
催化臭氧化技术在应用过程中可以将臭氧中的强氧化性和强化剂的自身吸附性与催化性实现有效融合,这对于水处理中问题的解决将起到显著帮助。
为此,本文笔者就将站在催化剂角度上,对国内外的催化臭氧化技术进行研究,希望对这项工作的开展提供更有效的理论指导。
一、均相催化剂在水处理中的应用催化剂和反应物质处于同相物质,由于没有相界,所以产生的反应被称作为均相催化作用,在工作中可以和均相催化剂产生作用的催化剂也被称作为均相催化剂。
据研究发现,均相金属催化臭氧化主要具备两种反应,其一,金属离子对臭氧的分解作用,之后在反应中生成·OH,借助·OH的高活性作用进行有机物的氧化。
其二,金属离子和有机物的融合,最终实现臭氧氧化。
一般均相催化臭氧化技术中采用的催化剂是过渡性金属,过渡元素是具备专业性质的电子元素,所以可以在电子传递环节中实现氧化还原反应。
[1]比如在对五氯酚进行催化氧化实验过程中,研究者就将重点放在了臭氧氧化特性上,最终实验结果证明,PCP在反应五分钟后,基本可以实现完全分解,但是TOC 在反应半小时后杂质去除率只有百分之五十,这也意味TOC难以实现彻底氧化分解。
因为在对PCP进行氧臭氧催化氧化过程中,需要先对其进行脱氯处理,在实验反应推进过程中,物质中的苯环将被打开,最终分解为二氧化碳和水。
高级氧化处理有机污水技术进展

高级氧化处理有机污水技术进展高级氧化处理有机污水技术进展近年来,随着工业化进程的加快和城市化过程的推进,我国面临着日益严峻的水污染问题。
有机污水是其中的一个重要组成部分,含有大量的有机物质和污染物,对水体环境造成严重的破坏。
传统的有机污水处理方法在处理效果和经济性方面存在一定的局限性,在此背景下,高级氧化处理有机污水技术应运而生。
高级氧化处理有机污水技术是利用氧化剂,如臭氧、过氧化氢、高价铁离子等,在适宜的条件下将有机物质氧化分解为低分子物质或无害物质的技术。
该技术具有高效、低毒性、无需使用大量化学药剂等特点,因此被广泛应用于有机污水处理领域。
高级氧化处理有机污水技术主要包括臭氧氧化技术、高级氧化过程(包括Fenton反应、光催化氧化等)和超声波氧化降解等。
这些技术在不同条件下具有一定的适用性和优势,广泛应用于有机污水处理工程中。
臭氧氧化技术是利用臭氧分子中高能量的氧原子与有机物质发生氧化反应,将有机物质降解为简单的无机化合物。
臭氧氧化技术具有高度选择性、高氧化效率、无需添加化学药剂等特点。
特别是在含有难以生物降解的氯代有机物或芳香族有机物的有机废水处理领域表现出良好的应用前景。
Fenton反应是一种通过加入氢氧化物和过氧化氢的方式产生羟基自由基进一步氧化有机物质的技术。
Fenton反应具有反应速度快、氧化还原电位宽、自身生成滴定剂等优势。
但是,由于Fenton反应需要保持酸性条件,增加了处理过程中对废水的调节难度。
光催化氧化技术是利用半导体材料的光催化性能,将有机物质氧化分解为无害物质。
光催化氧化技术具有高效、无污染、易于操作等特点,尤其适用于处理难降解有机物质。
超声波氧化降解技术是利用超声波的机械振动效应和声化学效应,实现有机物质的氧化降解。
超声波氧化降解技术具有高效、无需添加化学试剂、操作简便等优点。
然而,超声波氧化降解技术受到设备成本高和能耗大的限制,尚需进一步开发和完善。
高级氧化处理有机污水技术尚存在一些挑战和亟待解决的问题。
水处理多相催化臭氧氧化技术研究现状
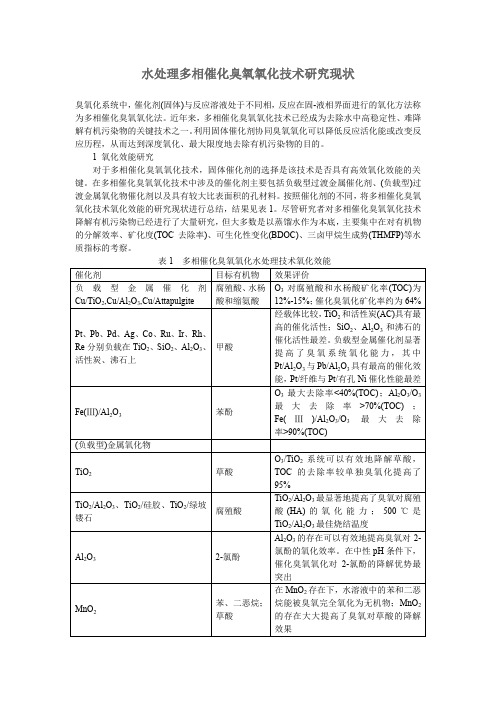
水处理多相催化臭氧氧化技术研究现状臭氧化系统中,催化剂(固体)与反应溶液处于不同相,反应在固-液相界面进行的氧化方法称为多相催化臭氧氧化法。
近年来,多相催化臭氧氧化技术已经成为去除水中高稳定性、难降解有机污染物的关键技术之一。
利用固体催化剂协同臭氧氧化可以降低反应活化能或改变反应历程,从而达到深度氧化、最大限度地去除有机污染物的目的。
1 氧化效能研究对于多相催化臭氧氧化技术,固体催化剂的选择是该技术是否具有高效氧化效能的关键。
在多相催化臭氧氧化技术中涉及的催化剂主要包括负载型过渡金属催化剂、(负载型)过渡金属氧化物催化剂以及具有较大比表面积的孔材料。
按照催化剂的不同,将多相催化臭氧氧化技术氧化效能的研究现状进行总结,结果见表1。
尽管研究者对多相催化臭氧氧化技术降解有机污染物已经进行了大量研究,但大多数是以蒸馏水作为本底,主要集中在对有机物的分解效率、矿化度(TOC去除率)、可生化性变化(BDOC)、三卤甲烷生成势(THMFP)等水质指标的考察。
2 实际应用效能研究多相催化臭氧氧化技术的大量研究工作是以蒸馏水作为本底,侧重于考察固体催化剂的催化活性。
作为一种新型水处理技术,多相催化臭氧氧化技术在实际水处理工程中的应用仍处于起步阶段;为提供实际应用基础试验数据,近年来研究者对应用多相催化臭氧氧化技术处理实际水体中有机污染物的效能进行了考察,其中对饮用水水源和污水中有机污染物的处理效果均有所涉及。
2000年,Gracia等利用TiO2/Al2O3催化臭氧氧化技术降解西班牙Ebro河水中的有机污染物(预处理去除悬浮颗粒物,初始TOC质量浓度=4.46mg/L;UV254=0.067)。
研究发现,催化剂的存在提高了TOC的去除率;多相催化臭氧氧化后用氯消毒,其副产物少于臭氧氧化后用氯消毒;多相催化臭氧氧化过程对有机污染的去除存在最佳臭氧投量。
