药物化学(第二版)第2章 药物设计的基本原理和方法
药物化学
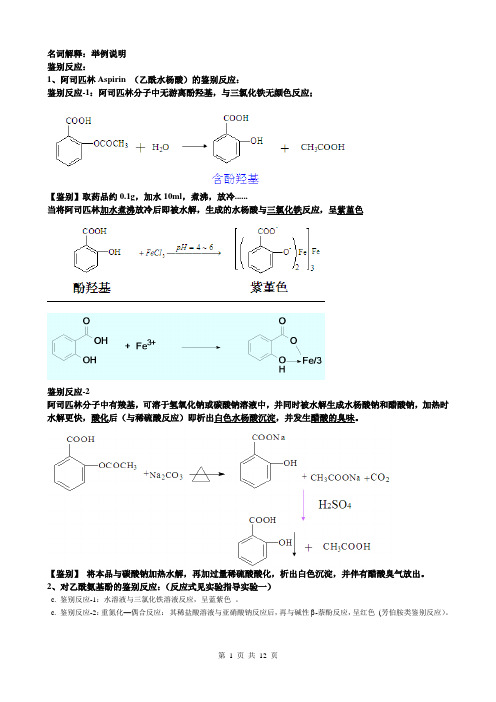
名词解释:举例说明鉴别反应:1、阿司匹林Aspirin (乙酰水杨酸)的鉴别反应:鉴别反应-1:阿司匹林分子中无游离酚羟基,与三氯化铁无颜色反应;【鉴别】取药品约0.1g,加水10ml,煮沸,放冷......当将阿司匹林加水煮沸放冷后即被水解,生成的水杨酸与三氯化铁反应,呈紫堇色鉴别反应-2阿司匹林分子中有羧基,可溶于氢氧化钠或碳酸钠溶液中,并同时被水解生成水杨酸钠和醋酸钠,加热时水解更快,酸化后(与稀硫酸反应)即析出白色水杨酸沉淀,并发生醋酸的臭味。
【鉴别】将本品与碳酸钠加热水解,再加过量稀硫酸酸化,析出白色沉淀,并伴有醋酸臭气放出。
2、对乙酰氨基酚的鉴别反应:(反应式见实验指导实验一)c. 鉴别反应-1:水溶液与三氯化铁溶液反应,呈蓝紫色。
c. 鉴别反应-2:重氮化—偶合反应:其稀盐酸溶液与亚硝酸钠反应后,再与碱性 -萘酚反应,呈红色(芳伯胺类鉴别反应)。
3、羟基保泰松(3,5-吡唑烷二酮类)的鉴别反应: 酸水解后重排,呈芳伯氨反应,与亚硝酸钠试液作用生成黄色重氮盐,再与β-萘酚偶合生成橙色沉淀4、 吲哚美辛的鉴别反应:本品的氢氧化钠溶液与重铬酸钾溶液和硫酸反应,呈紫色 ;(芳基乙酸类) 与亚硝酸钠和盐酸反应,呈绿色,放置后渐变黄色。
5、吗啡的鉴别反应:(1) 吗啡+中性FeCl 3 →蓝色 (2)吗啡+铁氰化钾→伪吗啡 + 亚铁氰化钾亚铁氰化铁 —— 蓝色可待因无此反应,可用作鉴别(3) 吗啡+甲醛硫酸试液→蓝紫色(Marquis 反应)(4) 吗啡+钼酸铵硫酸溶液→紫色,继变蓝色,最后变为绿色(Fröhde 反应)(5)对吗啡要进行杂质的限量检查6、肾上腺素(Epinephrine )的鉴别反应:(1)溶于稀盐酸后,与过氧化氢试液反应被氧化,显血红色。
(2)在pH3-3.5时与碘试液反应,再加硫代硫酸钠试液使过量碘的颜色消退,溶液呈红色。
(3)与三氯化铁试液反应,即显翠绿色(酚羟基与铁离子络合呈色);再加氨试液后变为紫色,最后变为紫红色。
药物化学药物设计的基本原理和方法共89页PPT

71、既然我已经踏上这条道路,那么,任何东西都不应妨碍我沿着这条路走下去。——康德 72、家庭成为快乐的种子在外也不致成为障碍物但在旅行之际却是夜间的伴侣。——西塞罗 73、坚持意志伟大的事业需要始终不渝的精神。——伏尔泰 74、路漫漫其修道远,吾将上下而求索。——屈原 75、内外相应,言行相称。——韩非
药物化学药物设计的基本原理和方法
31、园日涉以成趣,门虽设而常关。 32、鼓腹无所思。朝起暮归眠。 33、倾壶绝余沥,窥灶不见烟。
34、春秋满四泽,夏云多奇峰,秋月 扬明辉 ,冬岭 秀孤松 。 35、丈夫志四海,
药物化学-02-第二章_新药研究与开发概论
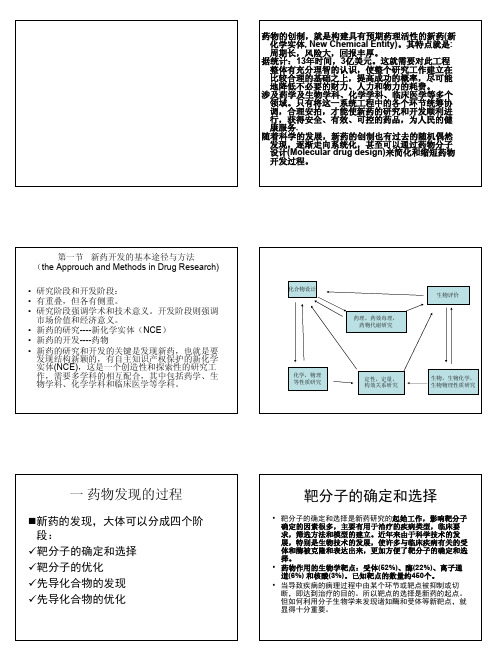
第二章新药研究与开发概论药物的创制,就是构建具有预期药理活性的新药(新化学实体, New Chemical Entity)。
其特点就是: 周期长,风险大,回报丰厚。
据统计:13年时间,3亿美元。