Li等研究了AC/O3多相催化臭氧氧化与生物活性炭技术(AC/O3/BAC)联用去除密云水库中难降解有机物的效能。
臭氧催化氧化深度处理石油化工废水存在问题研究

工业用水与废水INDUSTRIAL WATER &WASTEWATERVol .51No .4Aug.,2020杨志林,程传,程志刚,郭磊,张乐乐,于海(江苏方洋水务有限公司,江苏连云港222046)基金项目:江苏省科技基础设施建设计划项目(BM2016229)石油化工废水是石化工业在生产过程中产生的废水,典型的石油化工废水中含有石油类、COD、氨氮、硫、酚、氰化物等常规污染物。
同时,不同的企业因产品不同,所产生废水中还含有多种与其有机化学产品相关的特征污染物,如苯系物、酯类、杂环化合物、有机酸等,从而造成废水不仅水质复杂,而且有毒物质多。
国内石油化工废水二级处理主要是以水解酸化、厌氧、A/O 等生化处理工艺为主,随着GB 31571—2015《石油化学工业污染物排放标准》的实施,石化废水处理厂原有处理工艺不能达到排放标准。
原因主要是石油化工废水中除含有可以生物降解的有机物外,同时含有部分生物不能降解物质。
针对处理出水不达标情况,我国石化废水处理厂进行了技术升级改造,多数废水处理厂建成了以臭氧/臭氧催化氧化为核心的深度处理工艺单元[1-6],臭氧催化氧化对石化废水处理厂二级生化出水中难降解有机物的去除效果明显,但是随着装置的长期运行,也存在一些问题需要总结,这对于完善该技术在石油化工废水深度处理中的应用具有参考价值,也可以为其他类似工程提供借鉴。
1应用现状臭氧催化氧化是一种高级氧化水处理技术,臭氧催化氧化工艺是在臭氧氧化体系中加入过渡金属离子,能对臭氧产生明显的催化效果,可以催化臭氧在水中的自分解,产生羟基自由基(·OH),其氧化还原电位高达2.8eV,能够迅速氧化分解有机物,反应中生成的自由基可继续参加·OH 的链式反应,摘要:随着GB 31571—2015《石油化学工业污染物排放标准》的实施,石化废水处理厂的技术升级改造多数采用以臭氧催化氧化为核心的深度处理工艺单元,但在实际运行过程中出现了一些问题。
Fe-Mn-MCM-41分子筛催化臭氧氧化控制含溴水中溴酸盐的生成

Fe-Mn-MCM-41分子筛催化臭氧氧化控制含溴水中溴酸盐的生成薛颖;李旭凯;吴颖;李来胜【摘要】采用水热法合成铁、锰双金属掺杂MCM-41(Fe-Mn-MCM-41),并将其用于控制催化臭氧氧化含溴水体中溴酸盐的生成,研究了初始pH、叔丁醇(TBA)、磷酸盐等对溴酸盐抑制效果的影响.结果表明:当溶液初始pH为5.0~9.0时,溴酸盐生成量随pH的升高而增加,pH=5.0时催化剂对溴酸盐的抑制率达到85.9%.TBA 的加入使单独臭氧氧化与催化臭氧氧化体系中溴酸盐的生成量明显降低,当加入0.1 mmol/L TBA后,溴酸盐分别减少67.7%和81.1%.磷酸盐的加入(1、5、10 mg/L)会降低溴酸盐的生成量,当加入1 mg/L磷酸盐时,单独臭氧氧化与催化臭氧氧化体系中溴酸盐的抑制率分别达到29.6%和82.5%.此外,还研究了体系中生成的HOBr 与H2 O2的浓度,结果表明:单独臭氧氧化体系中次溴酸的浓度高于催化臭氧氧化过程,说明催化臭氧氧化过程是通过阻止Br-氧化生成HOBr/OBr-抑制溴酸盐的生成;Fe-Mn-MCM-41/O3中的H2 O2浓度高于O3过程,而H2 O2是一种溴酸盐抑制物,证明了催化剂的加入可以提高对溴酸盐的抑制率.因此,Fe-Mn-MCM-41是一种可用于控制含溴水体中溴酸盐生成的臭氧氧化催化剂.【期刊名称】《华南师范大学学报(自然科学版)》【年(卷),期】2019(051)002【总页数】6页(P56-61)【关键词】Fe-Mn-MCM-41;分子筛;臭氧;催化臭氧氧化;溴酸盐【作者】薛颖;李旭凯;吴颖;李来胜【作者单位】华南师范大学化学与环境学院,广州510006;华南师范大学化学与环境学院,广州510006;华南师范大学化学与环境学院,广州510006;华南师范大学化学与环境学院,广州510006【正文语种】中文【中图分类】X703.1近年来水体中消毒副产物( DBPs) 由于具有致癌、致畸、致突变的危害性而引起广泛关注,其中溴酸盐() 已被列为2B 级致癌物质[1],具有一定的DNA 和染色体遗传毒性,它在饮用水及生活用水中普遍存在.溴酸盐的产生与臭氧氧化过程密切相关,当使用臭氧对水体进行消毒且水体中溴离子质量浓度超过50 μg/L 时,臭氧可将其氧化生成溴酸盐[2-3].目前控制溴酸盐的主要方法包括吸附[2-3]、还原[4]、光催化[5]和光电催化[6]等,但它们的效率低、效果差、耗能大、费用高.催化臭氧氧化是目前饮用水深度处理研究中的热点技术,它不仅能够增强对有机物的去除效果,还能有效控制含溴水体中溴酸盐的生成[7].因此,本文采用催化臭氧氧化技术来研究控制溴酸盐的方法[7].尽管硅基介孔分子筛( MCM-41) 具有较大的比表面积和较规整的结构[8],但其晶格缺陷少,催化活性低,因此可采用金属掺杂的方法修饰其结构以提高催化活性[9-10],掺杂的金属一般为过渡金属( Fe、Mn、Cu、Ce 等) .本课题组前期研究结果表明: Fe 和Mn 改性的MCM-41具有较好的催化活性,能够用于控制臭氧氧化过程中溴酸盐的生成.近年来有研究者发现双金属掺杂能够增加催化剂表面活性位点,促进反应体系中电子的转移,达到更好的催化效果[11].因此本文主要研究Fe-Mn-MCM-41 在催化臭氧氧化含溴水体过程中溴酸盐的生成机制.1 实验部分1.1 试剂与仪器主要试剂:硅酸钠( Na2SiO3·9H2O) 与十六烷基三甲基溴化铵( 上海国药集团化学试剂有限公司) 、乙酸锰( MnC4H6O4·4H2O) ( 上海阿拉丁生化科技有限公司) 、硝酸铁( Fe( NO3)3·9H2O) ( 天津科密欧化学试剂有限公司) ,均为分析纯; 溴酸钠( NaBrO3) 与溴化钾( KBr) ( 上海阿拉丁生化科技有限公司) 、草酸( 天津科密欧化学试剂有限公司) 均为分析标准品.主要仪器:臭氧发生器( ANSEROS) 、紫外可见分光光度计( 上海美谱达仪器有限公司) 、离子色谱仪( DIONEX) 、高效液相色谱仪( HPLC,SHIMADZU,日本) 、臭氧浓度检测仪( DOZ7600,杭州陆恒生物科技有限公司) .1.2 实验方法1.2.1 催化剂的制备称取28.42 g Na2SiO3·9H2O溶于65 mL 去离子水中,用机械搅拌器搅拌,再将一定质量的Fe( NO3)3·9H2O 和MnC4H6O4·4H2O 溶解在10 mL去离子水中,混合搅拌30 min 后逐滴加入2 mol/L H2SO4,调节溶液pH 为11 时,出现凝胶,继续搅拌30 min;称取7.28 g 十六烷基三甲基溴化铵溶于25 mL 去离子水中,用磁力搅拌器在60 ℃下搅拌30 min 后,与上述溶液混合搅拌30 min;将所得溶液置于聚四氟乙烯内衬的反应釜中,在烘箱中145 ℃下晶化48 h;冷却至室温后抽滤、洗涤、烘干,将所得产物置于马弗炉中550 ℃下煅烧6 h,即可得到Fe-Mn-MCM-41.1.2.2 催化氧化实验实验中所用反应装置如图1所示,采用了1.2 L 柱形双层玻璃反应器.臭氧由臭氧发生器产生,通过反应器底部的多孔筛板进入柱状玻璃器内,实验中多余的臭氧用硫代硫酸钠溶液吸收.通过臭氧发生器的旋钮调节臭氧流速为100 mg/h,气体流量为1.0 min/L.反应温度由恒温水浴器控制在298 K.使用1 mg/L KBr 水溶液作为模拟含溴废水,在溶液初始pH、叔丁醇、磷酸盐以及催化臭氧氧化控制溴酸盐生成的机理实验中使用的反应溶液均为1 mg/L KBr 溶液;催化剂的质量浓度为1.0 g/L.实验中使用0.1 mol/L NaOH 和0.1 mol/L HCl 调节pH,如果无特别说明,实验的初始pH 均为7.0.图1 催化实验装置Figure 1 The schema of the experimental apparatus for ozonation( 1) 溶液初始pH 的影响: 用0.1 mol/L NaOH和0.1 mol/L HCl 分别调节KBr 水溶液( 1 mg/L) pH为5.0、7.0、9.0,在反应进行到0、5、15、30、45 和60 min时对单独臭氧氧化和催化臭氧氧化中溴酸盐的生成量进行研究.(2) 叔丁醇的影响:向1 L 1 mg/L KBr 溶液中分别加入0.1、0.5 mmol/L TBA,臭氧流速为100 mg/h,在反应进行到60 min 时,对体系中产生的溴酸盐进行分析.( 3) 磷酸盐的影响: 向1 L 的1 mg/L KBr 溶液中分别加入1、5、10 mg/L 磷酸盐溶液,臭氧流速为100 mg/h,在反应进行到0、5、15、30、45 和60 min时,对体系中产生的溴酸盐( 取样时间为60 min) 和过氧化氢( 取样时间为0、5、15、30、45 和60 min) 的质量浓度进行分析.( 4) Fe-Mn-MCM-41 催化臭氧氧化控制溴酸盐生成的机理探究:向1 L 1mg/L KBr 溶液中投加流速为100 mg/h 的臭氧,当反应进行到0、5、15、30、45 和60 min 时,对体系中产生的次溴酸根和臭氧的质量浓度进行分析.( 5) Fe-Mn-MCM-41 对草酸的去除效果: 向反应器中加入1 L 草酸( 1 mg/L) 溶液后,调节臭氧流速为100 mg/h,迅速加入1g/L Fe-Mn-MCM-41 催化剂,在反应进行到0、5、15、30、45 和60 min 时,对体系中草酸的质量浓度进行分析.1.2.3 分析方法 ( 1) 所有水样都经过0.45 μm 的滤膜过滤.液相臭氧浓度使用臭氧浓度检测仪测定.(2)离子和Br-的质量浓度通过美国Dionex 产ICS-900 型离子色谱仪( 配电导检测器) 测定,色谱柱为AS18( 250 mm×4 mm) . ( 3) HOBr/OBr-的质量浓度采用液相色谱仪检测,色谱柱为C18 柱( 150 mm×4.6 mm) ,测定条件: 流动相为VA∶VB=0.4∶0.6的混合液,其中A 为含乙酸水溶液( 体积分数为0.2%) ,B 为甲醇,流速设置为1.0 mL/min,检测波长为225 nm,进样量为100 μL.溴离子与溴酸根采用离子色谱测定;次溴酸与次溴酸根的检测需与苯酚反应后采用高效液相色谱检测. ( 4) 过氧化氢质量浓度的检测在波长551 nm 处采用DPD 分光光度法检测[12]. ( 5) 草酸质量浓度的检测采用液相色谱仪,色谱柱为C18 柱( 150 mm×4.6 mm) .