这就需要对此工程整体有充分理智的认识,使整个研究工作建立在比较合理的基础之上,提高成功的概率,尽可能地降低不必要的财力、人力和物力的耗费。
涉及药学及生物学科、化学学科、临床医学等多个领域。
只有将这一系统工程中的各个环节统筹协调,合理安拍,才能使新药的研究和开发顺利进行,获得安全、有效、可控的药品,为人民的健康服务.随着科学的发展,新药的创制也有过去的随机偶然发现,逐渐走向系统化,甚至可以通过药物分子设计(Molecular drug design)来简化和缩短药物开发过程。
第一节新药开发的基本途径与方法(the Approuch and Methods in Drug Research)•研究阶段和开发阶段:•有重叠,但各有侧重。
•研究阶段强调学术和技术意义。
开发阶段则强调市场价值和经济意义。
•新药的研究----新化学实体(NCE)•新药的开发----药物•新药的研究和开发的关键是发现新药,也就是要发现结构新颖的,有自主知识产权保护的新化学实体(NCE),这是一个创造性和探索性的研究工作,需要多学科的相互配合,其中包括药学、生物学科、化学学科和临床医学等学科。
化合物设计药理,药效毒理,药物代谢研究化学,物理等性质研究定性,定量,构效关系研究生物,生物化学,生物物理性质研究生物评价药物研究中中各研究分支学科之间的相互作用和关系一药物发现的过程新药的发现,大体可以分成四个阶段:9靶分子的确定和选择9靶分子的优化9先导化合物的发现9先导化合物的优化靶分子的确定和选择•靶分子的确定和选择是新药研究的起始工作,影响靶分子确定的因素很多,主要有用于治疗的疾病类型,临床要求,筛选方法和模型的建立。
近年来由于科学技术的发展,特别是生物技术的发展,使许多与临床疾病有关的受体和酶被克隆和表达出来,更加方便了靶分子的确定和选择。
药物化学期末复习

绪论1、药物化学(Medicinal Chemistry)是关于药物的发现、发展和确证,并在分子水平上研究药物作用方式的一门学科。
2、药物是对疾病具有预防、治疗和诊断作用或用以调节机体生理功能的物质。
3、根据药物的来源和性质不同,可以分为中药或天然药物、化学药物和生物药物。
4、化学药物是一类既有药物的功效,同时又有确切的化学结构的物质。
5、药物化学的三个时期:以天然产物为主的发现时期、以合成药物为主的发展时期、药物分子设计时期。
6、1899年,阿司匹林上市,标志着药物化学学科的形成。
第一章:新药研究和开发概论1、新化学实体(New Chemical Entities)是指在以前的文献中没有报道过的新化合物。
而有可能成为药物的新化学实体则需要时能够以安全和有效的方法治疗疾病的新化合物。
2、通常新药的发现分为4个主要的阶段:靶分子的确定和选择、靶分子的优化、先导化合物的发现和先导化合物的优化。
3、药品质量的主要含义是:A、药物的疗效和毒副作用,B、药物的纯度。
4、药品质量标准中,有两个重要的指征:一是药物的纯度,即有效成分的含量;二是药物的杂质限度。
5、药物的商品名通常是针对药物的最终产品,即剂量和剂型已确定的含有一种或多种药物活性成分的药品。
含同样活性成分的同一药品,每个企业应有自己的商品名,不得冒用、顶替别人的药品商品名称。