测定条件: 使用超纯水作为流动相( 加入0.018 mol/L 磷酸二氢钾调节pH 至2.5) ,流速设置为1.0 mL/min,检测波长为210 nm,进样量为100 μL.2 结果与讨论2.1 Fe-Mn-MCM-41 催化臭氧氧化控制含溴水中溴酸盐生成的影响因素2.1.1 溶液初始pH 的影响由于溴酸盐在pH<5.0的情况下几乎不产生[1],因此分别对溶液初始pH=5.0、7.0、9.0 时单独臭氧氧化和催化臭氧氧化中溴酸盐的生成量进行研究.向1.0 L 反应溶液中加入1.0 g Fe-Mn-MCM-41 后,溴酸盐的生成量随pH 升高显著增加( 图2) .与单独臭氧氧化相比,催化臭氧氧化中溴酸盐生成量明显降低,抑制率在pH=5.0、7.0和9.0时分别为85.9%、67.7%和73.2%,说明在不同pH 条件下,当反应进行到60 min时,催化臭氧氧化过程都能够有效抑制溴酸盐的生成. pH通过改变臭氧稳定性和HOBr/OBr的存在形态来影响溴酸盐的生成.在酸性条件下,尽管臭氧更加稳定[13],但HOBr/OBr-主要以HOBr 的形式存在( pKa=9)[14]. HOBr 与臭氧的反应速率很慢,k<0.013 mol/( L·s)[14],而与羟基自由基的反应较快,因此由HOBr 反应溴酸盐生成的途径被阻止,导致溴酸盐生成量减少.而在碱性条件下,HOBr/OBr-则更多以OBr-的形式存在,OBr-既能够与臭氧反应,又能与羟基自由基反应,因此促进了溴酸盐的生成[15].图2 初始pH 对Fe-Mn-MCM-41/O3抑制溴酸盐生成质量浓度的影响Figure 2 The influence of the initial pH on inhibition of bromate formation during the Fe-Mn-MCM-41/O3process2.1.2 叔丁醇的影响叔丁醇( TBA) 是一种典型的羟基自由基捕获剂,为探究羟基自由基对Fe-Mn-MCM-41催化臭氧氧化过程抑制溴酸盐生成的影响,分别加入0.1、0.5 mmol/L TBA,体系中生成溴酸盐的质量浓度如图3 所示.图3 叔丁醇( TBA) 对单独臭氧氧化和催化臭氧氧化系统中溴酸盐生成质量浓度的影响Figure 3 The influence of TBA on bromate formation during theO3and Fe-Mn-MCM-41/O3processes在单独臭氧氧化和催化臭氧氧化2 种体系下投加TBA 均会大幅降低溴酸盐的生成量( 图3) .加入0.1 mmol/L TBA 后,单独臭氧氧化中溴酸盐的质量浓度减少67.7%,而催化臭氧氧化中溴酸盐质量浓度减少81.1%.溴酸盐的产生与臭氧和羟基自由基密切相关.当羟基自由基被TBA 捕获之后,溶液中自由基数量减少,通过间接途径产生的溴酸盐也随之减少.同时,催化剂的加入使溶液中臭氧更容易分解为羟基自由基,导致参与反应的臭氧浓度降低,阻碍了臭氧氧化溴离子生成HOBr/OBr-,进一步减少溴酸盐的生成.2.1.3 磷酸盐的影响磷元素通常以磷酸盐的形式存在于天然水体中,绝大部分是由于人类活动的加剧引入的,生活污水中无机磷质量浓度为4 ~15 mg/L [16].磷酸盐在催化臭氧氧化过程中对溴酸盐的影响是研究的关键.在反应体系中分别加入1、5、10 mg/L 磷酸盐,检测生成溴酸盐的质量浓度.如图4 所示,在单独臭氧氧化和催化臭氧氧化中,磷酸盐的加入均会减少溴酸盐的生成量,而且溴酸盐生成量随磷酸盐投加量的增加而减少.在单独臭氧氧化中,加入1、5、10 mg/L 磷酸盐,溴酸盐分别减少29.6%、44.6%和59.7%.而在催化臭氧氧化中,溴酸盐分别减少82.5%、86.4%和90.3%.由此可见,磷酸盐对单独臭氧氧化和催化臭氧氧化中溴酸盐的生成有抑制作用,并且抑制率随磷酸盐质量浓度的增加而升高.图4对单独臭氧氧化和催化臭氧氧化系统中溴酸盐生成质量浓度的影响Figure 4 The influence ofon bromate formation during the O3and Fe-Mn-MCM-41/O3processes为进一步研究磷酸盐对溴酸盐的抑制机理,对反应过程中H2O2浓度( 溴酸盐抑制物) 进行检测[17]( 图5) .如图5 所示,催化臭氧氧化过程中H2O2质量浓度高于单独臭氧氧化过程,加磷酸盐的情况下产生H2O2的质量浓度高于不加磷酸盐的情况.如反应5 min 时催化臭氧氧化过程中H2O2的质量浓度为242.85 μg/L,而单独臭氧氧化过程中H2O2的质量浓度仅98.76 μg/L,说明催化剂的加入会促进H2O2的产生. H2O2质量浓度随磷酸盐质量浓度的升高而增加,在单独臭氧氧化过程中加入1、5、10 mg/L磷酸盐反应5 min 时体系中分别产生了106.86、116.57、136.00 μg/L H2O2,而在催化臭氧氧化过程中H2O2的质量浓度分别为286.57、294.67、299.52 μg/L.以上结果说明,催化剂和磷酸盐的加入都会促进H2O2的生成:·OH+·OH→H2O2( 1)Fe-Mn-MCM-41 的加入会促进溶液中臭氧分解产生更多羟基自由基[17],从而加快H2O2的生成速率,降低臭氧浓度,减少在直接氧化过程生成的溴酸盐;另一方面H2O2还能够消耗更多HOBr[17]:H2O2+HOBr →H++Br-+H2O+O2( 2)H2O2先将其还原为溴离子,从而降低溴酸盐的生成[14].而磷酸盐会促进羟基自由基生成H2O2,同时消耗HOBr,减少溴酸盐的生成量.据报道,在高铁酸盐氧化溴离子生成溴酸盐的体系中[18],磷酸盐的加入也会促进H2O2的生成,与本实验结果一致.另外,对臭氧氧化过程中溴酸盐生成的抑制作用还可以从它与催化剂表面的作用机制分析,这可能与磷酸根和三价铁形成的络合物有关.据文献报道,磷酸根会与催化剂中的含铁物质( 如Fe( Ⅱ) 、Fe( Ⅲ) 或Fe( Ⅳ) ) 形成络合物,由于生成的络合物活性不同,会造成过氧化氢的累积和活性物质生成量减少,最终减少溴酸盐的生成量.图5 单独臭氧氧化和催化臭氧氧化系统过程中H2O2的质量浓度Figure 5 The concentration of H2O2in the O3and Fe-Mn-MCM-41/O3processes 2.2 Fe-Mn-MCM-41 催化臭氧氧化控制溴酸盐生成的机理探究2.2.1 次溴酸根的浓度变化为探究Fe-Mn-MCM-41 催化臭氧氧化过程对溴酸盐的抑制机理,对单独臭氧氧化和催化臭氧氧化中产生的HOBr/OBr-进行检测[16].据报道,由溴离子生成HOBr/OBr-的过程是反应的控制步骤,次溴酸一旦生成就会迅速被氧化为溴酸盐[19].因此,减少HOBr/OBr-的生成量是抑制溴酸盐的控制手段之一,而减少HOBr/OBr-生成量的方法通常是引入外加碳源,使其与溴离子争夺溶液中的臭氧以达到降低HOBr/OBr-生成量的目的[16],因此会引入二次污染.如图6 所示,加入Fe -Mn-MCM-41后,HOBr/OBr-的生成量比单独臭氧氧化中的少,进一步说明催化剂的加入会阻碍溴离子氧化生成次溴酸的过程.与其他方法相比,Fe-Mn -MCM-41 的加入既可以抑制溴酸盐的生成,又不会引起水体二次污染.图6 单独臭氧氧化和催化臭氧氧化系统中HOBr/OBr-的质量浓度Figure 6 The concentration of HOBr/OBr-in the O3and Fe-Mn-MCM-41/O3processes2.2.2 臭氧质量浓度的变化在非均相催化臭氧氧化反应中,臭氧在催化剂表面的吸附和分解对于形成羟基自由基是关键步骤.对于臭氧氧化含溴水生成的反应来说,臭氧在液相中的平衡浓度与羟基自由基的形成以及通过直接氧化生成的有关[20].臭氧的平衡浓度如图7 所示,加入Fe-Mn-MCM-41 后,液相中臭氧质量浓度明显低于单独臭氧氧化过程,这说明铁锰双金属掺杂MCM-41 对臭氧的分解有促进作用.据报道[21],掺杂金属后,更多臭氧会在Lewis 酸性位点上分解,电子转移也有利于·OH的生成.正是因为在Fe-Mn-MCM-41/O3中的臭氧质量浓度更低,使Br-→HOBr/OBr-和的过程受到抑制.图7 单独臭氧氧化和催化臭氧氧化过程中臭氧的质量浓度Figure 7 The concentration of ozone in the Fe-Mn-MCM-41/O3and O3processes 2.3 Fe-Mn-MCM-41 对草酸的去除效果溴酸盐的生成与体系的氧化能力有关.草酸是有机物降解过程中常见的中间产物,将草酸作为目标污染物,研究了Fe-Mn-MCM-41 催化臭氧氧化过程对草酸的去除效果( 图8) . Fe-Mn-MCM-41 的加入能够显著提高草酸的去除率,单独臭氧氧化对草酸的降解率为10.2%,催化臭氧氧化过程臭氧的降解率则升高至89.0%,证明Fe-Mn-MCM-41 不仅能够在臭氧氧化过程中抑制溴酸盐的生成,还能提高对草酸的去除率.图8 Fe-Mn-MCM-41/O3和O3过程对草酸的去除率Figure 8 The efficiency of oxalic acid removal in the Fe-Mn-MCM-41/O3andO3processes3 结论以水热法合成的Fe、Mn 双金属掺杂MCM-41( Fe-Mn-MCM-41) 为催化剂,研究了不同pH 对Fe-Mn-MCM-41 催化臭氧氧化过程抑制溴酸盐的影响以及对溴酸盐的抑制机理.( 1) 在初始pH 为5.0 ~9.0 的范围内,Fe-Mn-MCM-41 催化臭氧氧化过程表现出良好的溴酸盐抑制效果.( 2) 在单独臭氧氧化和催化臭氧氧化2 种体系中加入磷酸盐均会抑制溴酸盐的生成.( 3) Fe-Mn-MCM-41 的加入会减少HOBr/OBr-的生成量,同时加快臭氧分解为羟基自由基,进一步生成H2O2,而H2O2可以消耗HOBr 并将其还原为Br -,达到抑制溴酸盐生成的目的.参考文献:【相关文献】[1] RICHARDSON S D,PLEWA M J,WAGNER E D,et al.Occurrence,genotoxicity,and carcinogenicity of regulated and emerging disinfection by-products in drinking water:A review and roadmap for research[J]. Mutation Research/reviews in Mutation Research,2007,636( 1) :178-242.