6、药物的通用名:也称为国际非专利药品名称,是世界卫生组织推荐使用的名称,通常是指有活性的药物物质,而不是最终产品,因此是药学研究人员和医务人员使用的共同名称,所以一个药物只有一个药品通用名,比商品名使用起来更为方便。
第二章:药物设计的基本原理和方法1、目前新药设计的靶点集中在受体、酶、核酸、离子通道和基因等上。
2、先导化合物(Lead Compound):通过各种途径得到的具有一定生理活性的化学物质。
3、先导化合物的发现方法和途径:a、从天然产物活性成分中发现先导化合物;b、通过分子生物学途径发现先导化合物;c、通过随机机遇发现先导化合物;d、从代谢产物中发现先导化合物;e、从临床药物的副作用或者老药新用途中发现新药;f、从药物合成中间体中发现先导化合物;g、通过计算机辅助药物筛选寻找先导化合物。
药物化学第2章 新药研究的基本原理与方法题库

第2章新药研究的基本原理与方法选择题每题1分
第2章新药研究的基本原理与方法填空题1每空1分
填空题2 每空1分
填空题3每空1分
第2章新药研究的基本原理与方法概念题每题2分
第2章新药研究的基本原理与方法问答与讨论题每题6分
前列腺素E2(PGE2)为结晶固体,但室温稳定期短,几个月内可迅速分解,不稳定因素是C-11位羟基易在酸性条件下,发生消除反应生成前列腺素A2(PGA 2) 这也是其口服无效的主因。
请设计两种较为稳定的衍生物。
举例说明根据受体结构进行药物分子设计
HIV蛋白水解酶催化机理
根据催化机理设计的HIV蛋白水解酶抑制剂
第2章新药研究的基本原理与方法合成/代谢/反应/设计题每题6分。
药物设计药物设计原理和方法

第18页/共71页
适宜衍生化的功能基及衍生化物:
醇羟基或酚羟基可衍生成酯或活泼醚;羧基可衍生成酯或酰胺;氨基(包括胺、酰胺和亚胺)可衍生成烷氧羰酰胺
第19页/共71页
2.4.1 提高生物利用度的前药
若原药分子中含有羟基、羧基或磷酸基,会因极性强或带有电荷而难以吸收。应用前药原理引入酯键可提高脂溶性,改善了脂/水分配系数,有利肠道吸收,或克服了首过效应。
第23页/共71页
2.4.2 增加水溶性的前药
有些酸性、碱性或盐类药物因为水溶性很小,不便制成注射剂使用;有些则会影响吸收。一般可将其制成适当的水溶性盐类。
第24页/共71页
2.4.2.1 磺胺嘧啶钠:将水溶性差的磺胺嘧啶制成钠盐,可增大水溶性,供注射用。
第25页/共71页
2.4.2.2 强力霉素盐酸盐:强力霉素微溶于水,制成水溶性好的盐酸盐,供注射用。
第47页/共71页
软药:是指一类本身有治疗效用或生物活性的化学实体,当在体内呈现药效并达到治疗目的后,按预料的代谢途径和可控的代谢速率的代谢, 转变成无毒、无活性的代谢物。
第48页/共71页
软药通常是在局部呈现药理作用,若分布或扩散到其他部位时,会迅速代谢失活,从而避免出现不良反应和毒性。
软药所发生的失活过程是单一的低能量和高容量的酶促反应
3.3.1 脂肪族三价等排体最常见的三价电子等排体是-CH=与-N=的变换,广泛应用于新药设计中。
第1页/共71页
3.3.1.1 胆固醇胆固醇的C20和C25被N替换,得化合物(2)是体内胆固醇生物合成的强效抑制剂。
第2页/共71页
3.3.1.2 抗抑郁药地昔帕明(3)、去甲替林(4)和普罗替林(5)是三环类抗精神病药,相互之间是三价原子等排变换。
药物化学第二章化学结构与药理活性课件
肾小管壁、肾小球、血脑屏障等。
1、生物膜的结构特点: A、细胞膜均由含量各占一半左右的脂质和蛋白质组成,还有 少量的糖、核酸、金属离子等; B、膜中的脂质主要是磷脂,呈双分子层,起支架作用,头部 为亲水性,为磷酸甘油基团,向(膜)外表;未部为两条 尾巴的疏水性,为脂肪酸链,向(膜)内部; C、蛋白质镶嵌在脂质分子中,有亲水性基团(极性),向外 表;有疏水性集团(非极性),向内部; D、 膜上有孔道,贯穿膜内外; E、药物可以通过脂质、蛋白质或孔道而进行转运。
pKa
[HA] lg [A-]
pKa
弱酸(碱)在不同pH值时分子态、离子态所占的比例
pKa– pH 3 2 1 0 -1 -2 -3 [HA]%or[BH+]% 99.91 99.01 90.91 50.0 9.09 0.99 0.09 [A -]%or[B]% 0.09 0.99 9.09 50.0 90.91 99.01 99.91
1) 胃中pH为1~1.5,故多数弱酸性药物(pKa3 ~
7.5)在胃中以分子态存在,易于吸收。如阿司 匹林(pKa 3.5)为弱酸,在胃中99%以分子态 存在,故只在胃中吸收; 2) 肠道pH为7~8,故多数弱碱性药物(pKa7.5 ~ 10)在肠道吸收。如可待因( pKa 8.0),胃中 多以离子态存在而不吸收,只在肠道吸收; 3) 酸碱性很弱的药物或中性分子,在体内多以非离 子型存在,故易吸收而产生全身作用;而季铵碱 或两性药物,在胃肠道不易吸收,故一般作为胃 肠道用药或外用。
药剂相(Pharmaceutical Phase) 药物动力相( Pharmacokinetic Phase ) 药效相( Pharmacodynemic Phase )
1、药剂相:主要包括剂型的崩解,有效物质的释放或溶出。 给药剂量可被吸收的部分,是衡量药物利用度的尺度,它与 药物的剂型、配方、剂量和给药途径有关。 2、药代动力相:包括药物的吸收、分布、代谢和排泄。 给药剂量,进入总循环的部分,是衡量生物利用度的尺度。 3、药效相:包括在靶组织中药物与受体相互作用的过程, 由此产生的生化和生物物理的变化,最终导致药物的效 应,即其生物活性。 生物利用度——药物到达靶组织的浓度. 该过程主要取决于药物的理化性质
第二章 药物的构效关系 药物化学 课件
第二章 药物的构效关系
第四节 药物其它特性对药效的影响
二、电子云密度对药效的影响
各种元素的原子核对其核外电子的吸引力各不相同而显示 电负性的差异。由电负性不同的原子组成的化合物分子就存在 电子密度分布不均匀状态。药物分子的电子密度分布如果和酶 蛋白分子的电荷分布恰好相反,则有利于相互作用而结合,形 成复合物。
化学工业出版社
第二章 药物的构效关系
第一节 药物的基本结构和药效的关系
药物作用过程的三个阶段
过程分类 发生过程 研究目的
药剂相
药物的释放
优化处方和 给药途径
药物动力学
药效相
吸收、分布和消除 药物-受体在靶 (代谢及排泄) 组织的相互作用