[2] JI H,WU W,LI F,et al. Enhanced adsorption of bromate from aqueous solutions on ordered mesoporous Mg-Al layered double hydroxides ( LDHs) [J]. Journal of Hazardous Materials,2017,334:212-222.[3]张建琳,陈硕,全燮,等.介孔二氧化锰制备及其催化臭氧氧化草酸研究[J].大连理工大学学报. 2017,57( 5) :447-452.ZHANG J L,CHEN S,QUAN X,et al. Preparation of mesoporous manganese dioxide and its catalytic ozonation of oxalic acid[J]. Journal of Dalian University of Technology,2017,57( 5) :447-452.[4] XIE L,SHANG C. Effects of copper and palladium on the reduction of bromate by Fe( 0) [J]. Chemosphere,2006,64( 6) :919-930.[5] NIE Y,LI N,HU C. Enhanced inhibition of bromate formation in catalytic ozonation of organic pollutants over Fe-Al LDH/Al2O3[J]. Separation and Purification Technology,2015,151:256-261.[6] SELCUK H,SARIKAYA H Z,BEKBOLET M,et al. Bromate formation on the non-porous TiO2photoanode in the photoelectrocatalytic system[J]. Chemosphere,2006,62( 5) :715-721.[7] BING J,HU C,NIE Y,et al. Mechanism of catalytic ozonation inFe2O3/Al2O3@SBA-15 aqueous suspension for destruction of ibuprofen[J]. Environmental Science &Technology,2015,49( 3) :1690-1697.[8] LAHA S,KUMAR R. Promoter-induced synthesis of MCM-41 type mesoporous materials including Ti-and VMCM-41 and their catalytic properties in oxidation reactions[J]. Microporous and Mesoporous Materials,2002,53,163-177.[9]李来胜,谢燕华,潘兆琪,等. Ce-MCM-41 分子筛催化臭氧氧化水中腐殖酸[J].华南师范大学学报( 自然科学版) ,2017,49( 2) :73-79.LI L S,XIE Y H,PAN Z Q,et al. Mineralization of humic acid in water by catalytic ozonation with Ce-MCM-41[J]. Journal of South China Normal University( Natural Science Edition) ,2017,49( 2) :73-79.[10]唐莉莉,刘杰,黄瑞欢,等. Fe/MCM-41 催化臭氧氧化水中对氯苯甲酸的研究[J].华南师范大学学报( 自然科学版) ,2013,45( 2) :74-78.TANG L L,LIU J,HUANG R H,et al. Catalytic ozonation of p-chlorobenzoic acid in aqueous solution by Fe/MCM-41[J]. Journal of South China NormalUniversity( Natural Science Edition) ,2013,45( 2) :74-78.[11]CHEN W,LI X,LIU M,et al. Effective catalytic ozonation for oxalic acid degradation with bimetallic Fe-Cu-MCM-41:operation parameters and Mechanism [J]. Journal of Chemical Technology & Biotechnology,2017,92( 11) :2862-2869.[12]BADER H,STURZENEGGER V,HOIGN J. Photometric method for the determination of low concentrations of hydrogen peroxide by the peroxidase catalyzed oxidation of N,N-diethyl-p-phenylenediamine ( DPD) [J]. Water Research,1988,22( 9) :1109-1115.[13] KASPRZYKHORDERN B,ZIOLEK M,NAWROCKI J.Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment[J]. Applied Catalysis B:Environmental,2003,46( 4) :639-669.[14]VON G U,HOIGNE J. Bromate formation during ozonization of bromide-containing waters:interaction of ozone and hydroxyl radical reactions[J]. Environmental Science &Technology,1994,28(7) :1234-1242.[15]VON G U,OLIVERAS Y. Advanced oxidation of bromidecontaining waters? bromate formation mechanisms[J]. Environmental Science & Technology,1998,32( 1) :63-70.[16]SONG R,MINEAR R,WESTERHOFF P,et al. Bromate formation and control during water ozonation[J]. Environmental Technology,1996,17( 8) :861-868.[17] STAEHELIN J,HOIGNE J. Decomposition of ozone in water: rate of initiation by hydroxide ions and hydrogen peroxide[J]. Environmental Science and Technology,1982,16( 10) :676-681.[18]HUANG X,DENG Y,LIU S,et al. Formation of bromate during ferrate( VI) oxidation of bromide in water[J].Chemosphere,2016,155:528-533.[19]SAYATO Y,NAKAMURO K,HAYASHI M,et al. Formation of active bromine from bromide ion by aqueous ozonation[J]. Chemosphere,1990,20( 3) :309-315.[20] VON G U. Ozonation of drinking water: disinfection and by-product formation in presence of bromide,iodide or chlorine[J]. Water Research,2003,37( 7) :1469-1487.[21] ZHAO L,SUN Z,MA J. Novel relationship between hydroxyl radical initiation and surface group of ceramic honeycomb supported metals for the catalytic ozonation of nitrobenzene in aqueous solution[J]. Environmental Science & Technology,2009,43( 11) :4157-4163.。
臭氧催化剂(臭氧催化氧化)
臭氧催化剂臭氧是一种强氧化剂,常见应用于水处理,主要应用于水的杀菌、消毒,去除色度等功能,被社会所广泛接受并大范围工业化应用。
臭氧的水溶性很差,应用于废水处理的利用率一直很差,大部分臭氧未有得到充分的利用就直接排放到尾气破坏器。
为了提高臭氧的利用率,人们开始采取尽可能缩小臭氧气泡来提高与水的接触面积,通过大的比表面积进而提高臭氧利用率。
微孔曝气、微气泡、微纳米气泡等形式,但气泡的极限决定提高利用率需要进一步改进。
臭氧催化剂的发现与引入在微气泡基础上大幅度提高了臭氧利用率与臭氧的有机物矿化能力。
当前常见的臭氧催化剂按基体区分可分为:碳基、陶基、铝基、复合载体。
中文名臭氧催化剂外文名Ozone catalyst一、发展历史(1)催化剂的萌芽状态(碳基催化剂):最早期的臭氧催化剂采取比表面积较大的活性炭载体(碳基催化剂),此载体具有较大的比表面积,可以大幅度提升催化组分与水的接触面积,进而提升催化效果。
但由于碳基的先天强度差问题,造成基体的磨损损耗较大;物理结合对催化组分的固化作用力差,催化组分易流失;加上碳基自身具备的强大吸附效果,不易区分是催化还是吸附作用,致使很多项目设计失败或者失效,被市场慢慢所淘汰。
(2)催化剂的改进时期(陶基催化剂):因为碳基的强大吸附效果容易误导设计参数,后期引入陶粒载体催化剂(陶基),吸附作用得到了大大的排除,设计参数参考意义得到了提升,设计的稳妥性得到了改变与认可,激发了臭氧催化剂大范围使用的新时期。