优化生物利用度
优化所需的 生物效应
化学工业出版社
化学工业出版社
P=CO/CW
化学工业出版社
第二章 药物的构效关系
第二节 药物的理化性质和药效的关系
二、药物的解离度对药效的影响 多数药物为弱酸、弱碱及其盐类,体液中部分解离,
以离子型和非离子型(分子型)同时存在。药物常以分子型 通过生物膜,在膜内的水介质中解离成离子型,再起作用。 因此药物需有适宜的解离度。
胃肠道各部分的pH不同,不同pKa药物在胃肠道各部分 的吸收情况也就有差异。
化学工业出版社
第二章 药物的构效关系
第一节 药物的基本结构和药效的关系
三、药物的特异结构与非特异结构 (一)结构非特异性药物
药物活性主要取决于药物分子的各种理化性质,与化学结 构的关系不大。临床应用的非特异性药物较少,主要有全身吸 入麻醉药,酚类和长链季铵盐的杀菌药以及巴比妥的催眠药等。 (二)结构特异性药物
第3章 药物设计的基本原理和方法(2)
药物化学中的生物电子等排原理 法,称为药物化学中的生物电子等排原理。
生物电子等排原理为设计新药提供了一条相当有实用 价值的研究途径,在新药研究中占有重要地位, 价值的研究途径,在新药研究中占有重要地位,尤其 适合我国制药工业中现有的实际情况。 适合我国制药工业中现有的实际情况。
2011-12-14 11
1925年Grimm综合了 年 综合了Hinsberg和Hiickel的等价部分概念并加以 综合了 和 的等价部分概念并加以 扩大将电子等排体的理论广义化: 扩大将电子等排体的理论广义化:凡分子或原子团具有 相同数目的价电子,都称为电子等排体 如一CH3、一OH和一 电子等排体。 相同数目的价电子,都称为电子等排体。如一 和一 NH2,- ,-CH2一和-O-互为电于等排体。 一和- -互为电于等排体。 年提出“ 于1925年提出“氢化物替代规律” (hydride displacement 年提出 氢化物替代规律” law),指出自元素周期表第Ⅳ主族起,任何一种元素与一个或 ,指出自元素周期表第Ⅳ主族起, 几个氢原于结合形成的分子或基团, 几个氢原于结合形成的分子或基团,可以称之为假原于 (pseudoatom),同一元素与不同数目氢原子形成的假原子,具 ,同一元素与不同数目氢原子形成的假原子, 有不同的性质, 有不同的性质,但与一个氢原子结合形成的假原子的性质与比 它高一族的元素相似; 它高一族的元素相似;与两个氢原于结合形成的假原子性质与 2011-12-14 12 较其高两族的元素相似
2011-12-14
4
③合理药物设计
合理药物设计是指根据基因组学、 合理药物设计是指根据基因组学、蛋白质组学等确定的靶点结 构,基于药物与受体作用的所谓“锁—钥’模型,利用计算机 基于药物与受体作用的所谓“ 钥 模型, 进行药物设计的一种方法。 进行药物设计的一种方法。 合理药物设计由于设计目的明确, 合理药物设计由于设计目的明确,可以大大地减少筛选化合物 的数目,缩短药物研发周期和降低研发成本,因而成为目前寻 的数目,缩短药物研发周期和降低研发成本, 找新药的一种非常重要的手段。 找新药的一种非常重要的手段。
药物设计原理和方法PPT课件
.