开始中等水量应用于污水厂的脱色,去除有机物。
(3)催化剂的兴盛时期(铝基催化剂)陶基催化剂优化了碳基基体的吸附问题,但碳基载体、陶基载体与催化组分都是物理结合,都存在对催化组分固化不强,易流失问题;且载体强度都相对较差,损耗较大,年补充量高达20%-50%,甚至需要每年更换一次,催化成本一直居高不下。
铝基催化载体的发现改变了传统的载体与催化组分物理结合状态,都为金属类物质,实现了载体与催化组分的合金化学键结合,催化组分稳固不易流失;金属结构也大大提高了载体强度,实现了更高的填充高度,节约了设备与土地投资。
一种新型的水处理高级氧化技术——金属催化臭氧化
W ATER REATⅣIN I 一 Ⅱ AL —CATA T 】 ' I I C 0Z0NATI ON
F NG T o L U n —b , U Yu E a , I Ho g o S N e—f g e n
fco EvomnSecadTc og,i iUiry , 307,7 J h lf nr e i e n h lyr qn nei, 咖 002C S oo in tcn e no a vs t l
样 ,金属催化臭氧化技术也是利用反应过程中产 生氧化性 自由基 ( 羟基 自由基 ) 来氧化分解水中的 有机物从而达到水质净化。根据所用催化剂物相 的不同, 此项技术大致 可分为两类 : 利用溶液中金 属 ( 离子态 )的均相金属催化臭氧化和以固体金
染料废水 的臭氧化脱色过程。 nr z 晤 在研究 A de z 等 o i
金属催化臭氧化技术是近年发展起来的一种
1 均相金属催化臭氧化
A d [ 等对染料废水进行研究 时发现 硝酸 bo 1 7 银、硫酸锌或硫酸铜和三氧化二铬的加入加快了
在常温常压下将那些难 以用臭氧单独氧化的有机 物氧化降解的新型高级氧化技术 。同其它高级氧
化技 术 如 0/ 2 2 V 0 、 i l 和 U /  ̄ 一 3H 0 、 / 3TO Ⅳ / U V HO2
程 中反应机理 、 影响因素的研 究等。 关键词 : 催化臭氧化 ; 催化剂; 水处理 中图分类号: 73 X 0 文献标识码 : A 文章编号 : 0 .7920 ) .03 0 1 6 85 (060 02 —4 0 4
臭氧催化氧化在工业废水处理中的应用进展
臭氧催化氧化在工业废水处理中的应用进展臭氧催化氧化在工业废水处理中的应用进展一、引言随着工业化的快速发展,工业废水污染问题日益突出。
为了减少工业废水对环境的影响,保护生态环境,提升水质安全,各国纷纷加大了工业废水处理技术研究的力度。
臭氧催化氧化作为一种高效能、低成本的工业废水处理技术,近年来得到了广泛关注和应用。
本文将重点讨论臭氧催化氧化在工业废水处理中的应用进展。
二、臭氧催化氧化的原理臭氧催化氧化利用臭氧高强氧化能力,通过催化剂的作用,将废水中的有机污染物氧化为无害的物质。
臭氧分解产生的氧化自由基能够迅速与有机物接触并氧化分解,达到净化水质的目的。
催化剂作为活性剂,能够提高臭氧催化氧化的效果,减少能源消耗。
三、臭氧催化氧化在工业废水处理中的应用1. 有机废水处理有机废水中的有机物如苯系物质、酚类化合物等,对环境具有潜在的危害性。
臭氧催化氧化技术可将有机废水中的有机物完全氧化为二氧化碳和水,达到高效净化的效果。
此外,催化剂的使用还可以提高臭氧消耗效率,降低处理成本。
2. 重金属废水处理重金属废水是工业废水中常见的一种类型,其含有高浓度的重金属离子,对环境和生物体有严重的毒害作用。
臭氧催化氧化技术能够将重金属离子与氧化自由基进行络合,使其转化为难溶于水的沉淀物,提高重金属废水的处理效果。
3. 染料废水处理染料废水中的有机染料含有毒性、耐酸碱、难降解的特点,对水体造成严重的污染。
臭氧催化氧化技术能够通过氧化分解有机染料,将其转化为无害物质。
此外,臭氧催化氧化还具有剩余臭氧消灭能力,可以将处理后的废水中的剩余臭氧进行降解,确保排放的废水符合排标要求。
4. 脂肪酸废水处理脂肪酸废水中的酸性物质含量较高,对地下水和生态环境造成潜在威胁。
臭氧催化氧化技术通过氧化酸性物质,将其转变为无害物质,有效净化脂肪酸废水。
四、臭氧催化氧化的优势和挑战1. 优势臭氧催化氧化技术具有高效、低成本、无二次污染等优势。
臭氧氧化效率高,氧化后产物无二次污染,能够有效净化废水。
aop臭氧催化氧化
aop臭氧催化氧化
AOP(Advanced Oxidation Processes)是一种将氧化作用用于水处理的技术。
在AOP中,臭氧催化氧化是一种常用的方法之一。
臭氧催化氧化利用臭氧与反应物接触,产生氧化反应以去除有机物、有毒物质和微污染物。
在催化氧化反应中,臭氧分子中的活性氧被释放出来,与污染物发生反应,从而将其转化为无害的产物。
催化剂通常用于提高臭氧分解和反应速率,以增加氧化能力和效果。
臭氧催化氧化在污水处理、废水处理、水源净化和空气净化等领域中被广泛应用。
它可以有效去除水中的有机物、有毒物质和微污染物,提高水质的净化效果。
此外,臭氧催化氧化也可以用于空气净化,可以去除空气中的有害气体和恶臭。
然而,臭氧催化氧化也存在一些挑战和限制,如高能耗、催化剂的选择和稳定性以及副产物的处理等。
因此,在实际应用中需要综合考虑各种因素,选择适当的AOP方法和操作条件,以获得最佳的氧化效果。