19
➢适宜衍生化的功能基及衍生化物:
✓醇羟基或酚羟基可衍生成酯或活泼醚; ✓羧基可衍生成酯或酰胺; ✓氨基(包括胺、酰胺和亚胺)可衍生成烷氧 羰 ✓酰胺
.
20
➢2.4.1 提高生物利用度的前药
✓若原药分子中含有羟基、羧基或磷酸基,会因极 性强或带有电荷而难以吸收。应用前药原理引入 酯键可提高脂溶性,改善了脂/水分配系数,有利 肠道吸收,或克服了首过效应。
H3 C O H
+
H3 CN C H3
C O O-
H3 C N H2
+
H3 CN
C O O-
C H3
H3 C N H2 C O O-
H3 C
C H3
10
11 .
12 9
➢3.5 环和非环的电子等排体 ➢雌激素激动剂 ➢雌二醇的开环类似物已烯雌酚同样具有雌激素活性。为了 维持两个酚基和两个乙基在空间适宜的配置,以便与雌激素 受体结合,已烯雌酚分子中间的反式双键是非常重要的。
✓帕瑞昔布(Parecoxib)是戊地昔布(Valdecoxib) 的水溶性非活性前体药物,系非肠道给药的选择性 COX-2抑制剂,用于治疗手术后疼痛。
NH2SO2
CH3
O N
Na CH3CH2CO-N-SO2
CH3
O N
Valdecoxib .
Valdecoxib 29
➢2.4.2.5 UR-14048
OH
HO
HO
.
HO
11
§2.4 前 药 原 理
.
12
➢前药(Prodrug):是指体外活性较 小或无活性,在体内经酶促或非酶化 学反应,释放出活性物质而发挥药理 作用的化合物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
先导化合物的优化可分为:
(1)传统的化学方法 (2)现代的方法: 包括CADD和3DQSAR(在第三章介绍)
先导化合物优化的一般方法
一、烷基链或环的结构改造
二、生物电子等排原理
三、前药原理
四、软药
五、硬药
六、孪药
七、定量构效关系研究
一、烷基链或环的结构改造
1、成环或开环 镇痛药吗啡→哌替啶
N
4 -苯 基 哌 啶 类
2、插烯原理(vinylogues)(烷基链局部减少双键 或引入双键) 抗癫痫药胡椒碱→桂皮酰胺类的衍生物
O O 胡 椒 碱 N O 桂 皮 酰 胺 衍 生 物 X O N H R
3、烃链的同系化原理 利福平(甲基哌嗪)→利福喷汀(环戊基哌嗪)
二、生物电子等排(Bioisosteris)
安西他滨(环胞苷)是阿糖胞苷的中间体, 后发现安西他滨不仅具有抗肿瘤作用,且 副作用轻,体内代谢比阿糖胞苷慢,故作 用时间长,治疗各种白血病。
C H O H O O H O H C H O H 2 H O N O O N N H
.H C l
. O N H 3H 2
N H 2 N H O O N O H O
发展为理想的药物,这一过程称为先导化
合物的优化。
第一节
先导化合物发现的方法和途径
Approaches for lead discovery
一、从天然药物的活性成分中获得
(From Active Component of Natural Resources)
(1)植物来源 p17
青蒿素:来自于中药黄花蒿,以此为先导 物,发现蒿甲醚、青蒿琥酯等; 紫杉醇:来自于红豆杉树皮,以此为先导 物,发现多西他赛等。
(2)ADMET(吸收-分布-代谢-排泄-毒性)
(3)毒性筛选
(4)结构新颖性筛选
(5)与受体对接的研究
八、通过组合化学获得 (By Combinatorial Chemistry)
组合化学是利用一些基本的小分子单元如氨基 酸、单核苷酸、单糖及各种各样的有机小分子 化合物通过化学或生物合成的方法,系统地反 复以共价键装配成不同的组合,构建具有结构 多样性的化合物库。 组合化学能高效、大批量合成化合物,配合高 通量筛选,大大地加速了先导化合物发现的速 度。
CH3 H
N
CH3 H
破 D环 吗啡喃类 破 C环 N 破 C ,D 环 N E B A HO D O 吗啡 破 B ,C ,D 环 CH3 H C OH 苯并吗喃类 破 B环 N CH3 CH3
结构修饰 HO 左啡诺 CH3 N CH3 CH3 CH3 HO 喷他佐辛
结构修饰
O O CH3
结构修饰 N CH3 哌替啶
先导化合物的发现(lead discovery) 先导化合物的优化(lead optimization) 先导化合物(lead compound)简称先导物, 又称原型物,是通过各种途径得到的具有 一定生理活性的化学物质。
p16
由于先导化合物存在着某些缺陷,如活性 不够高,化学结构不稳定,毒性较大,选 择性不好,药代动力学性质不合理等等, 需要对其进行化学修饰,进一步优化使之
人类基因组学与新药设计
1990年10月1日,被誉为生命“登月计划” 的国际人类基因组计划(human genome
program)正式启动,我国作为六个参与国
家之一,承担了1%的测序任务。
人类基因组计划旨在阐明人类基因组30亿个
碱基对的序列,发现所有人类基因并搞清其在
染色体上的位置,破译人类全部遗传信息。
.H C l
M e O H
H O
H O
D 阿 拉 伯 糖
安 西 他 滨
阿 糖 胞 苷
七、通过计算机辅助药物筛选寻找 (By Virtual Screening)
利用计算机对虚拟化合物库进行筛选有可能发现 先导化合物。包括:
(1)类药筛选
类药五规则(Role of five, Lipinski规则) 是指如果一个化合物违背了下列规则中的任意两 条就很难被生物体吸收:分子量在500以下,分 配系数ClogP值小于5,氢键的给体不超过5个, 氢键的接受体不超过10个。
组胺:体内生物活性物质,以此为先导物,发 现了替丁类抗溃疡药物。
N N H
C H C H N H 2 2 2
N N H
N C N C H S C H C H N HCN H C H 2 2 2 3 C H 3 西 咪 替 丁
组 胺
三、通过随机机遇发现 (From Accidentally discover )
第二章 药物设计的基本原理和方法
(Basic Principles of Drug Design )
第二章 药物设计的基本原理和方法
p16
* 早期寻找新药的方法多是基于经验和尝 试,通过大量化合物的筛选与偶然发现。 * 随着生命科学的相关学科在上世纪后半 期的迅速发展,定量构效关系、合理药物 设计、计算机辅助药物设计、组合化学、 高通量筛选等新技术、新方法不断涌现, 新药设计学也应运而生。
7 6
H3C
美伐他汀
洛伐他汀
(3)动物来源
九肽替普罗肽(谷-色-脯-精-脯-谷-亮-脯脯):来源于巴西毒蛇,以此为先导物,发 现了ACE抑制剂卡托普利,开创了一类新 的影响重大的抗高血压药物。
CH3 HS O N COOH
卡托普利
(4)海洋生物来源
海葵毒素:来自于海葵,为肽类毒素,具 有强心作用,以此为先导物,得到一些重 组蛋白。
青蒿素系列
C H H 3 H C 3 O OO H O H C 3 H H C H 3 C H 3 H O OO H O H C 3 C H 3 H O OO H O
O 青 蒿 素
H H C H 3 O C H 3
H H C H 3 O C O C H C H C O O H 2 2 青 蒿 琥 酯
C O N H N H 2 C H 3 C O N H N HCC H 3 H
N
N
异 烟 肼 ( 抗 结 核 药 ) 可 抑 制 单 胺 氧 化 酶
异 丙 烟 肼 ( 抗 抑 郁 药 ) 单 胺 氧 化 酶 抑 制 剂 类 抗 抑 郁 药
又如: 安定作用,SAR研究,发现了抗精神病
药物氯丙嗪。
(1)异丙嗪是抗过敏药,但有严重的镇静和
蒿 甲 醚
(2)微生物来源
p18
美伐他汀和洛伐他汀,来源于青霉菌属、红
曲霉菌和土曲霉菌,羟甲戊二酰辅酶A还原
酶抑制剂的 Lead Compound 。后开发了 人工合成的阿托伐他汀(No. 1)
HO O H3C H3C H O H O H CH3 H3C H3C
3
O O O
1
HO O H H
O
CH3
一价 二价 三价 四价 环内等排体
F,OH,NH2,CH3 Cl, SH,PH2 Br I
-O-S-Se-Te-
-N= -P= -As= -Sb= -CH=
=C= =N+= =P+= =As+= =Sb+=
-CH=CH-S-O-NH-
非经典的电子等排体范围较广,包括
(1)能产生相似或相拮抗生理作用的生物电 子等排体; (2)疏水性、电性和空间效应等重要参数类 似的电子等排体。
药物在体内经过生物转化后,有些药物代谢
产物降低或失去了活性,称为代谢失活;有
些药物的代谢产物正好相反,可能使活性升
高,称为代谢活化。代谢活化得到的药物代
谢产物,可直接作为药物使用,也可作为先
导化合物,进行进一步的结构修饰和优化。
例如百浪多息→磺胺,再以磺胺为先导物开发 了大量的磺胺类药物。
百浪多息 体外无抗 菌活性 体内抑制 葡萄球菌
(2)西地那非---一种磷酸二酯酶抑制剂类药物。
最初作为心血管疾病药物投入临床,后来发现它 的副作用大于治疗作用,于是决定终止临床试验、 收回药品。结果,不少试用者拒绝交回,……
六、从药物合成的中间体中发现 (From Synthetic Intermediates)
一些药物合成的中间体,由于与目的化 合物结构上有相似性,经过筛选也可发 现先导化合物。 阿糖胞苷 →环胞苷(中间体)
九、其他新发展的方法 (New Methods for drug discover )
1、反义寡核苷酸 2、综合技术平台
第二节 先导化合物的优化 (Lead Optimization)
p25
对先导化合物进行合理的结构修饰,这 种过程和方法称为先导化合物的优化 (Lead Optimization)。
药物靶标
据估计人类基因组计划为我们提供了大量
潜在的蛋白质药物靶标,其中至少有1万个
可以作为寻找新药的靶标。
药物设计方法
p16
以受体为靶点,可分别设计受体的激动 剂和拮抗剂 以酶为靶点,设计酶抑制剂
以离子通道为靶点,则可分别设计钠、 钾和钙离子通道的激活剂(开放剂)或 阻断剂(拮抗剂)。
药物设计的两个阶段(p16)
1、 1929年青霉素的发现
2、异丙肾上腺素:β -受体激动剂,结构改造, 发现β -受体阻断剂--普萘洛尔,
C H 3 O H H O H O H N C H 构 改 造 3 结 O O H N H C H 3
C H 3
异 丙 肾 上 腺 素
普 萘 洛 尔
四、从药物代谢产物中发现 ( From Metabolites )
N CN , CN C
CH N
羧基: COO H